Potential Activity of Fevicordin-A from Phaleria macrocarpa (Scheff) Boerl. Seeds as Estrogen Receptor Antagonist Based on Cytotoxicity and Molecular Modelling Studies

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
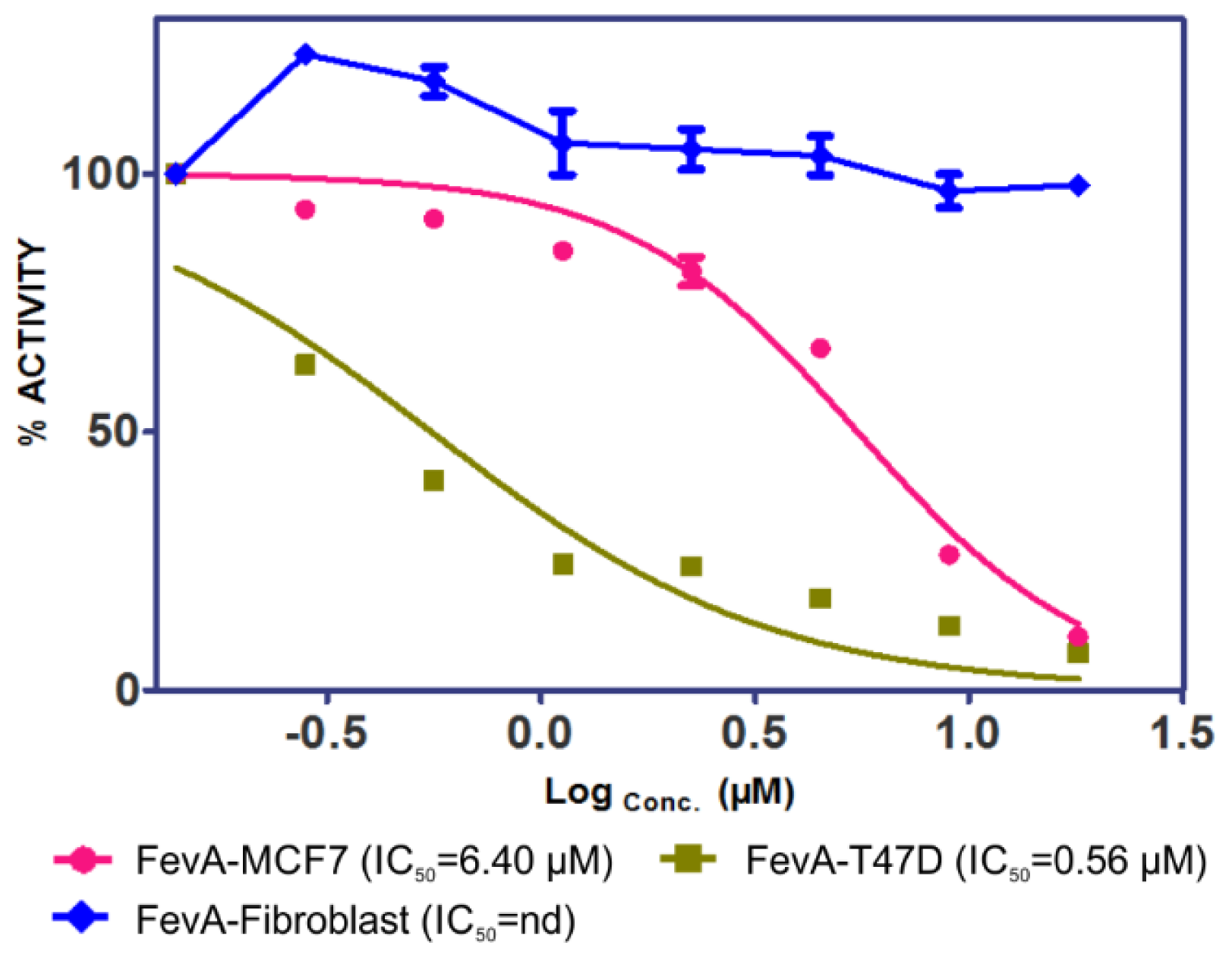
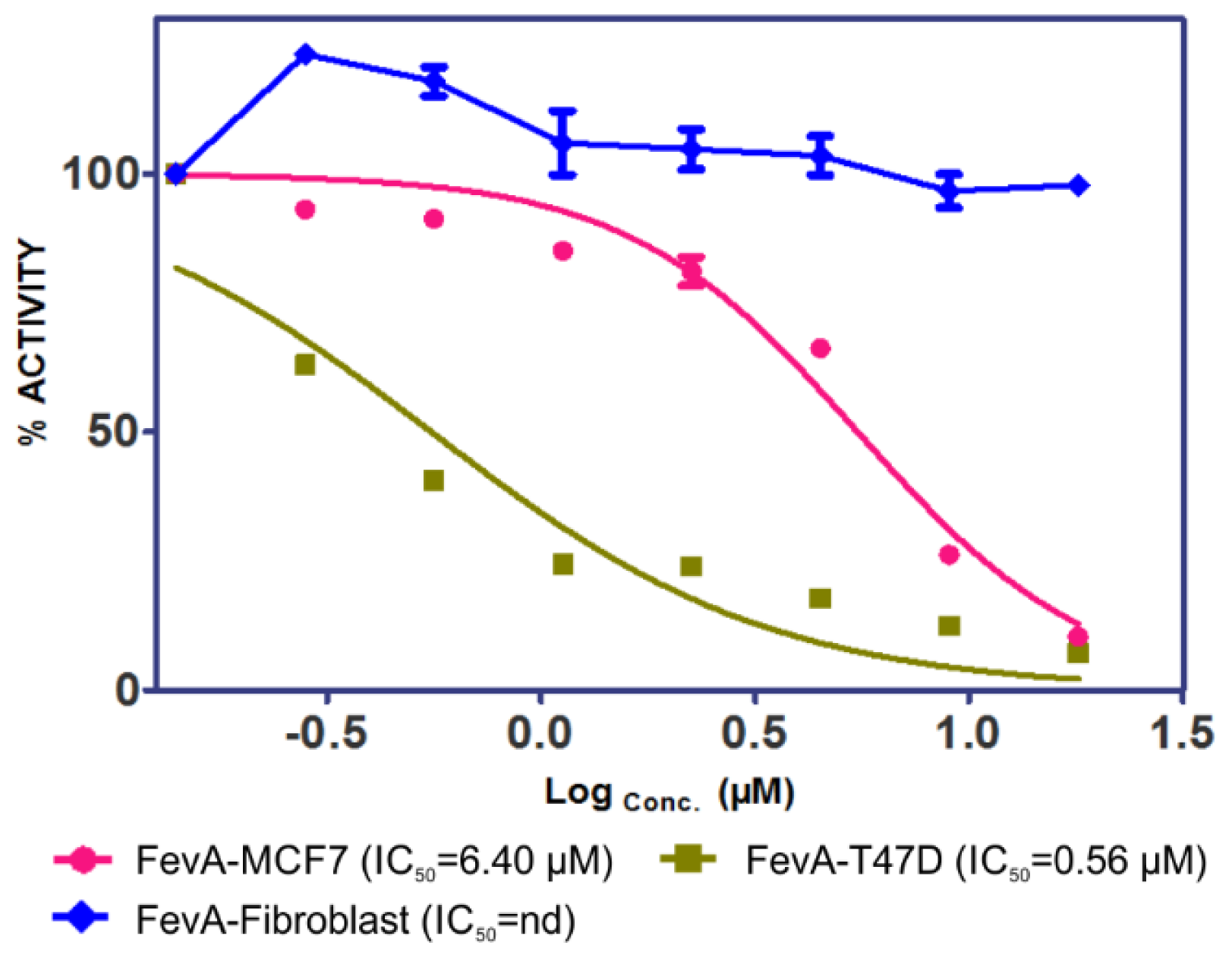
