Molecular Recognition of Agonist and Antagonist for Peroxisome Proliferator-Activated Receptor-α Studied by Molecular Dynamics Simulations

Abstract
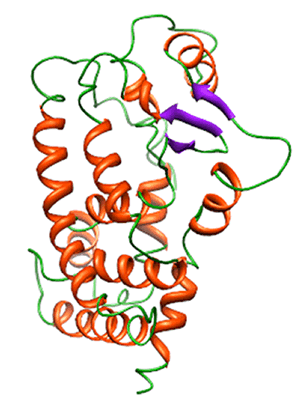
:
1. Introduction
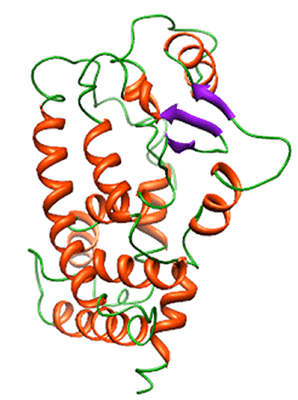
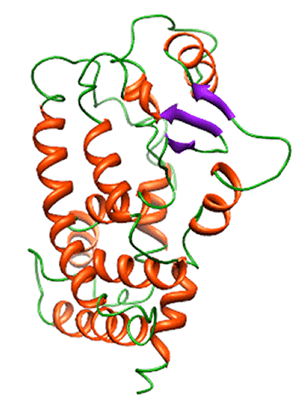
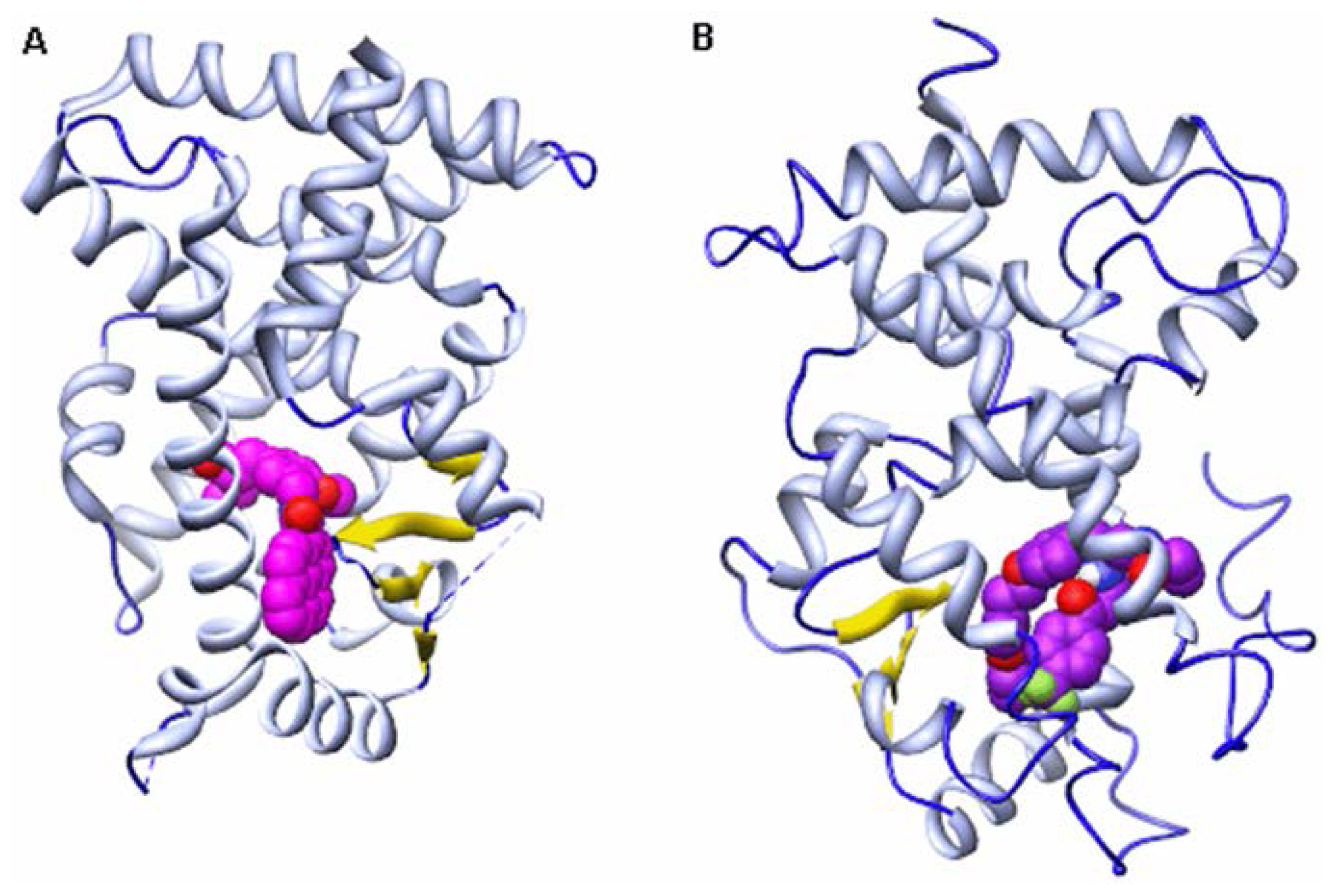
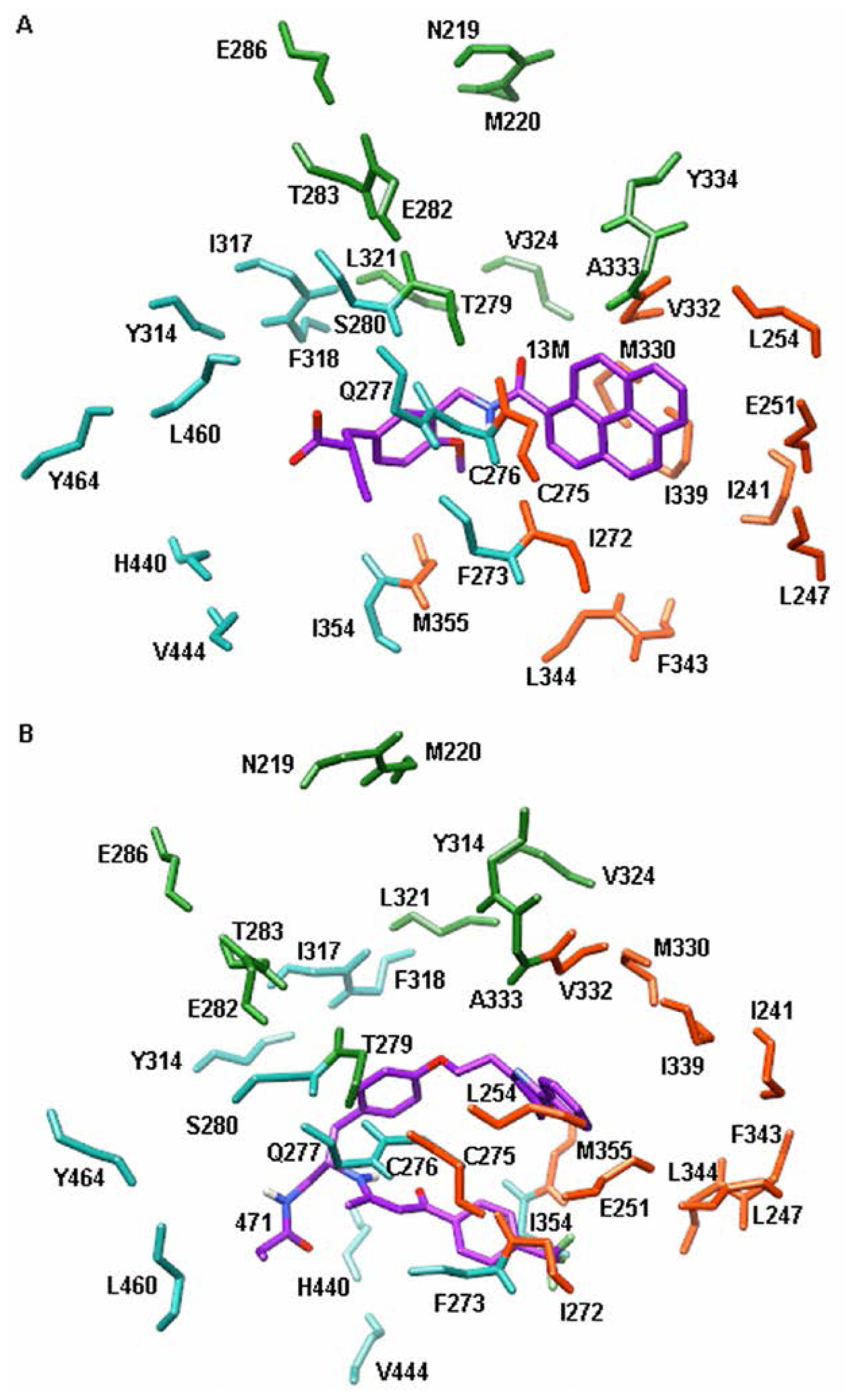
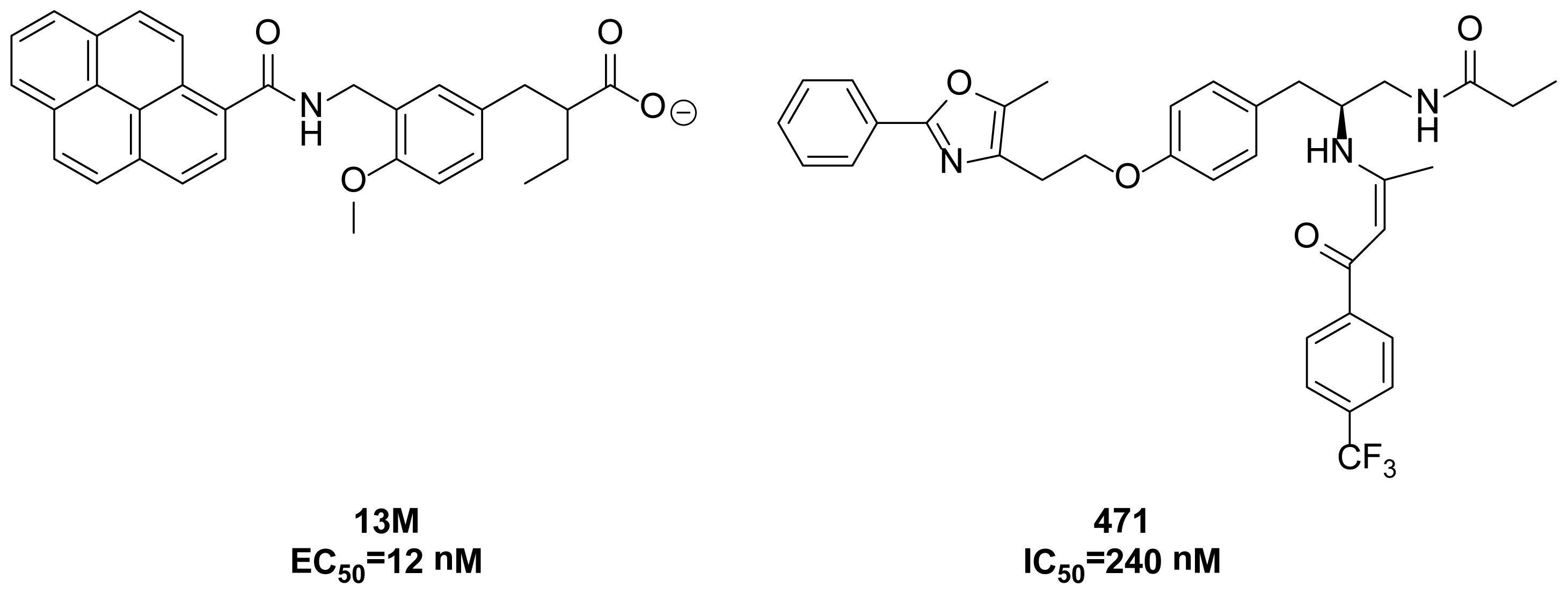
