Synthesis, Preliminary Bioevaluation and Computational Analysis of Caffeic Acid Analogues

Abstract
:1. Introduction
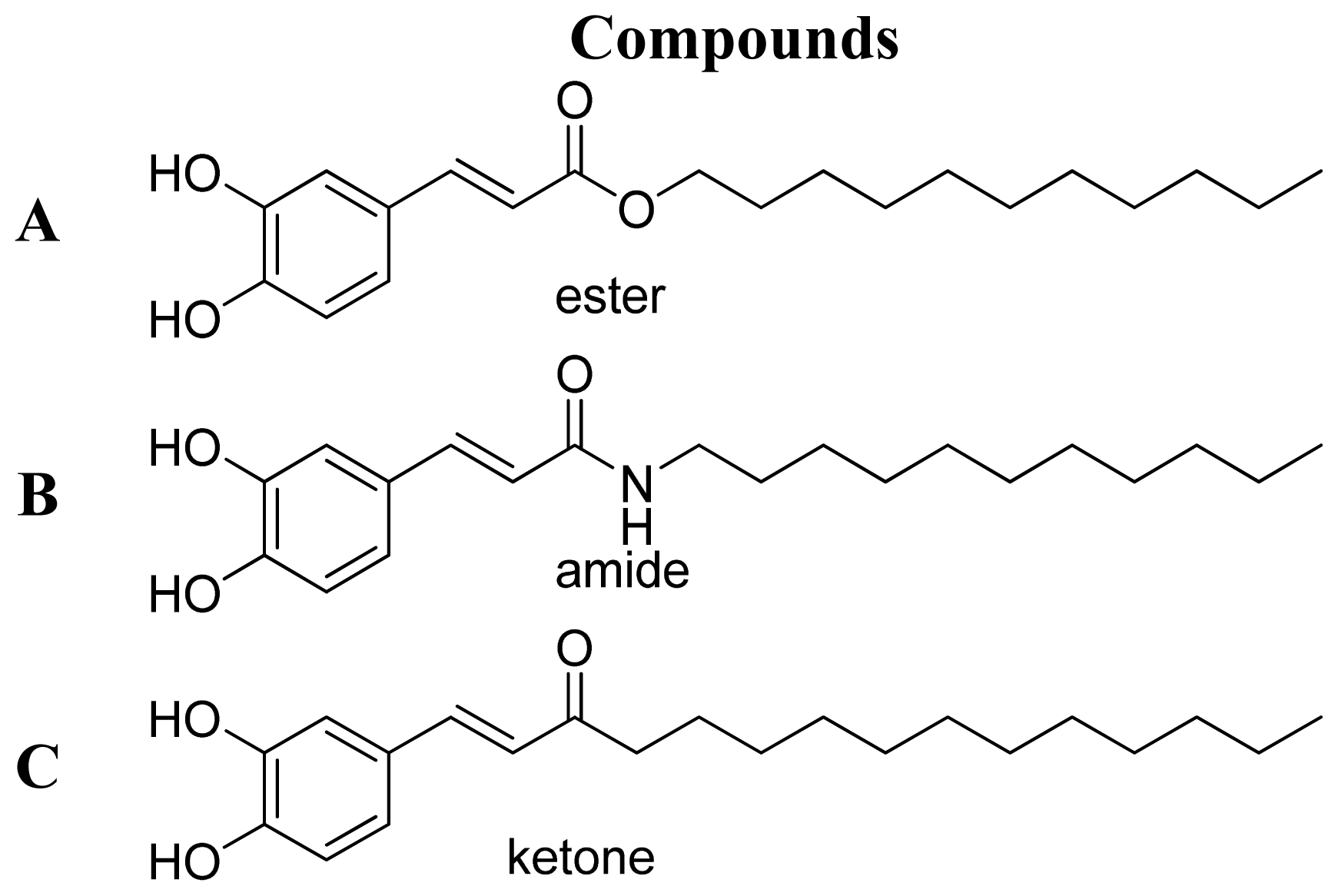
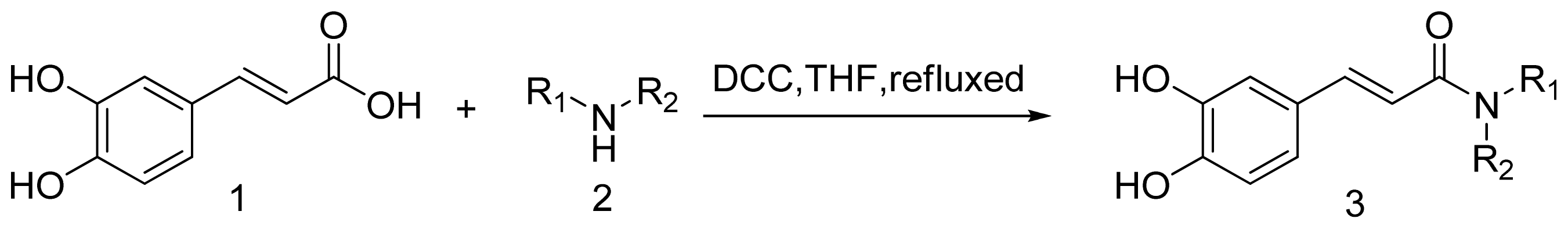
2. Results and Discussion