2.1. Cytotoxicity of FevA on MCF-7 and T-47D Human Breast Cancer Cell Lines
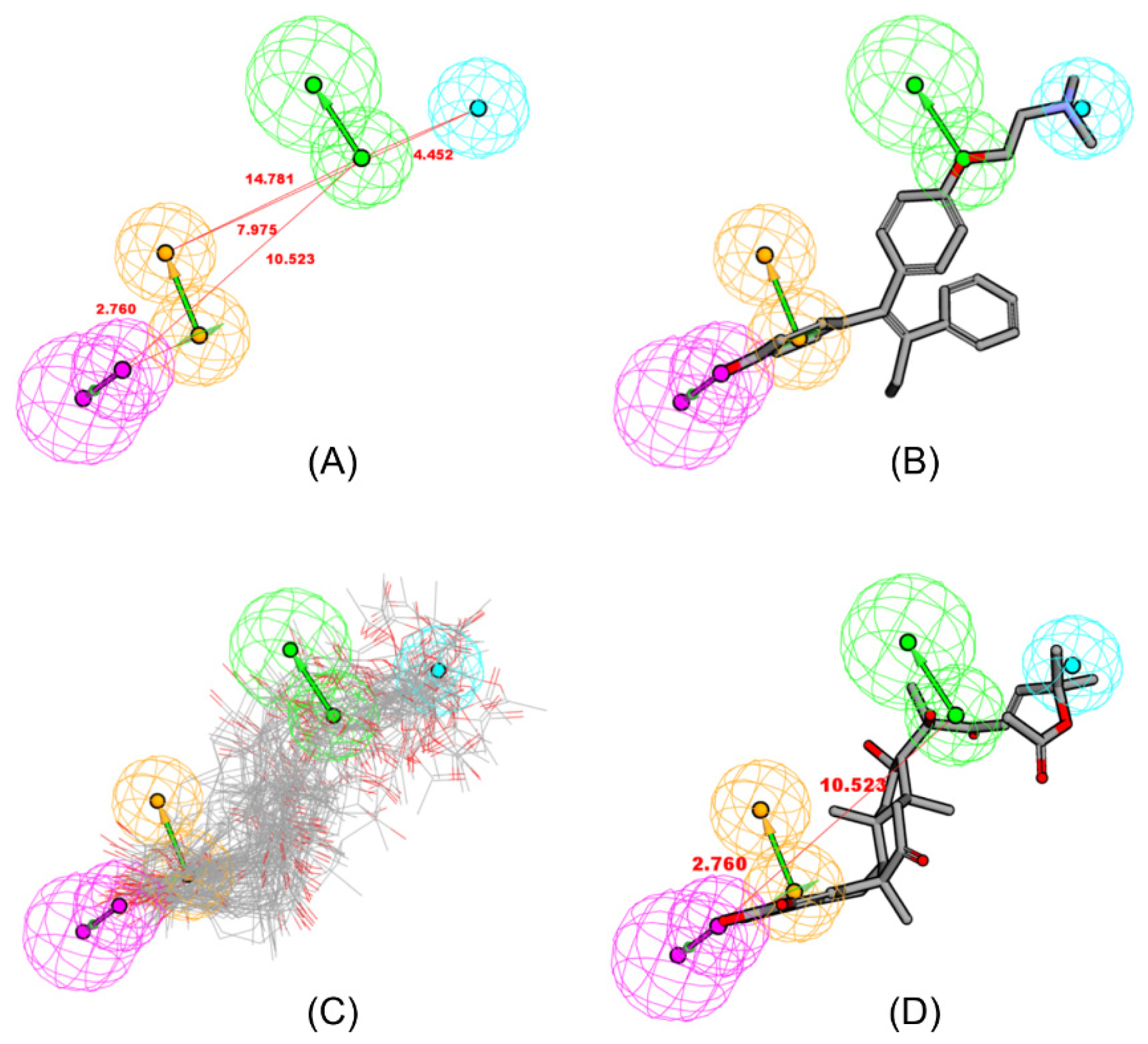
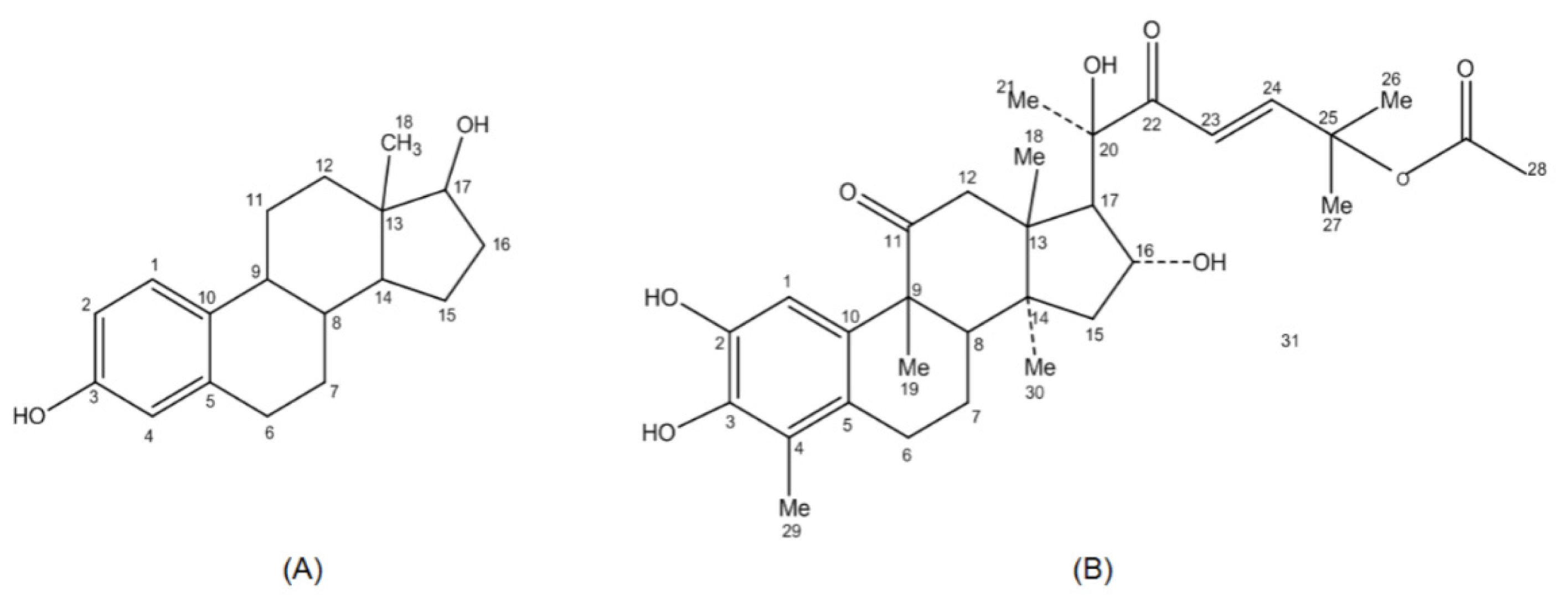