2. Results and Discussion
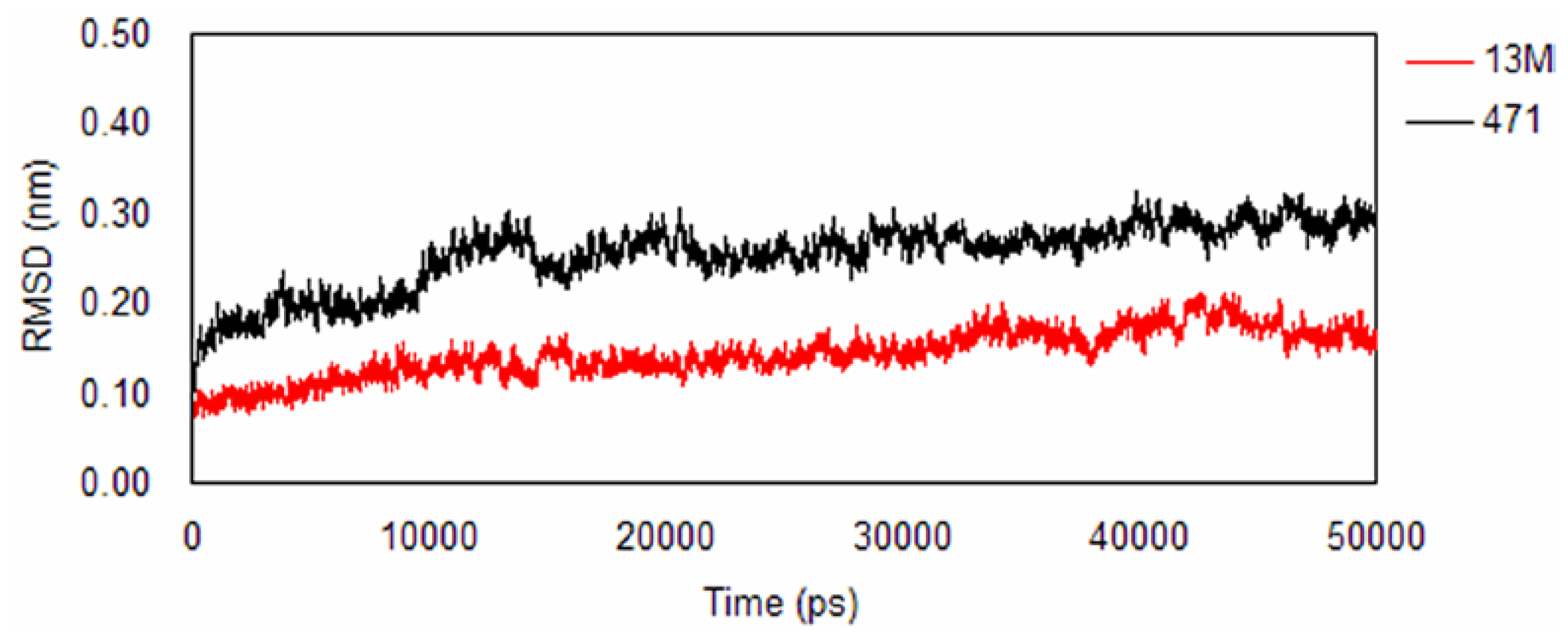
2.1. Backbone Stability
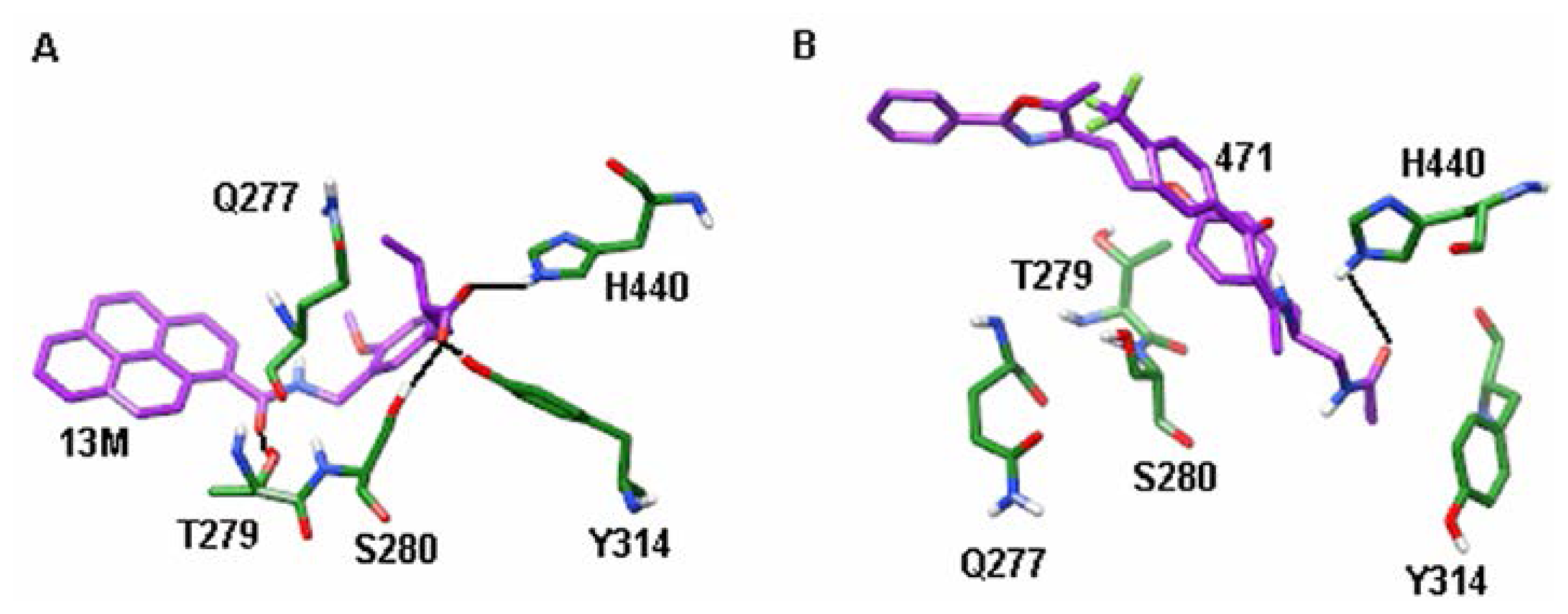
2.2. Hydrogen Bond Analysis
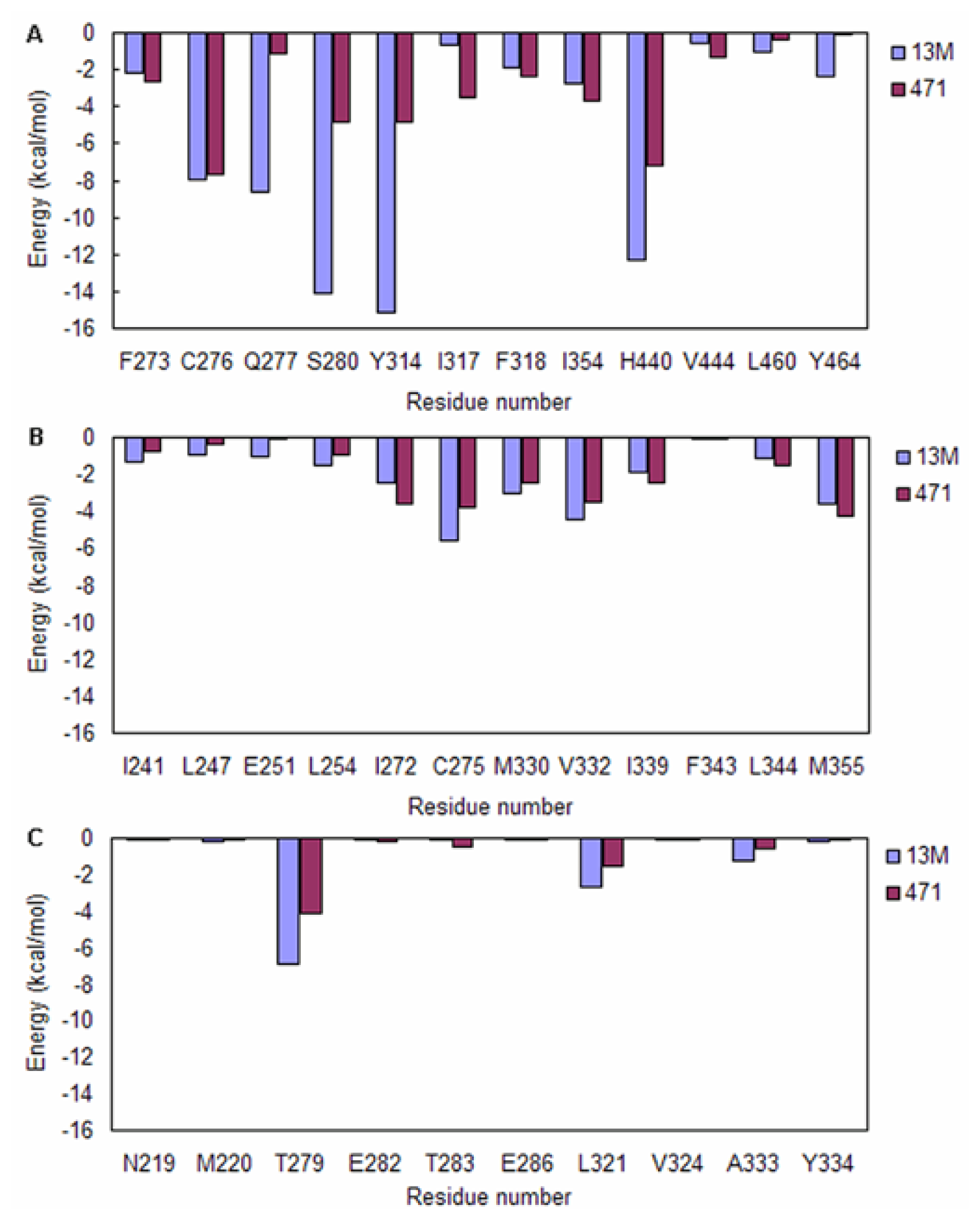
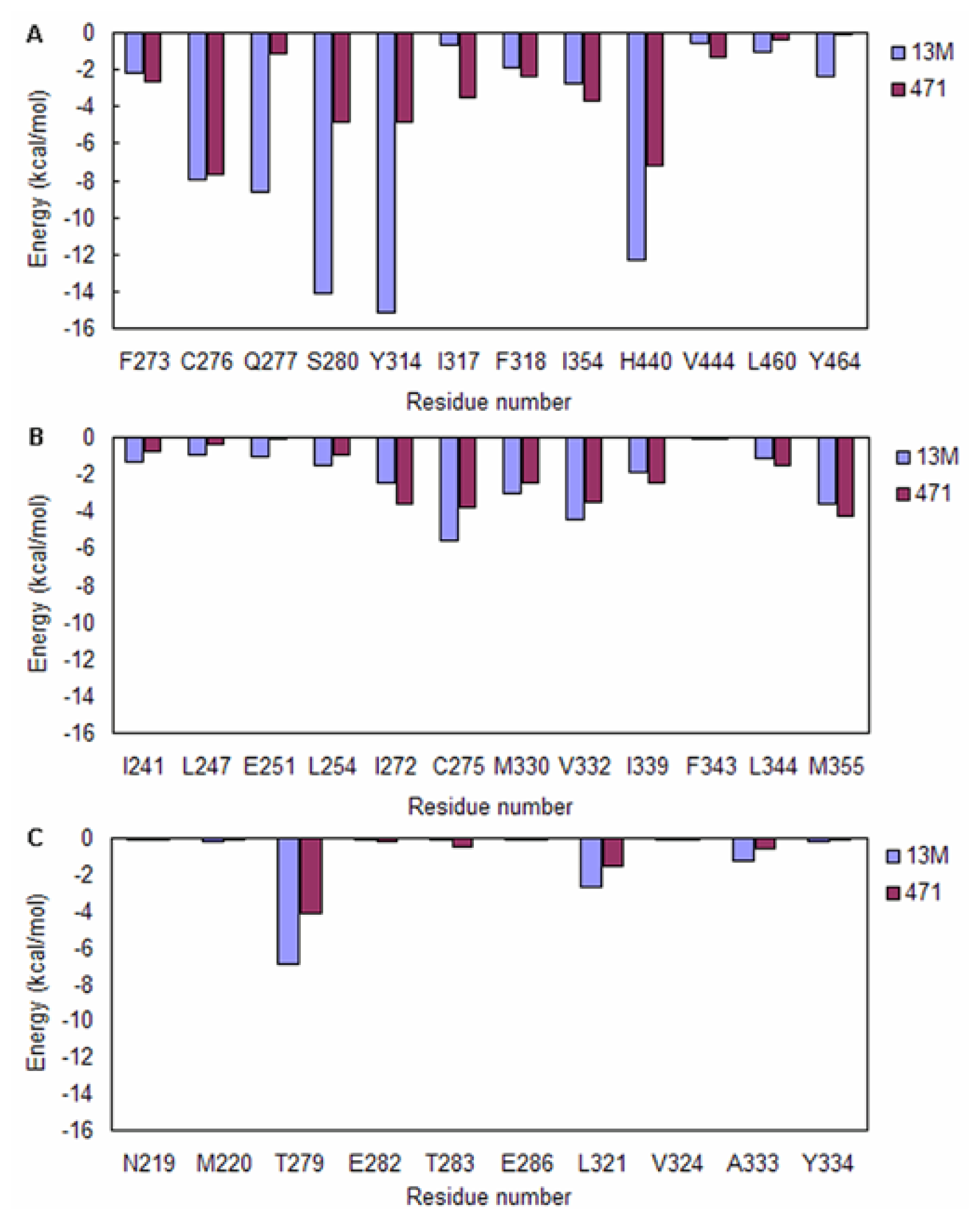
2.3. Energy Decomposition Analysis
3. Experimental Section
3.1. System Preparation
3.2. Molecular Dynamics Simulations
3.3. Hydrogen Bond Analysis
3.4. Energy Decomposition Analysis
4. Conclusions
Conflicts of Interest
- Author ContributionsMengyuan Liu wrote the manuscript and carried out molecular dynamics simulations. Lushan Wang, Xian Zhao and Xun Sun designed the experiments and revised manuscript.