2.1. Biological Studies
2.2. Pharmacophore Model
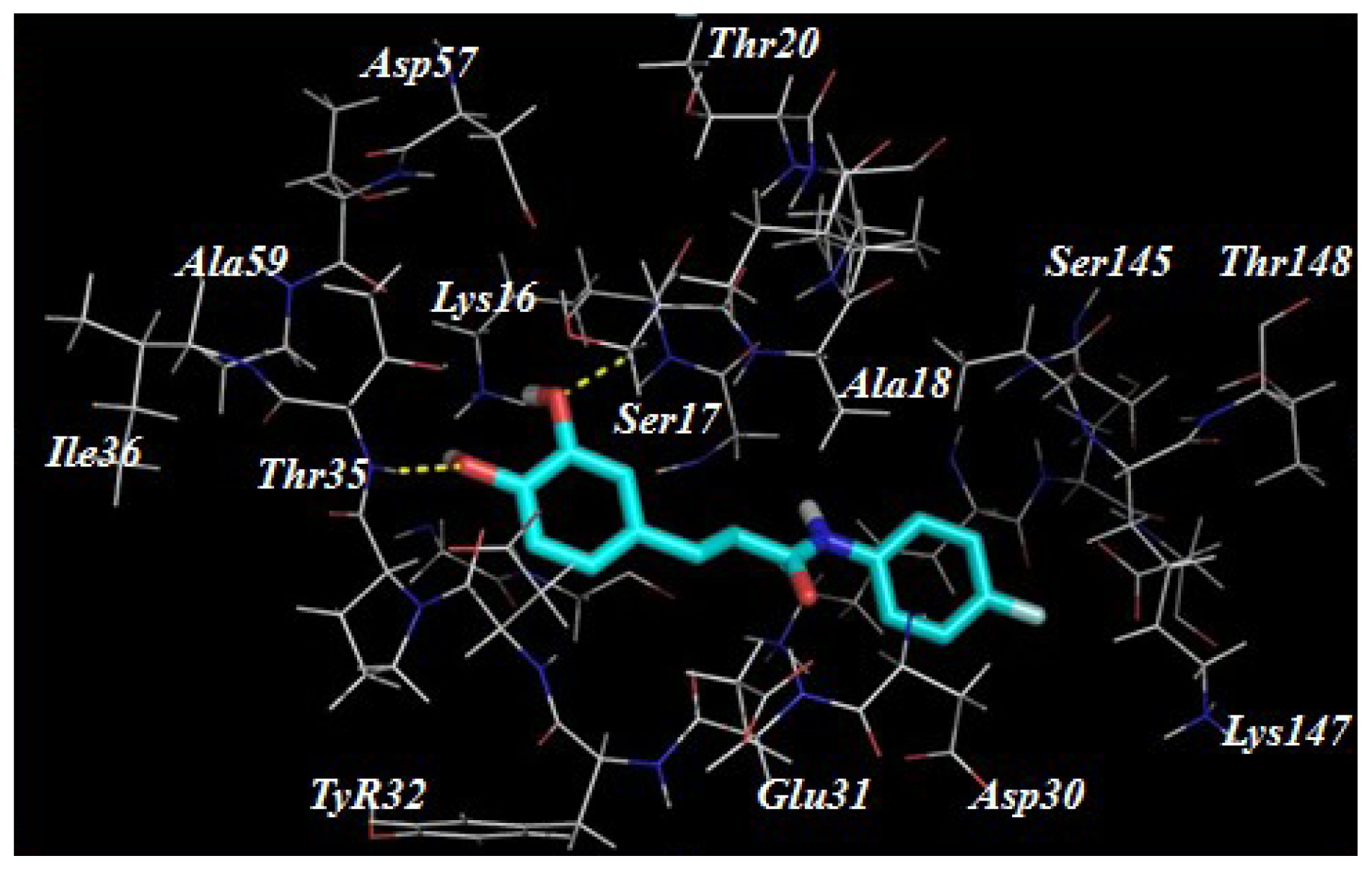
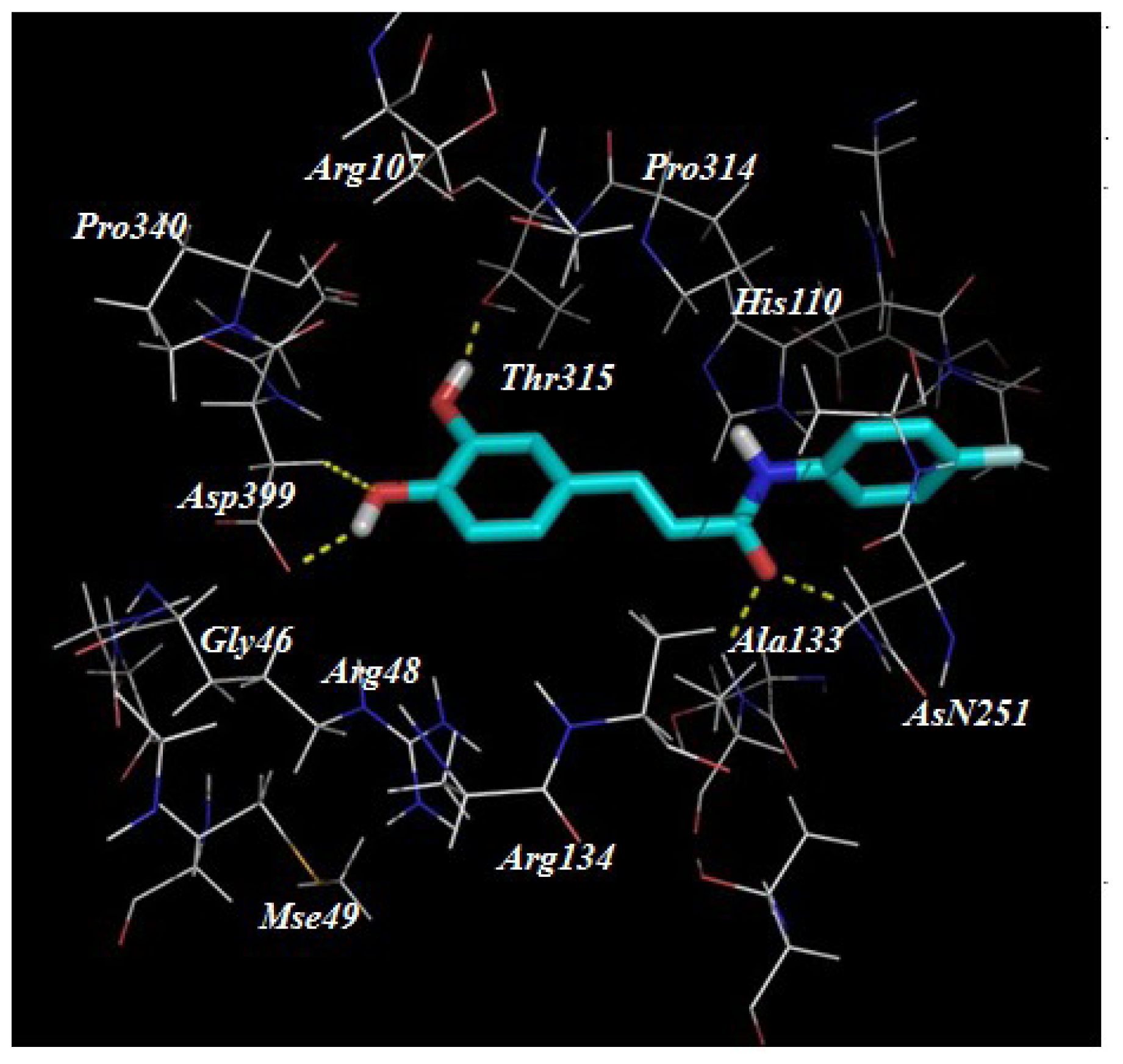
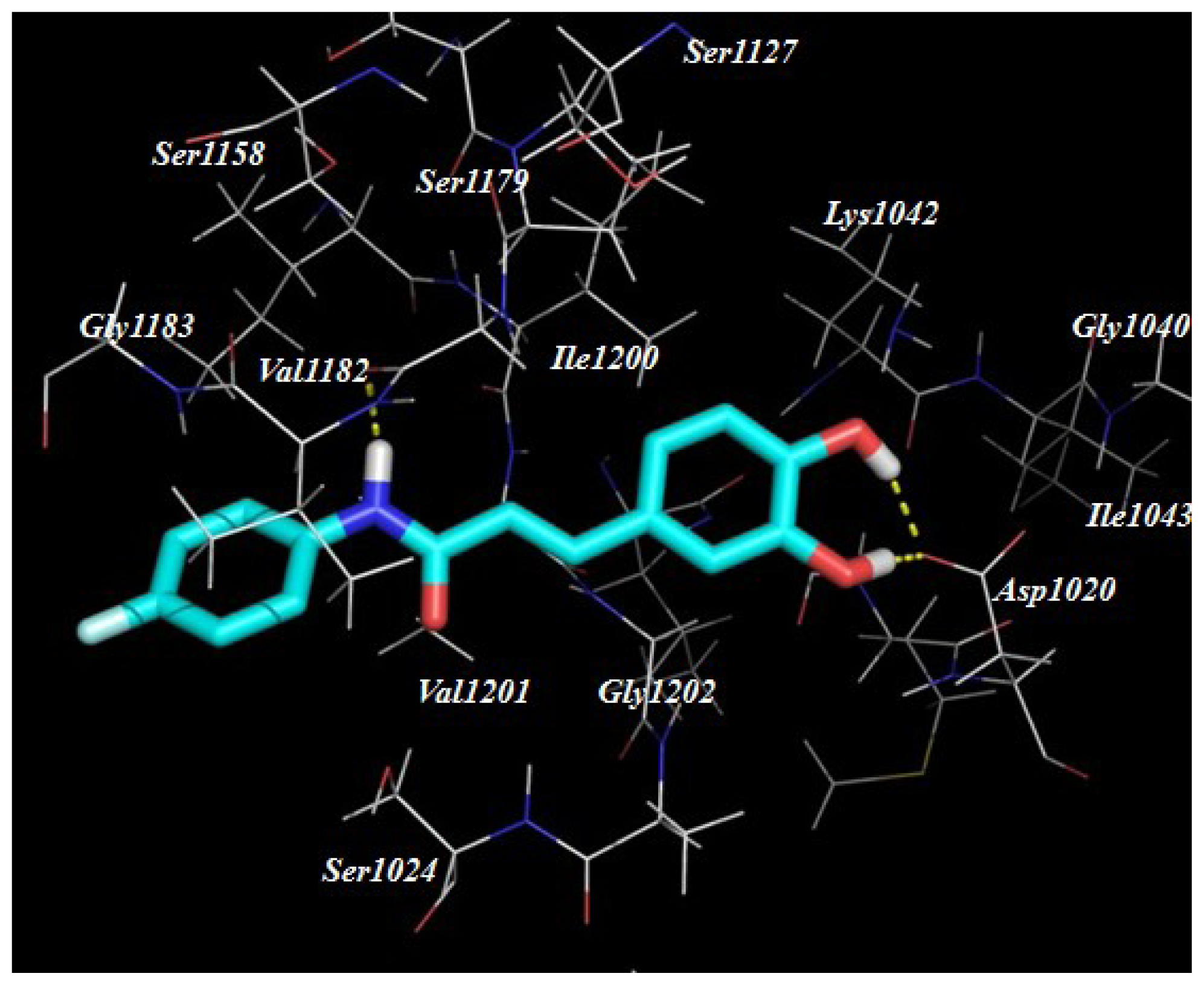
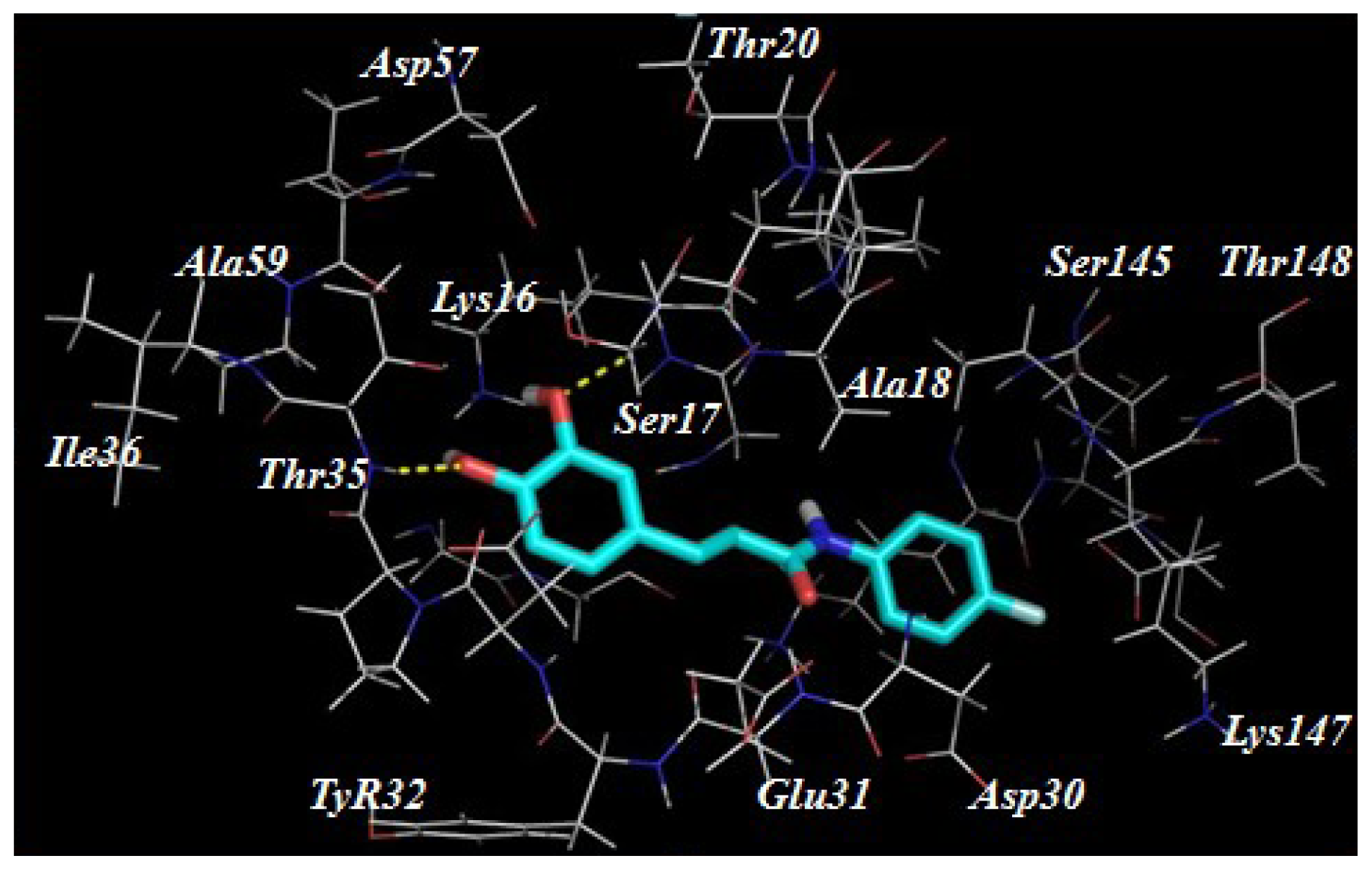