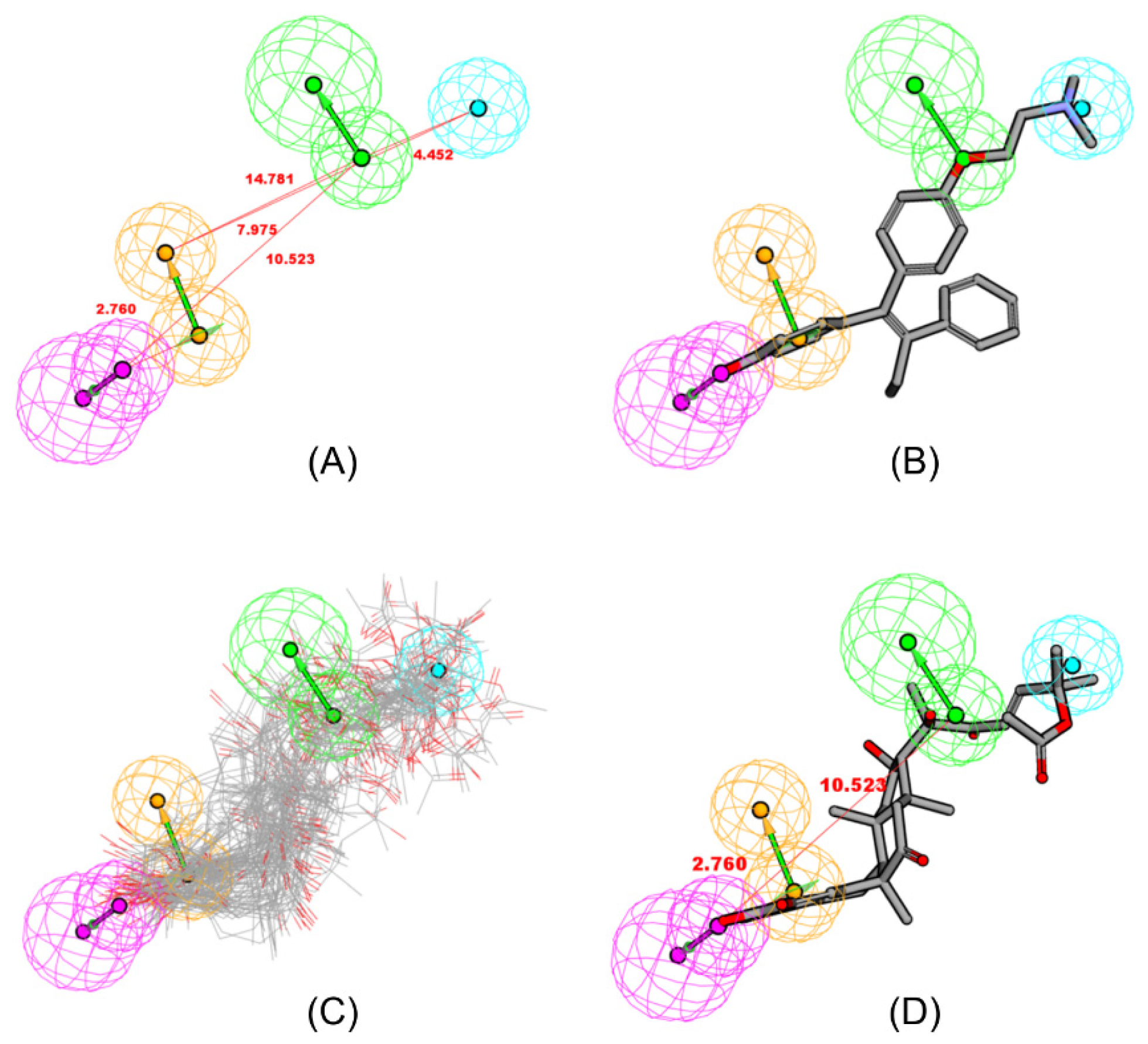
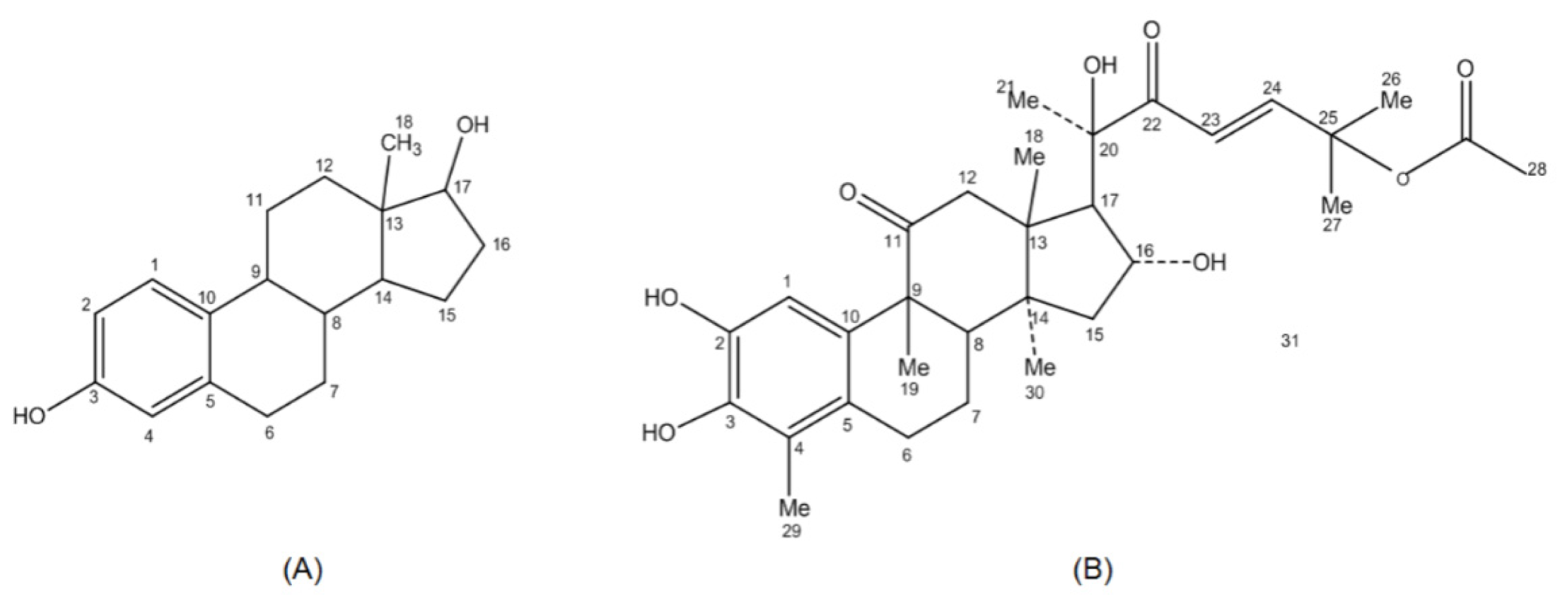
2.2. Pharmacophore Mapping
2.3. Molecular Dynamics of FevA on to ERα