References
- Ammazzalorso, A.; Giancristofaro, A.; D’Angelo, A.; Filippis, B.D.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R. Benzothiazole-based N-(phenylsulfonyl)amides as a novel family of PPARα antagonists. Bioorg. Med. Chem. Lett 2011, 21, 4869–4872. [Google Scholar]
- Dhoke, G.V.; Gangwal, R.P.; Sangamwar, A.T. A combined ligand and structure based approach to design potent PPAR-α agonists. J. Mol. Struct 2012, 1028, 22–30. [Google Scholar]
- Ricote, M.; Huang, J.T.; Welch, J.S.; Glass, C.K. The peroxisome proliferator-activated receptor (PPARγ) as a regulator of monocyte/macrophage function. J. Leukoc. Biol 1999, 66, 733–739. [Google Scholar]
- Jiang, X.; Lee, G.T.; Prasad, K.; Repič, O. A practical synthesis of a diazepinylbenzoic acid, a retinoid X receptor antagonist. Org. Process Res. Dev 2008, 12, 1137–1141. [Google Scholar]
- Pingali, H.; Jain, M.; Shah, S.; Makadia, P.; Zaware, P.; Goel, A.; Patel, M.; Giri, S.; Patel, H.; Patel, P. Design and synthesis of novel oxazole containing 1,3-dioxane-2-carboxylic acid derivatives as PPARα/γ dual agonists. Bioorg. Med. Chem 2008, 16, 7117–7127. [Google Scholar]
- Tsao, T.; Kornblau, S.; Safe, S.; Watt, J.C.; Ruvolo, V.; Chen, W.; Qiu, Y.; Coombes, K.R.; Ju, Z.; Abdelrahim, M.; et al. Role of peroxisome proliferator-activated receptor-γ and its coactivator DRIP205 in cellular responses to CDDO (RTA-401) in acute myelogenous leukemia. Cancer Res 2010, 70, 4949–4960. [Google Scholar]
- Julan, L.; Guan, H.; van Beek, J.P.; Yang, K. Peroxisome proliferator-activated receptor δ suppresses 11β-hydroxysteroid dehydrogenase type 2 gene expression in human placental trophoblast cells. Endocrinology 2005, 146, 1482–1490. [Google Scholar]
- Liu, X.-Y.; Wang, R.-L.; Xu, W.-R.; Tang, L.-D.; Wang, S.-Q.; Chou, K.-C. Docking and molecular dynamics simulations of peroxisome proliferator activated receptors interacting with pan agonist sodelglitazar. Protein Pept. Lett 2011, 18, 1021–1027. [Google Scholar]
- Han, H.O.; Kim, S.H.; Kim, K.-H.; Hur, G.-C.; Yim, H.J.; Chung, H.-K.; Woo, S.H.; Koo, K.D.; Lee, C.-S.; Koh, S.-J.; et al. Design and synthesis of oxime ethers of α-acyl-β-phenylpropanoic acids as PPAR dual agonists. Bioorg. Med. Chem. Lett 2007, 17, 937–941. [Google Scholar]
- Zhang, H.; Ryono, D.E.; Devasthale, P.; Wang, W.; O’Malley, K.; Farrelly, D.; Gu, L.; Harrity, T.; Cap, M.; Chu, C.; et al. Design, synthesis and structure-activity relationships of azole acids as novel, potent dual PPARα/γ agonists. Bioorg. Med. Chem. Lett 2009, 19, 1451–1456. [Google Scholar]