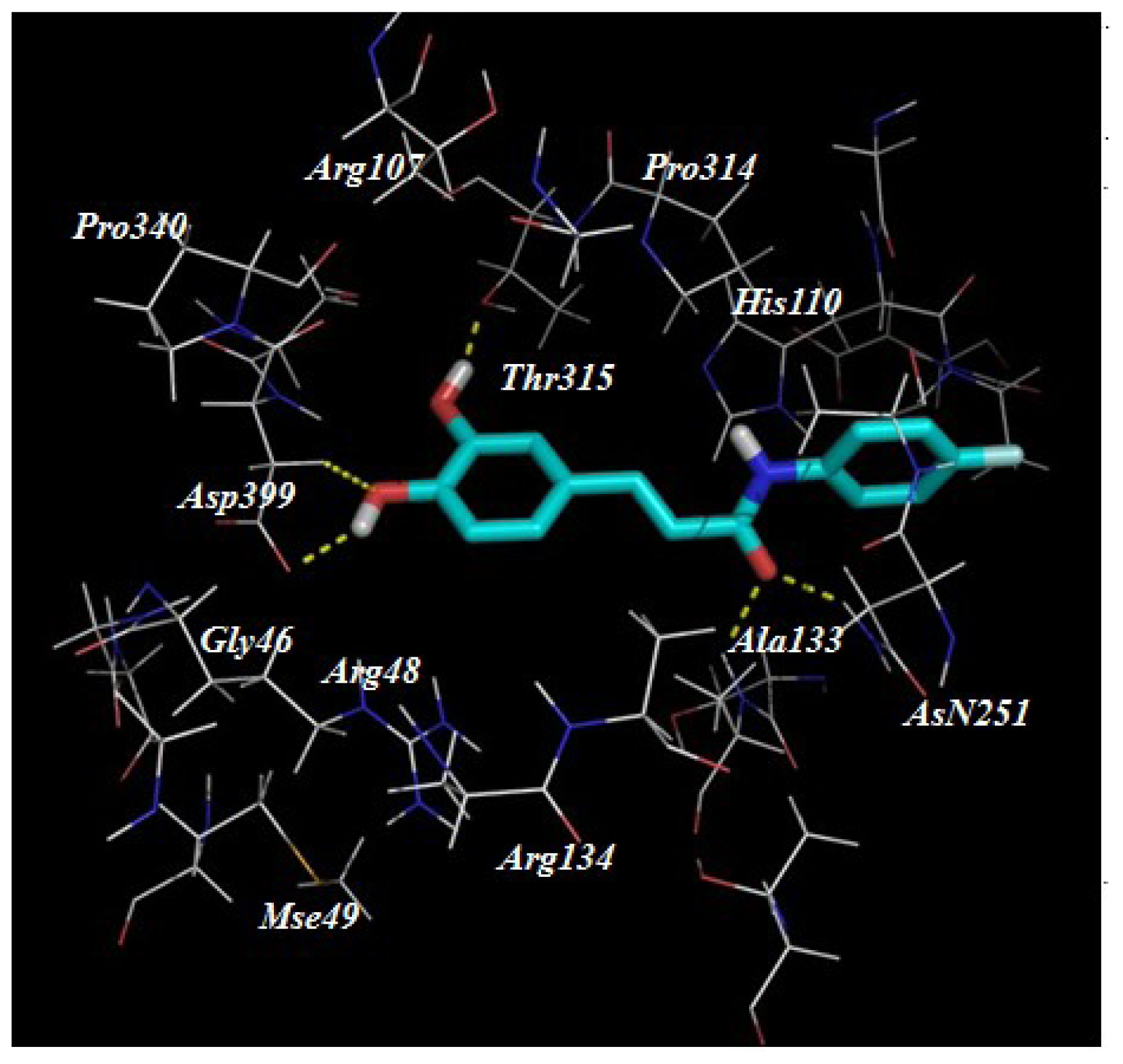
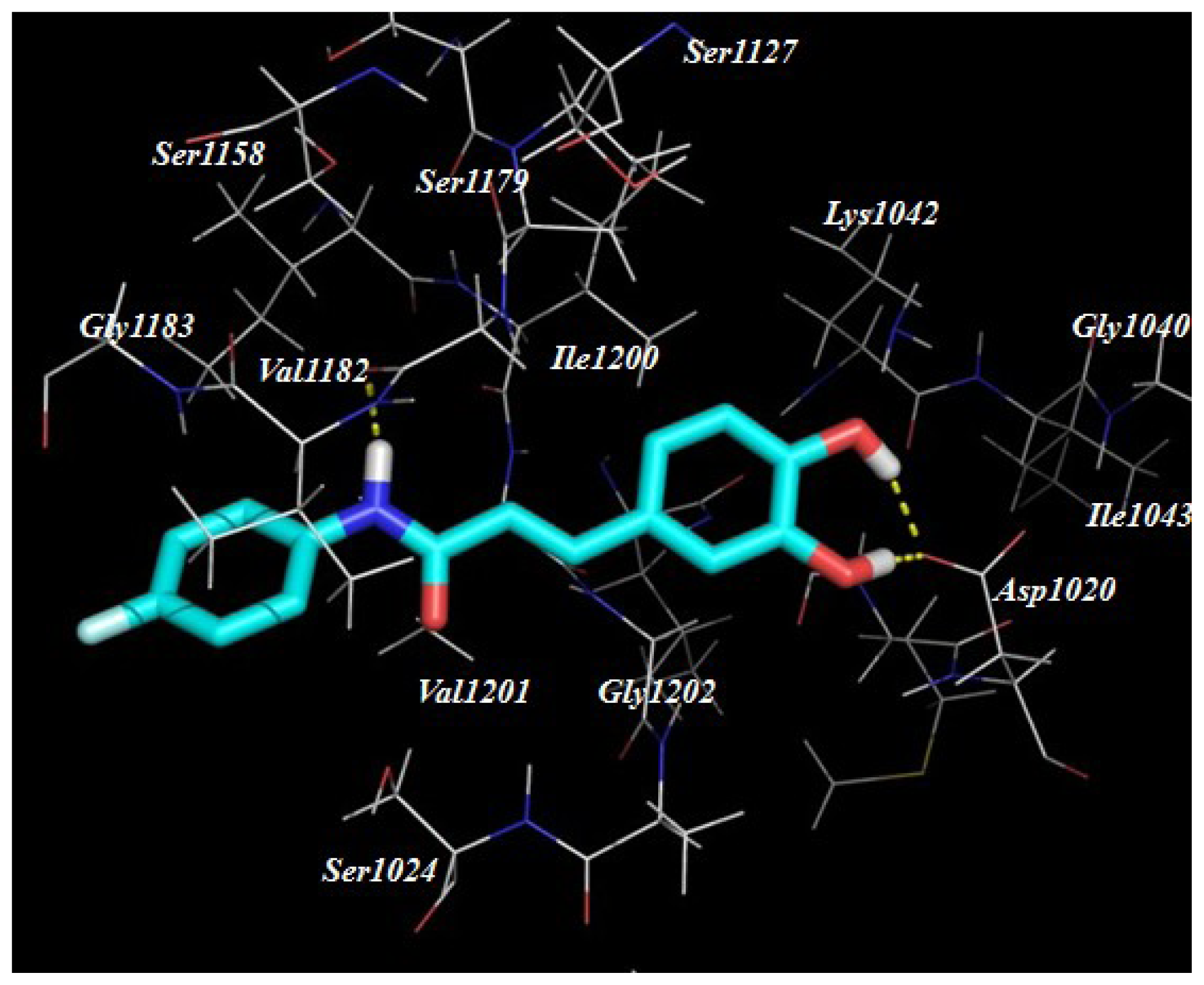
2.3. Target Predication and Molecular Docking
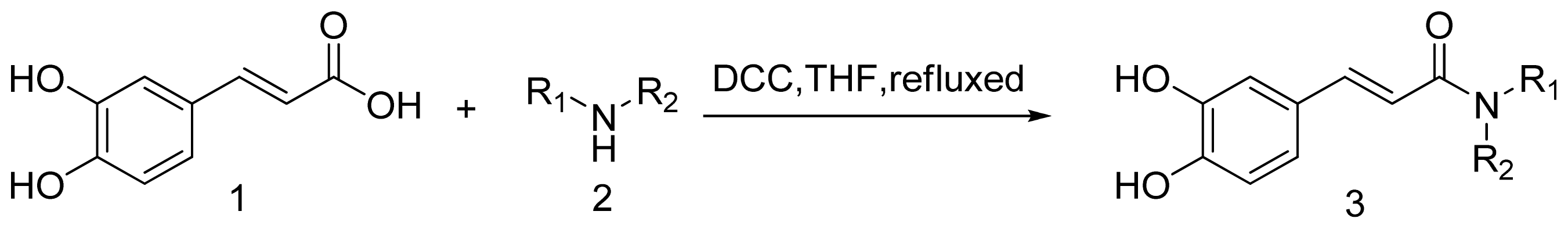
3. Experimental Section
3.1. Chemistry
3.1.1. General Procedure for the Preparation of Amine (3a–3t)
3.2. Biology
3.2.1. Cell Culture
3.2.2. Measurement of Nitric Oxide
3.3. Computational Protocols
3.3.1. Pharmacophore Generation
3.3.2. Molecular Docking Study
4. Conclusions
Supplementary Information
ijms-15-08808-s001.pdfAcknowledgments
Conflicts of Interest
- Author ContributionsZhiqian Liu: acquisition of data; analysis and interpretation of data; and drafting of the manuscript; Jianjun Fu, Lei Shan and Qingyan Sun funding obtained and study supervision. All authors read and approved the final manuscript. Weidong Zhang administrative support; and study supervision; and review of the manuscript.