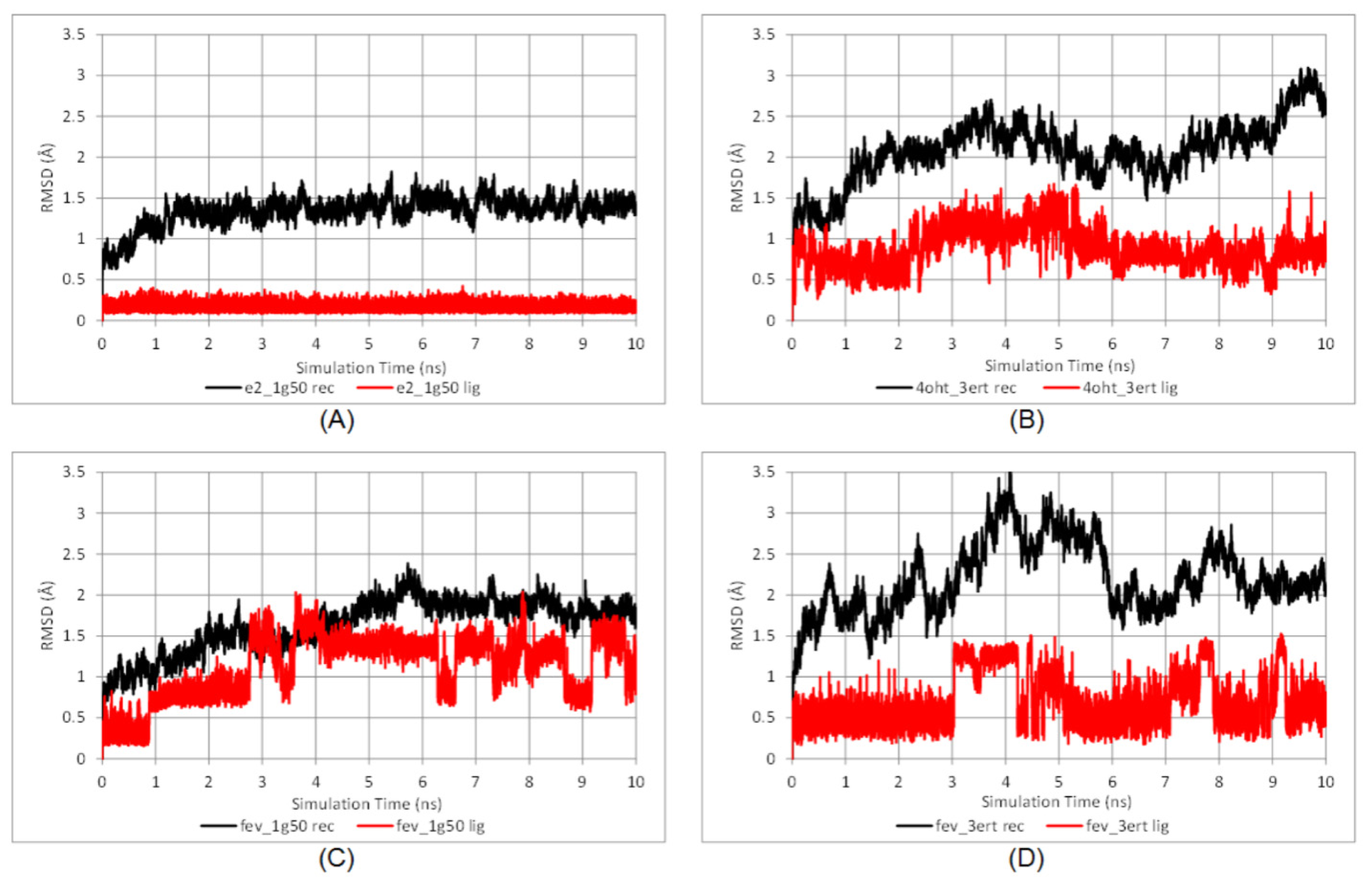
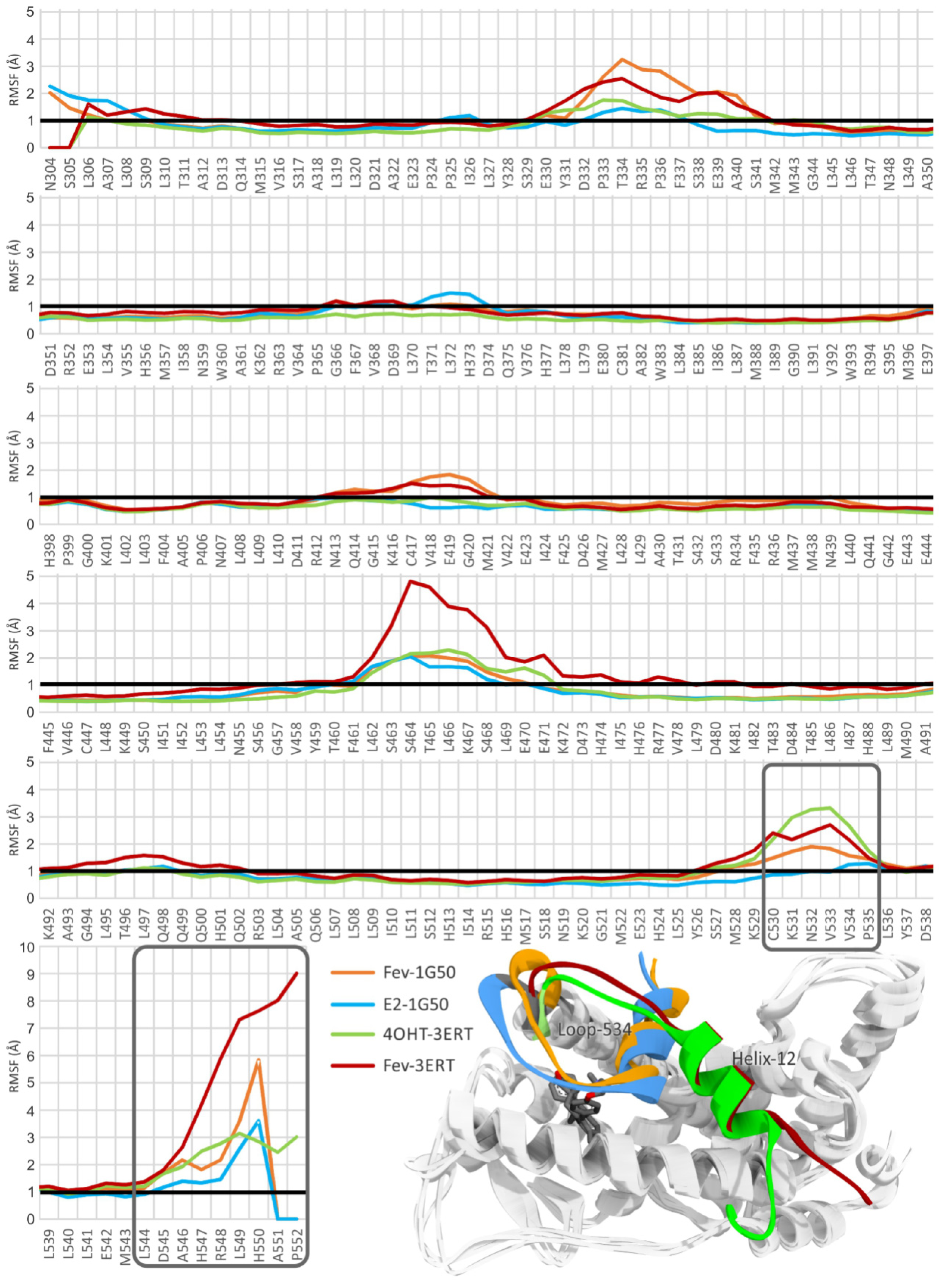
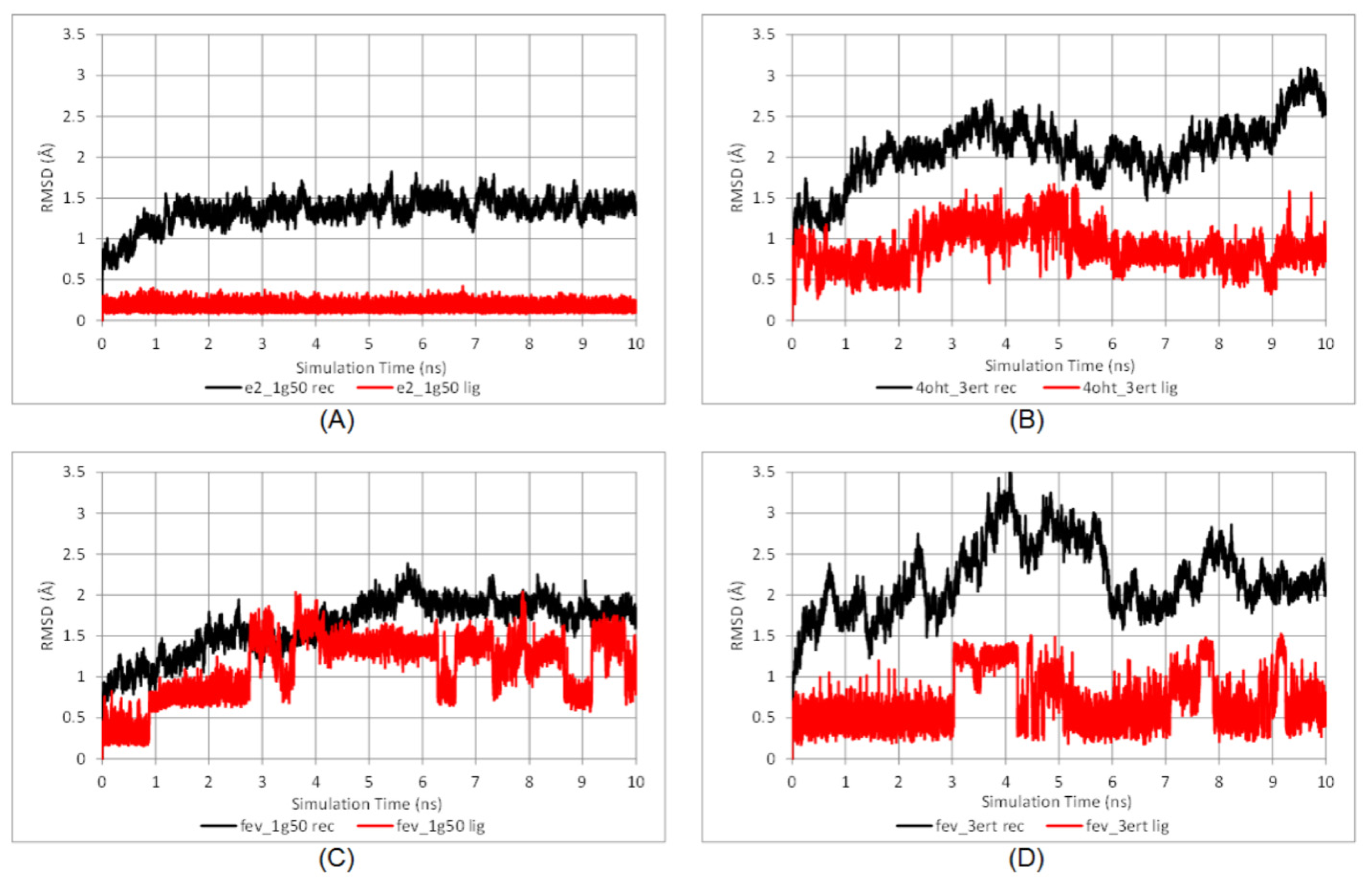
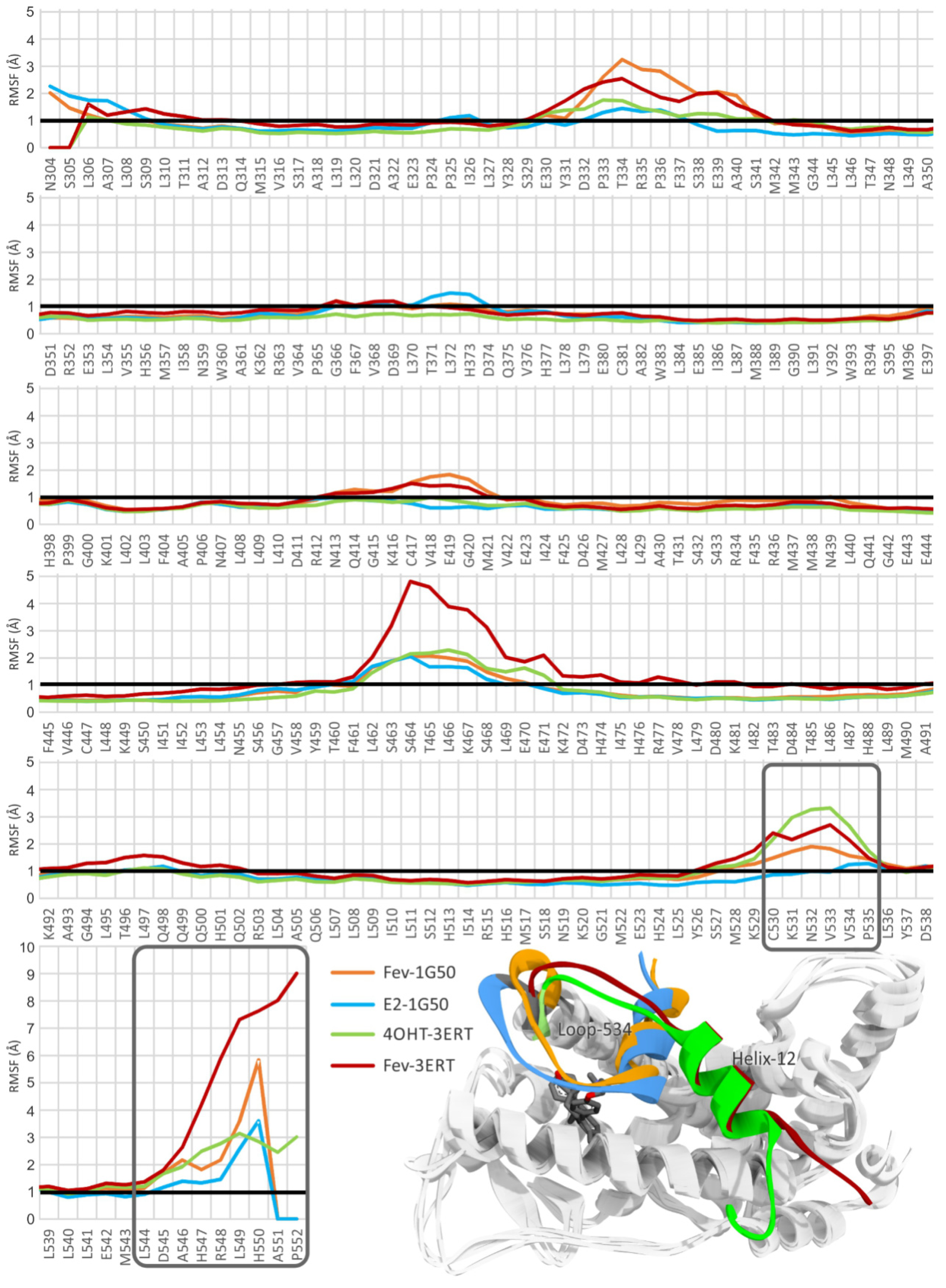