- Ichihara, S.; Obata, K.; Yamada, Y.; Nagata, K.; Noda, A.; Ichihara, G.; Yamada, A.; Kato, T.; Izawa, H.; Murohara, T.; et al. Attenuation of cardiac dysfunction by a PPAR-α agonist is associated with down-regulation of redox-regulated transcription factors. J. Mol. Cell. Cardiol 2006, 41, 318–329. [Google Scholar]
- Guerre-Millo, M.; Gervois, P.; Raspé, E.; Madsen, L.; Poulain, P.; Derudas, B.; Herbert, J.-M.; Winegar, D.A.; Willson, T.M.; Fruchart, J.-C.; et al. Peroxisome proliferator-activated receptor α activators improve insulin sensitivity and reduce adiposity. J. Biol. Chem 2000, 275, 16638–16642. [Google Scholar]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar]
- Cronet, P.; Petersen, J.F.W.; Folmer, R.; Blomberg, N.; Sjöblom, K.; Karlsson, U.; Lindstedt, E.-L.; Bamberg, K. Structure of the PPARα and γ ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure 2001, 9, 699–706. [Google Scholar]
- Li, J.; Kennedy, L.J.; Shi, Y.; Tao, S.; Ye, X.-Y.; Chen, S.Y.; Wang, Y.; Hernández, A.S.; Wang, W.; Devasthale, P.V.; et al. Discovery of an oxybenzylglycine based peroxisome proliferator activated receptor α selective agonist 2-((3-((2-(4-chlorophenyl)-5-methyloxazol-4-yl)methoxy)benzyl) (methoxycarbonyl)am ino)acetic acid (BMS-687453). J. Med. Chem 2010, 53, 2854–2864. [Google Scholar]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar]
- Sagar, D.R.; Kendall, D.A.; Chapman, V. Inhibition of fatty acid amide hydrolase produces PPAR-α-mediated analgesia in a rat model of inflammatory pain. Br. J. Pharmacol 2008, 155, 1297–1306. [Google Scholar]
- Cwinn, M.A.; Jones, S.P.; Kennedy, S.W. Exposure to perfluorooctane sulfonate or fenofibrate causes PPAR-α dependent transcriptional responses in chicken embryo hepatocytes. Comp. Biochem. Phys. C 2008, 148, 165–171. [Google Scholar]
- Donnelly, L.E.; Newton, R.; Kennedy, G.E.; Fenwick, P.S.; Leung, R.H.; Ito, K.; Russell, R.E.; Barnes, P.J. Anti-inflammatory effects of resveratrol in lung epithelial cells: Molecular mechanisms. Am. J. Physiol.-Lung C 2004, 287, L774–L783. [Google Scholar]
- Kuwabara, N.; Oyama, T.; Tomioka, D.; Ohashi, M.; Yanagisawa, J.; Shimizu, T.; Miyachi, H. Peroxisome proliferator-activated receptors (PPARs) have multiple binding points that accommodate ligands in various conformations: phenylpropanoic acid-type PPAR ligands bind to PPAR in different conformations, depending on the subtype. J. Med. Chem 2012, 55, 893–902. [Google Scholar]