References
- Hu, J.; Wang, Y.; Wei, X.; Wu, X.; Chen, G.; Cao, G.; Shen, X.; Zhang, X.; Tang, Q.; Liang, G.; et al. Synthesis and biological evaluation of novel thiazolidinone derivatives as potential anti-inflammatory agents. Eur. J. Med. Chem 2013, 64, 292–301. [Google Scholar]
- Uwai, K.; Osanai, Y.; Imaizumi, T.; Kanno, S.; Takeshita, M.; Ishikawa, M. Inhibitory effect of the alkyl side chain of caffeic acid analogues on lipopolysaccharide-induced nitric oxide production in RAW264.7 macrophages. Bioorg. Med. Chem 2008, 16, 7795–7803. [Google Scholar]
- O’Sullivan, L.A.; Liongue, C.; Lewis, R.S.; Stephenson, S.E.; Ward, A.C. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol. Immunol 2007, 44, 2497–2506. [Google Scholar]
- Jacques, C.; Gosset, M.; Berenbaum, F. The role of IL-1 and IL-1Ra in joint inflammation and cartilage degradation. Vitam. Horm 2006, 74, 371–403. [Google Scholar]
- Reber, L.; da Silva, C.A.; Frossard, N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur. J. Pharmacol 2006, 533, 327–340. [Google Scholar]
- Flaster, H.; Bernhagen, J.; Calandra, T.; Bucala, R. The macrophage migration inhibitory factor-glucocorticoid dyad: Regulation of inflammation and immunity. Mol. Endocrinol 2007, 21, 1267–1280. [Google Scholar]
- Dalli, J.; Norling, L.V.; Renshaw, D.; Cooper, D.; Leung, K.Y.; Perretti, M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood 2008, 112, 2512–2519. [Google Scholar]
- De Lucca, G.V. Recent developments in CCR3 antagonists. Curr. Opin. Drug Discov. Devel 2006, 9, 516–524. [Google Scholar]
- Chen, J.F.; Pedata, F. Modulation of ischemic brain injury and neuroin-flammation by adenosine A2A receptors. Curr. Pharm. Des 2008, 14, 1490–1499. [Google Scholar]
- Luger, T.A.; Brzoska, T. Alpha-MSH related peptides: A new class of anti-inflammatory and immunomodulating drugs. Ann. Rheum. Dis 2007, 66, 52–55. [Google Scholar]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol 1998, 16, 225–260. [Google Scholar]
- Li, L.; Zeng, H.W.; Liu, F.; Zhang, J.G.; Yue, R.C.; Lu, W.Q.; Yuan, X.; Dai, W.X.; Yuan, H.; Sun, Q.Y.; et al. Target identification and validation of (+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate, An anti-inflammatory natural product. Eur. J. Inflamm 2012, 10, 297–309. [Google Scholar]
- Santos, S.A.; Freire, C.S.; Domingues, M.R.; Silvestre, A.J.; Pascoal, N.C. Characterization of phenolic components in polar extracts of Eucalyptus globulus Labill bark by high-performance liquid chromatography-mass spectrometry. J. Agric. Food Chem 2011, 59, 9386–9393. [Google Scholar]
- Choudhary, M.I.; Naheed, N.; Abbaskhan, A.; Musharraf, S.G.; Siddiqui, H. Atta-Ur-Rahman. Phenolic and other constituents of fresh water fern Salvinia molesta. Phytochemistry 2008, 69, 1018–1023. [Google Scholar]
- Lee, Y.S.; Kang, Y.H.; Jung, J.Y.; Lee, S.; Ohuchi, K.; Shin, KH.; Kang, I.J.; Park, J.H.; Shin, H.K.; Lim, S.S. Protein Glycation Inhibitors from the Fruiting Body of Phellinus linteus. Biol. Pharm. Bull 2008, 31, 1968–1972. [Google Scholar]
- Nagaoka, T.; Banskota, A.H.; Tezuka, Y.; Midorikawa, K. Caffeic acid phenethyl ester (CAPE) analogues: Potent nitric oxide inhibitors from the Netherlands propolis. Biol. Pharm. Bull 2003, 26, 487–491. [Google Scholar]