2.3.1. Stability of the System
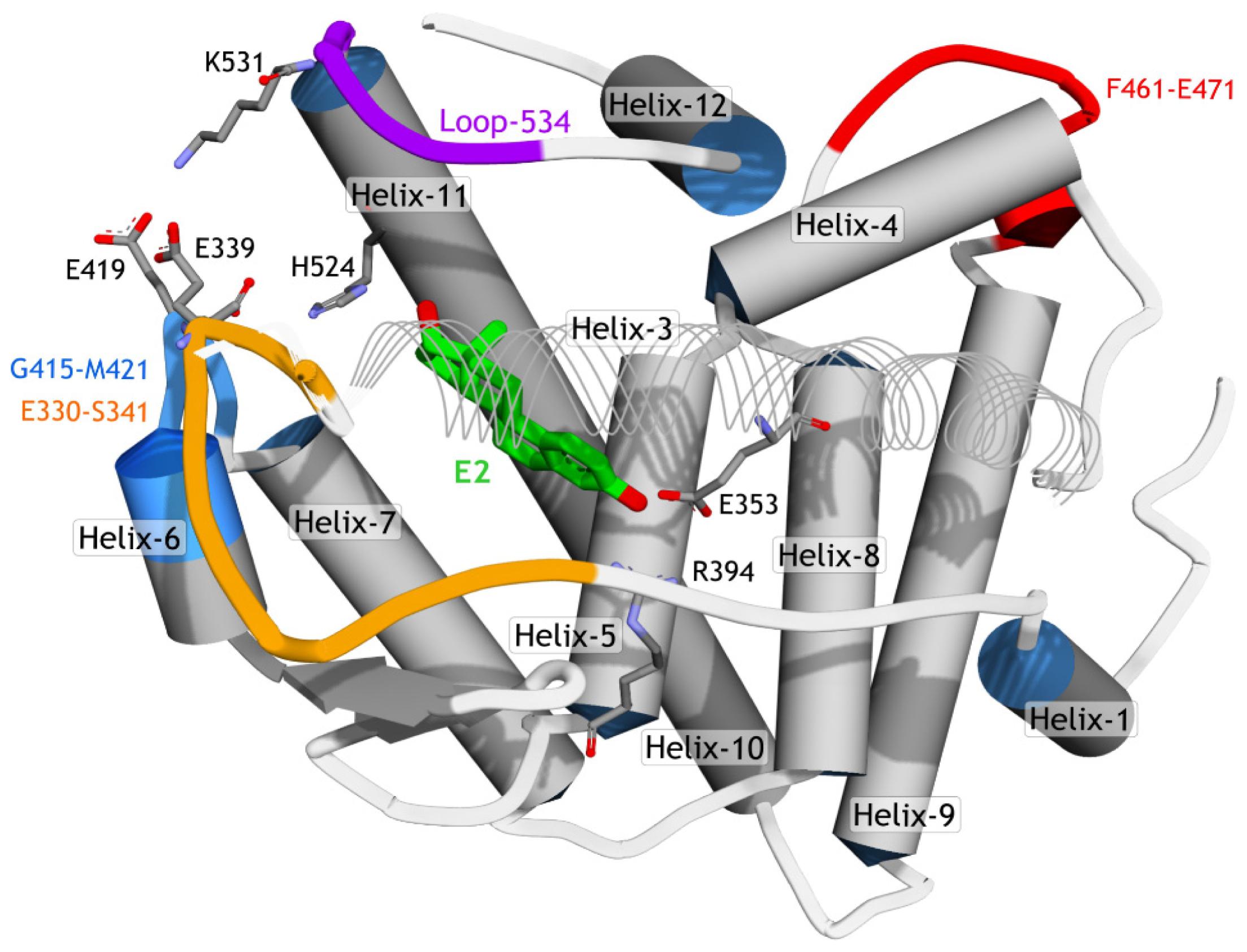
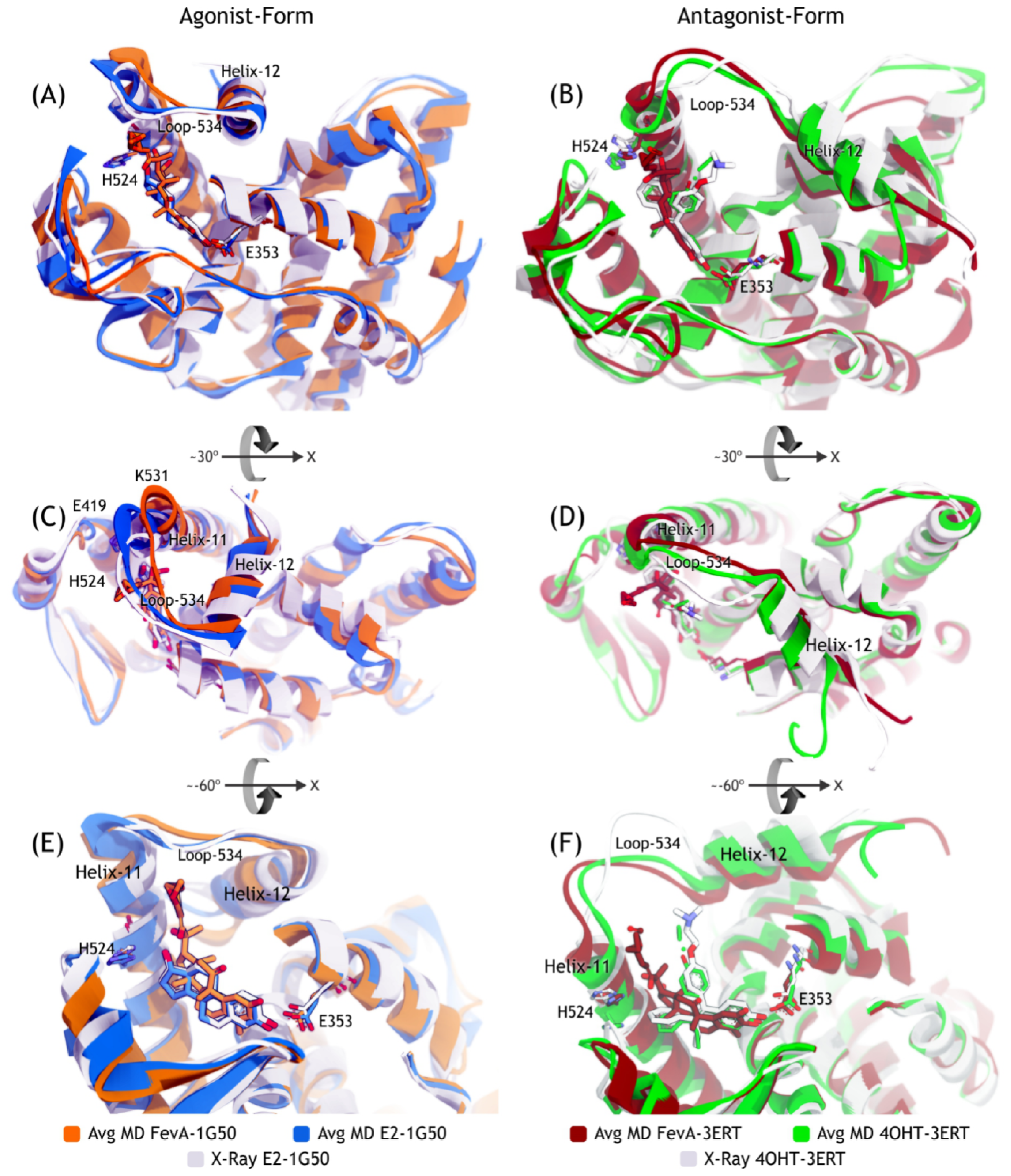
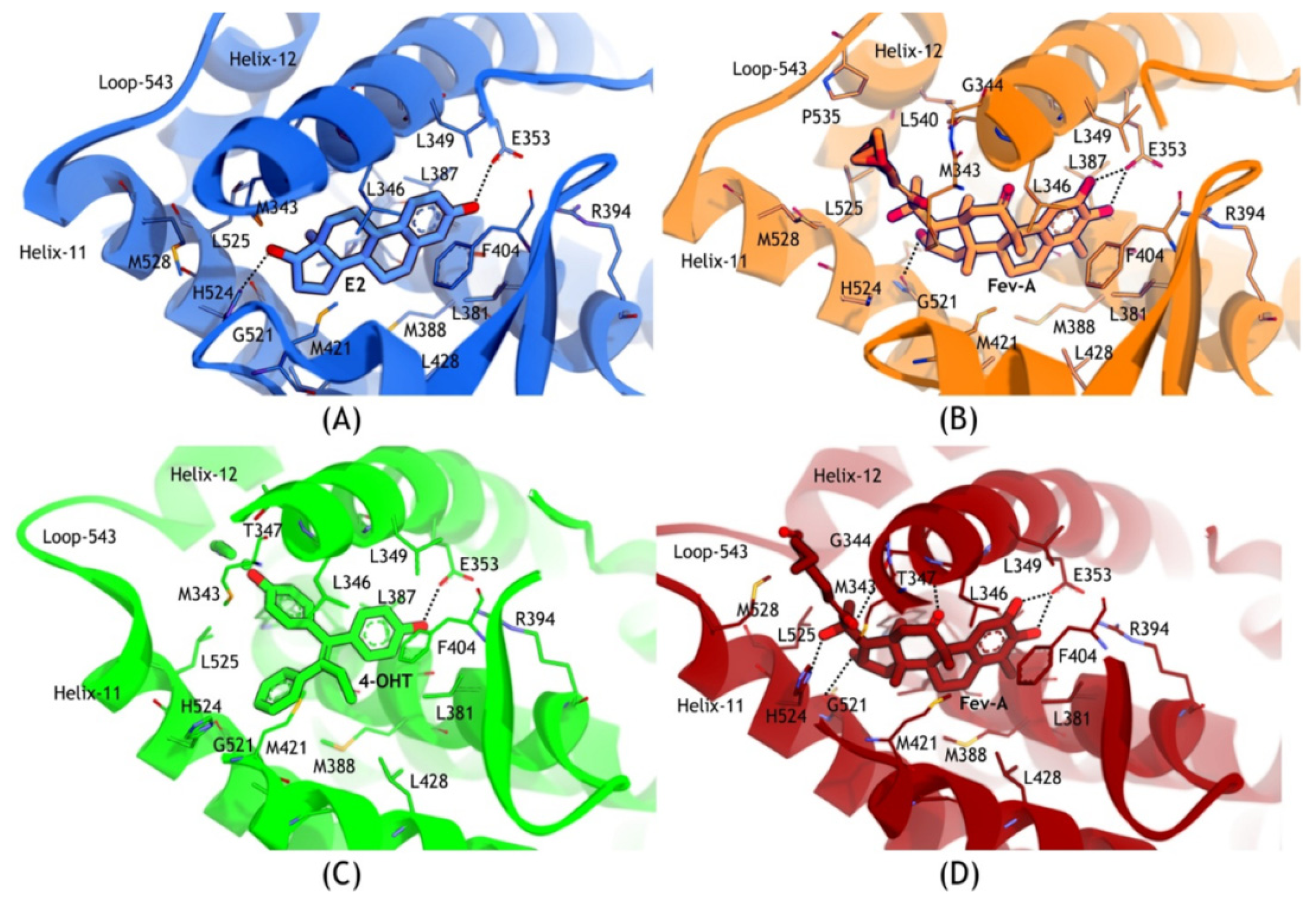