- Xu, H.E.; Stanley, T.B.; Montana, V.G.; Lambert, M.H.; Shearer, B.G.; Cobb, J.E.; McKee, D.D.; Galardi, C.M.; Plunket, K.D.; Nolte, R.T.; et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARα. Nature 2002, 415, 813–817. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 2008, 4, 435–447. [Google Scholar]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 1995, 91, 43–56. [Google Scholar]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem 2005, 26, 1701–1718. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [Google Scholar]
- Zielkiewicz, J. Structural properties of water: Comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J. Chem. Phys 2005, 123, 104501. [Google Scholar]
- Abraham, M.J.; Gready, J.E. Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J. Comput. Chem 2011, 32, 2031–2040. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput 2008, 4, 116–122. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys 2007, 126, 014101. [Google Scholar]
- Cosentino, U.; Pitea, D.; Moro, G.; Saracino, G.A.A.; Villa, A. Conformational behaviour determines the low-relaxivity state of a conditional MRI contrast agent. Phys. Chem. Chem. Phys 2009, 11, 3943–3950. [Google Scholar]
- Wang, Q.; Gao, J.; Liu, Y.; Liu, C. Molecular dynamics simulation of the interaction between protein tyrosine phosphatase 1B and aryl diketoacid derivatives. J. Mol. Graph. Model 2012, 38, 186–193. [Google Scholar]
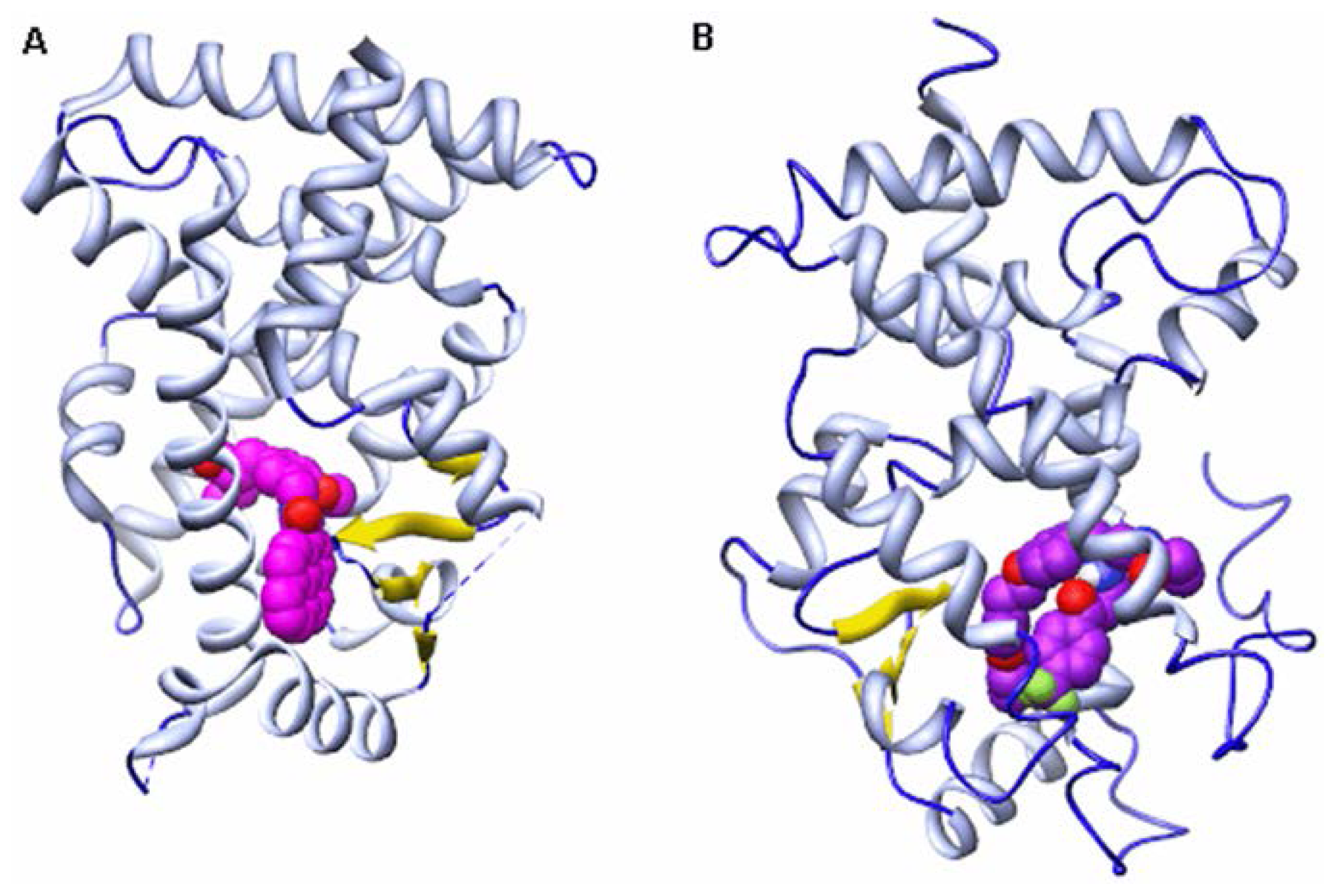

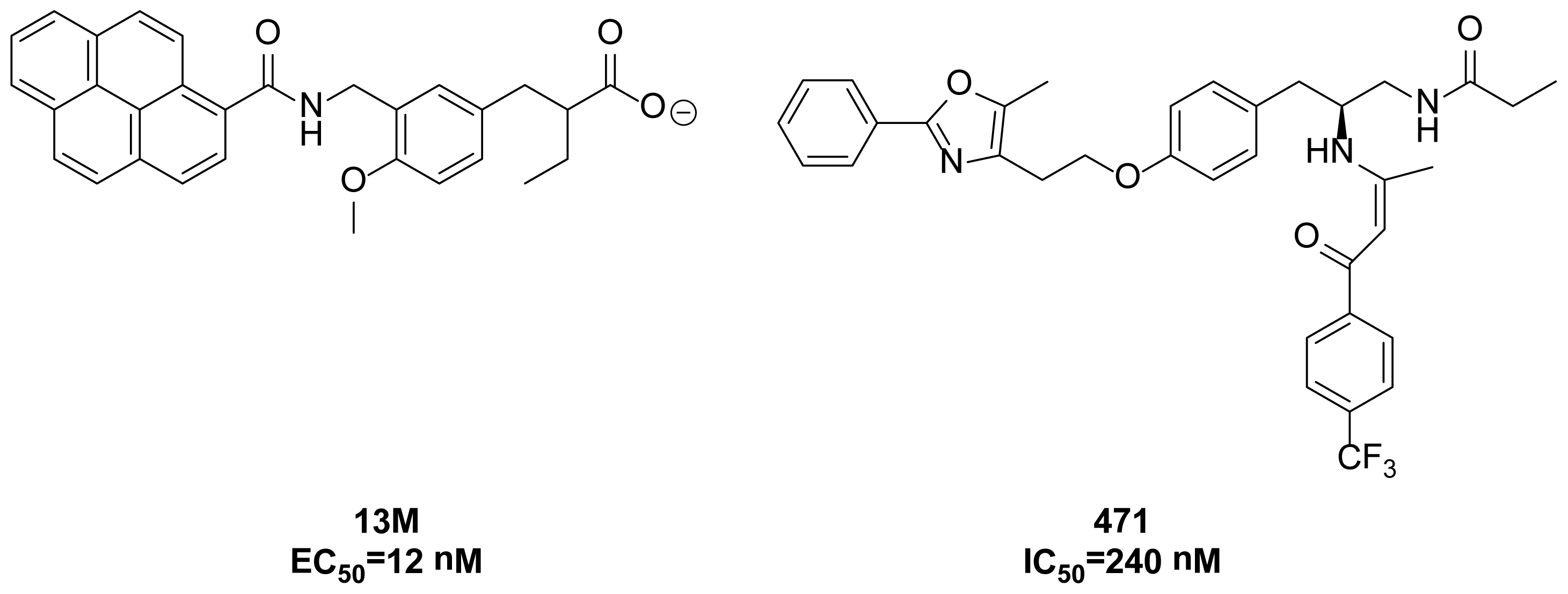



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residues | Ligands | |
|---|---|---|
| 13M | 471 | |
| Q277 | 31.1 | 0.0 |
| T279 | 78.5 | 0.1 |
| S280 | 99.7 | 19.1 |
| Y314 | 100.0 | 0.0 |
| H440 | 88.8 | 73.3 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, M.; Wang, L.; Zhao, X.; Sun, X. Molecular Recognition of Agonist and Antagonist for Peroxisome Proliferator-Activated Receptor-α Studied by Molecular Dynamics Simulations. Int. J. Mol. Sci. 2014, 15, 8743-8752. https://doi.org/10.3390/ijms15058743
Liu M, Wang L, Zhao X, Sun X. Molecular Recognition of Agonist and Antagonist for Peroxisome Proliferator-Activated Receptor-α Studied by Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2014; 15(5):8743-8752. https://doi.org/10.3390/ijms15058743
Chicago/Turabian StyleLiu, Mengyuan, Lushan Wang, Xian Zhao, and Xun Sun. 2014. "Molecular Recognition of Agonist and Antagonist for Peroxisome Proliferator-Activated Receptor-α Studied by Molecular Dynamics Simulations" International Journal of Molecular Sciences 15, no. 5: 8743-8752. https://doi.org/10.3390/ijms15058743