- Sherif, Y.E.; Fu, J.; Lotfy, M.; Zhu, H.L. QSAR study for newly caffeic acid amides with prominent antibacterial and antifungal activity. Der Pharma Chem 2010, 2, 105. [Google Scholar]
- Fu, J.; Cheng, K.; Zhang, Z.M.; Fang, R.Q.; Zhu, H.L. Synthesis, structure and structure–activity relationship analysis of caffeic acid amides as potential antimicrobials. Eur. J. Med. Chem 2010, 45, 2638–2643. [Google Scholar]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar]
- Nagarajan, S.; Choo, H.; Cho, Y.S.; Oh, K.S.; Lee, B.H.; Shin, K.J.; Pae, A.N. IKKbeta inhibitors identification part II: Ligand and structure-based virtual screening. Bioorg. Med. Chem 2010, 18, 3951–3960. [Google Scholar]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A Web server for potential drug target identification using pharmacophore mapping approach. Nucleic. Acids Res 2010, 38, 609–614. [Google Scholar]
- Hung, C.C.; Tsai, W.J.; Kuo, L.M.; Kuo, Y.H. Evaluation of caffeic acid amide analogues as anti-platelet aggregation and anti-oxidative agents. Bioorg. Med. Chem 2005, 13, 1791–1797. [Google Scholar]
- Pharmmapper. Available online: http://59.78.96.61/pharmmapper/index.php access on 23 June 2013.
- Zeng, H.; Liu, X.; Dou, S.; Xu, W.; Li, N.; Liu, X.; Zhang, W.; Hu, Z.; Liu, R. Huang-Lian-Jie-Du-Tang exerts anti-inflammatory effects in rats through inhibition of nitric oxide production and eicosanoid biosynthesis via the lipoxygenase pathway. J. Pharm. Pharmacol 2009, 61, 1699–707. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R1 | R2 | Nitric Oxide Inhibition/IC50 (μM) |
|---|---|---|---|
| f | n-butyl | H | 6.1 |
| 3b | cyclopropylmethanyl | H | >10 |
| 3c | –CH2)5– | –(CH2)5– | >10 |
| 3d | –(CH2)4– | –(CH2)4– | >10 |
| 3e | –(CH2)2– | –(CH2)2– | >10 |
| 3f | n-butyl | n-butyl | >10 |
| 3g | 3,5-bis(trifluoromethyl)phenyl | H | >10 |
| 3h | 3,5-difluorophenyl | H | 4.1 |
| 3i | 3-(trifluoromethyl)phenyl | H | 7.9 |
| 3j | 4-methoxyphenyl | H | 5.2 |
| 3k | 4-fluorophenyl | H | 3.7 |
| 3l | 2-(hydroxymethyl)phenyl | H | >10 |
| 3m | 2-acetylphenyl | H | >10 |
| 3n | 3-chlorophenyl | H | >10 |
| 3o | 3-bromophenyl | H | >10 |
| 3p | 4-methanylphenyl | H | >10 |
| 3q | 2-methanylphenyl | H | >10 |
| 3r | phenylmethanyl | H | >10 |
| 3s | 2-(1H-indol-3-yl)ethyl | H | 6.7 |
| 3t | 2-(benzo[d][1,3]dioxol-5-yl)ethyl | H | 5.0 |
| caffeic acid | - | - | 165 a |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Z.; Fu, J.; Shan, L.; Sun, Q.; Zhang, W. Synthesis, Preliminary Bioevaluation and Computational Analysis of Caffeic Acid Analogues. Int. J. Mol. Sci. 2014, 15, 8808-8820. https://doi.org/10.3390/ijms15058808
Liu Z, Fu J, Shan L, Sun Q, Zhang W. Synthesis, Preliminary Bioevaluation and Computational Analysis of Caffeic Acid Analogues. International Journal of Molecular Sciences. 2014; 15(5):8808-8820. https://doi.org/10.3390/ijms15058808
Chicago/Turabian StyleLiu, Zhiqian, Jianjun Fu, Lei Shan, Qingyan Sun, and Weidong Zhang. 2014. "Synthesis, Preliminary Bioevaluation and Computational Analysis of Caffeic Acid Analogues" International Journal of Molecular Sciences 15, no. 5: 8808-8820. https://doi.org/10.3390/ijms15058808