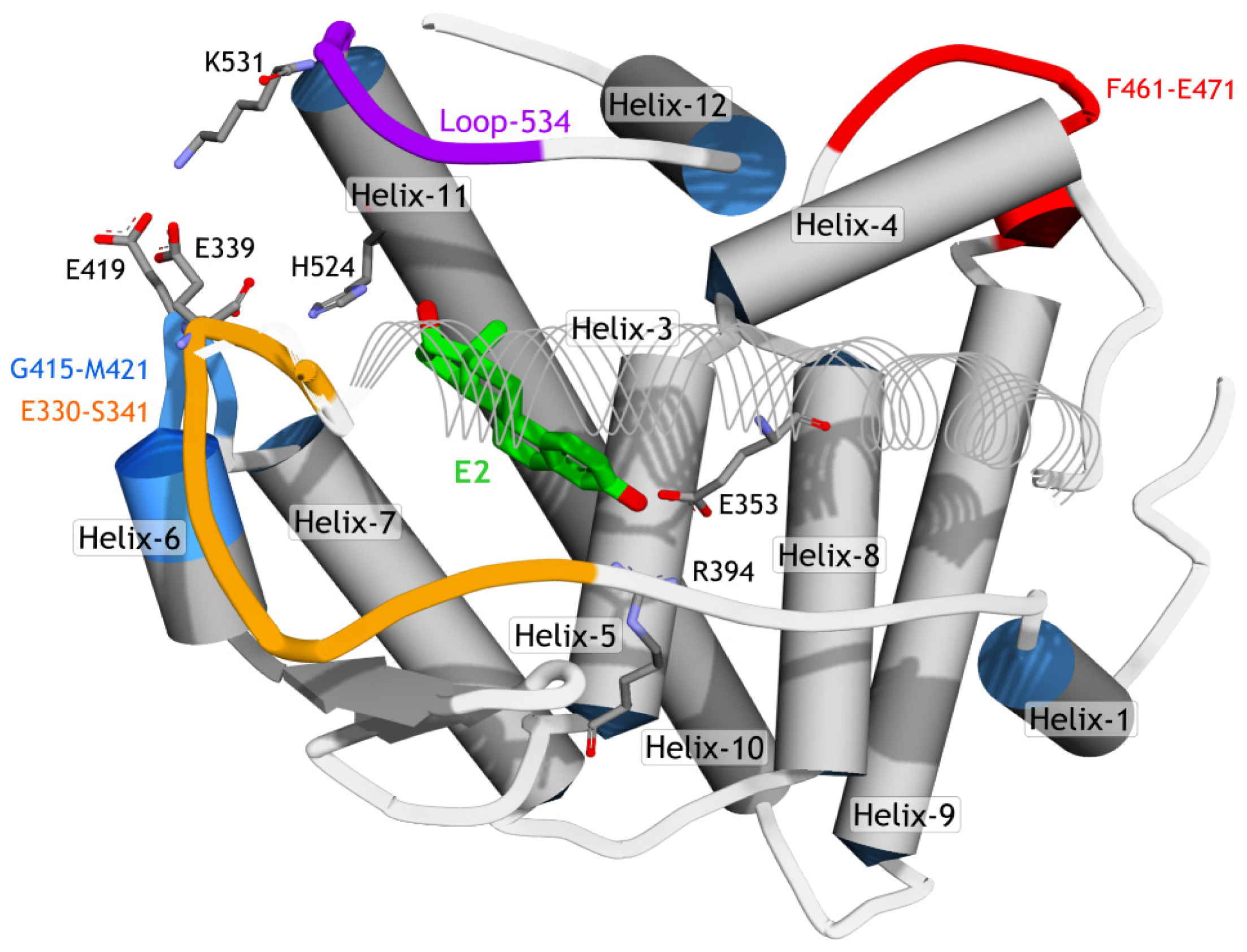
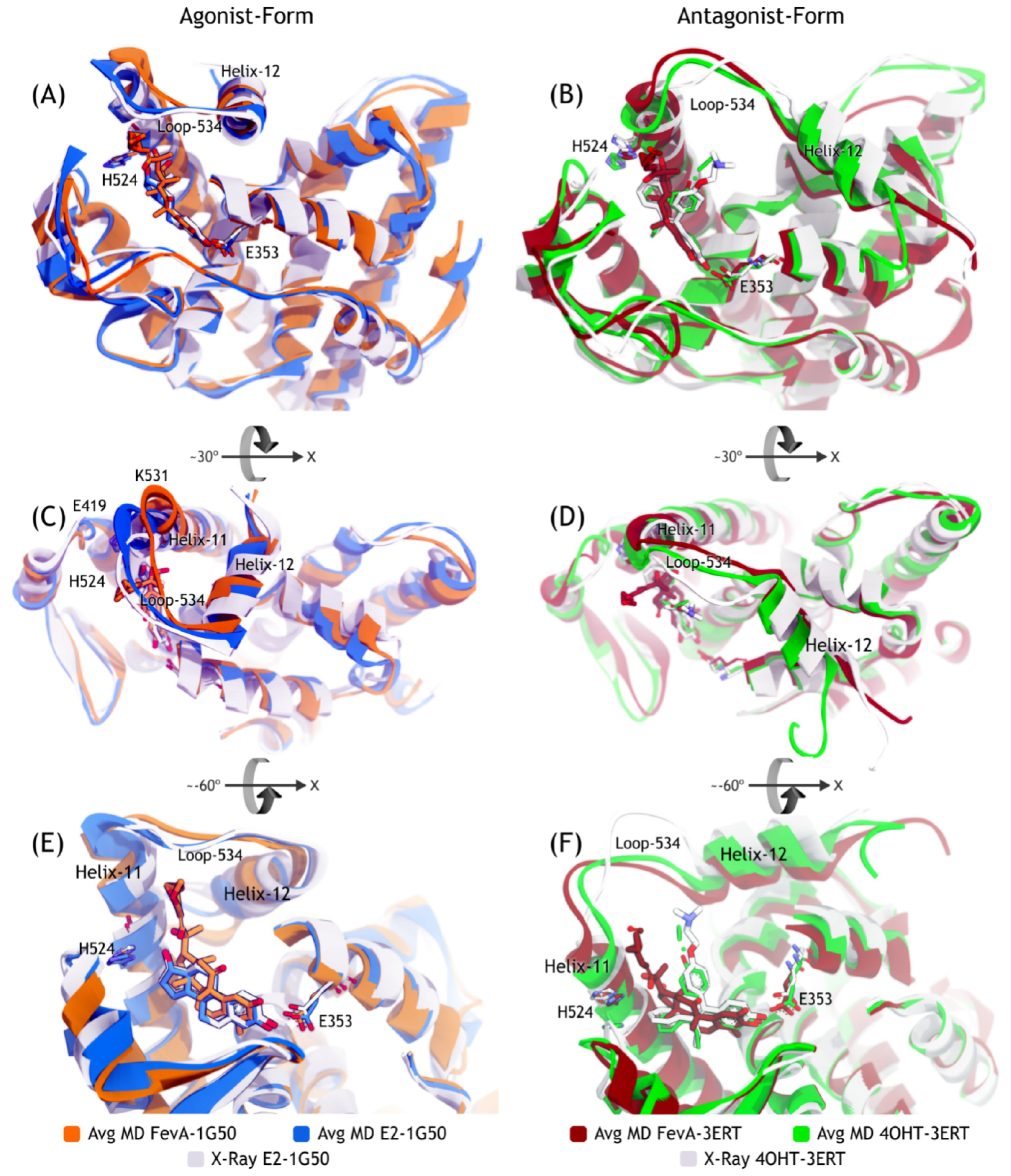
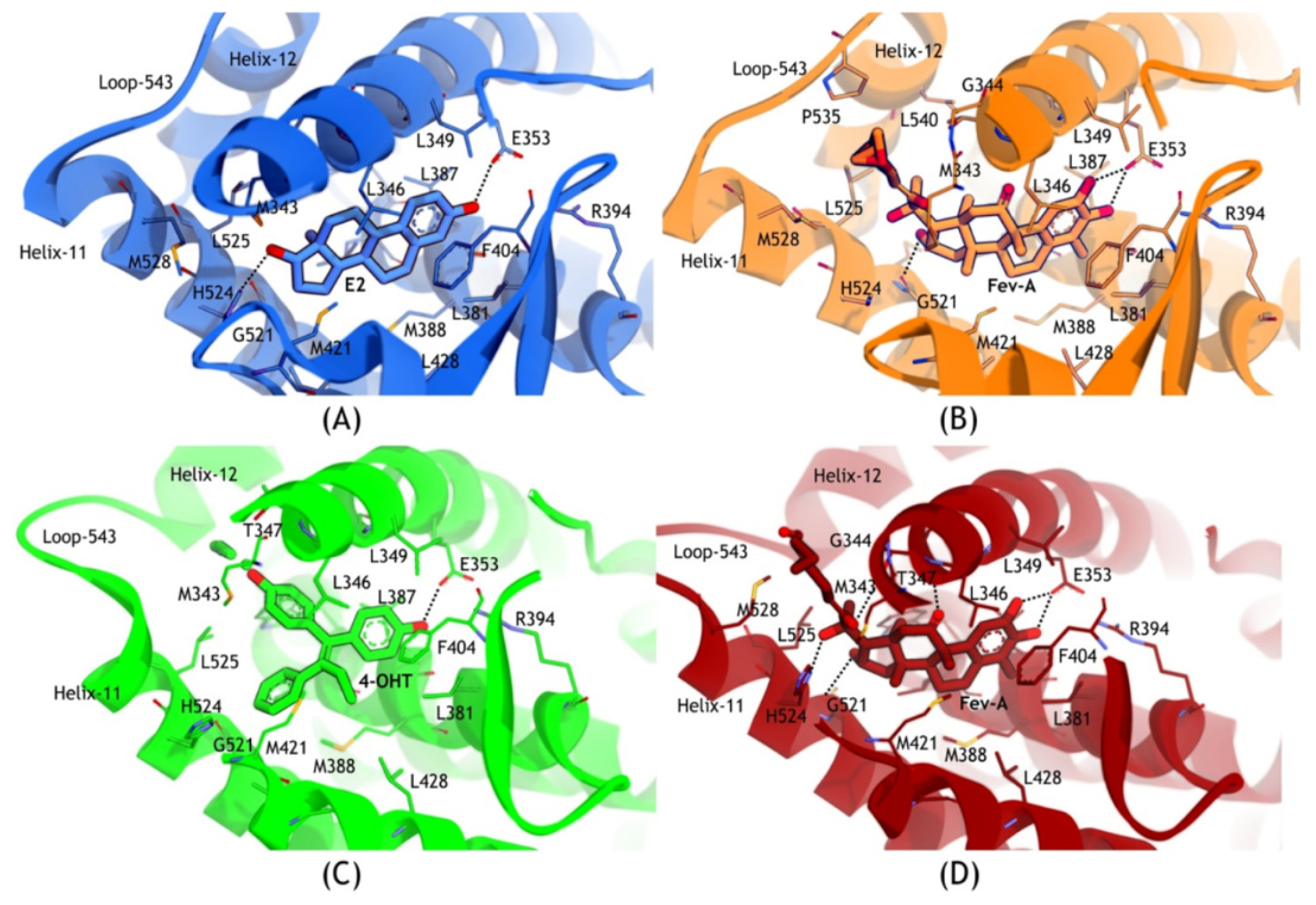
2.3.2. Mode of Ligand Binding Interactions
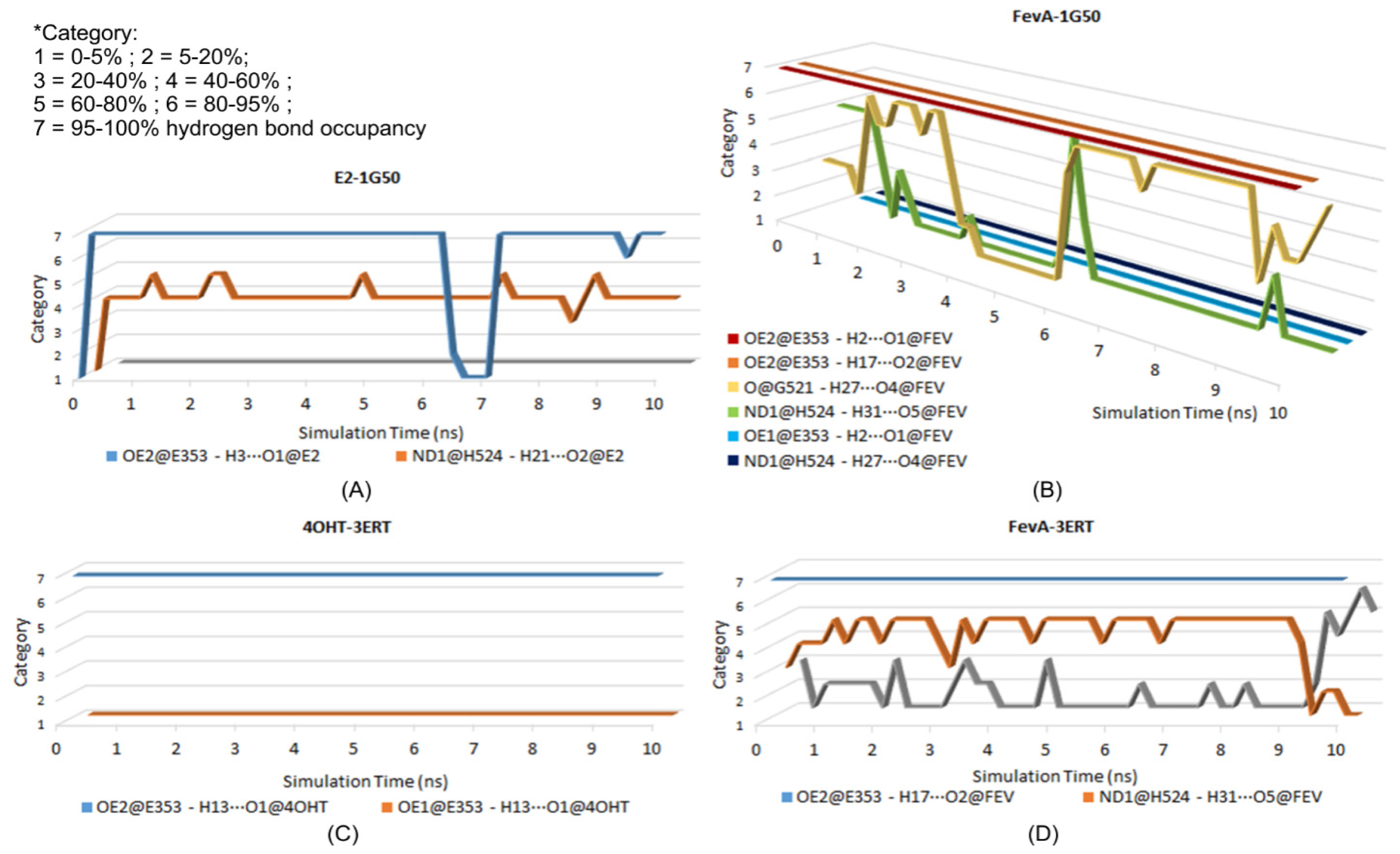
2.3.3. Hydrogen Bonding Analysis
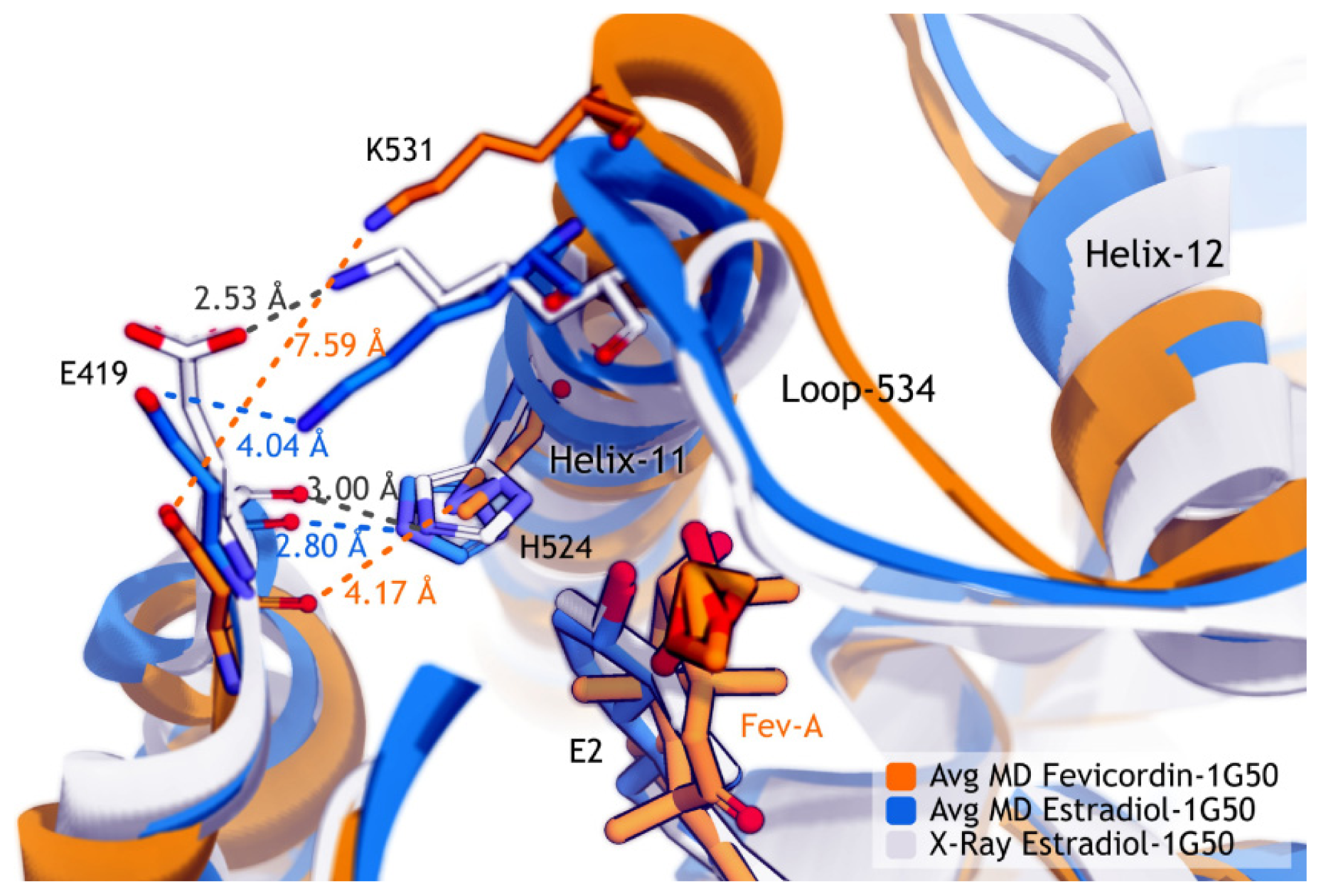
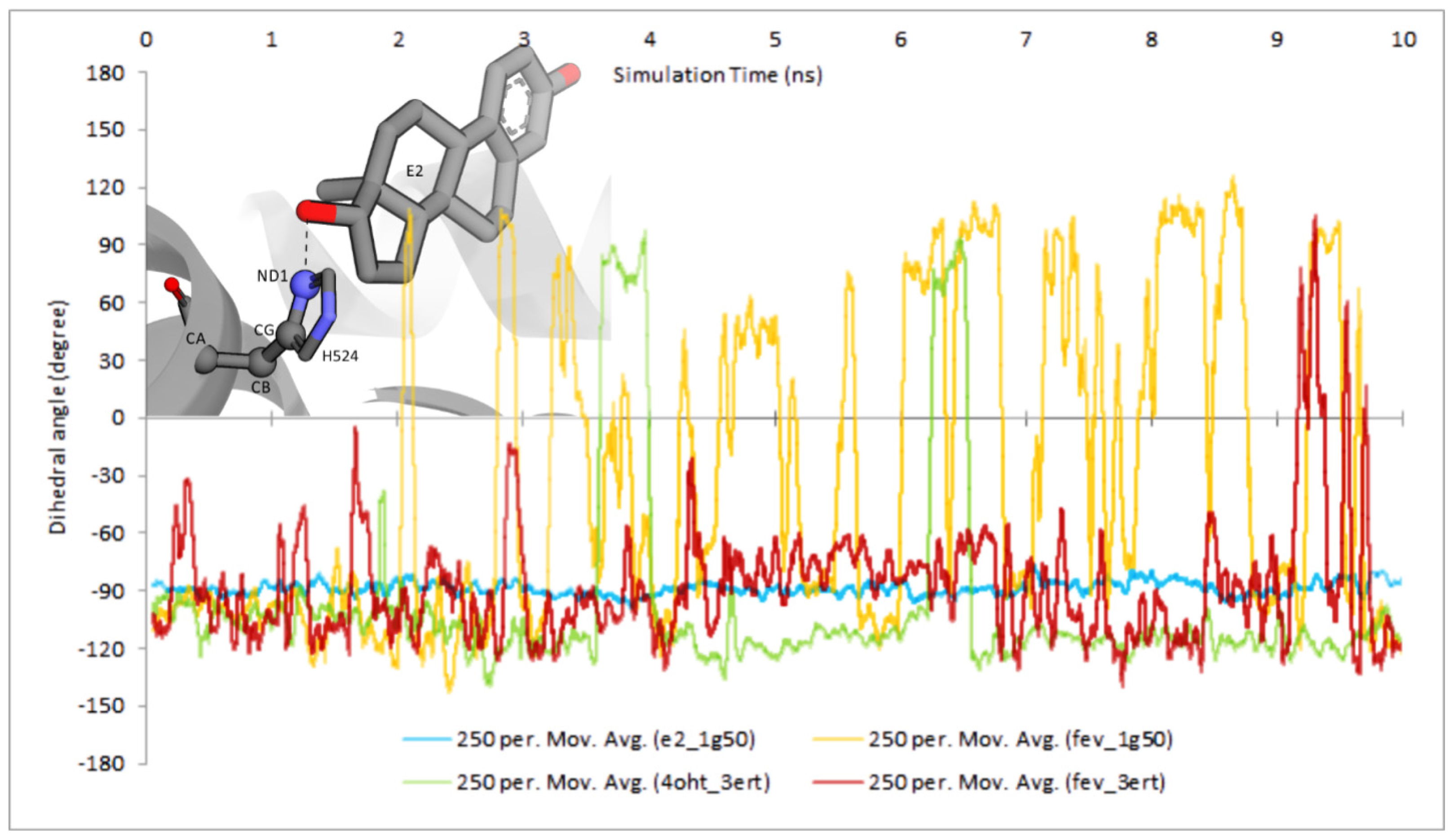
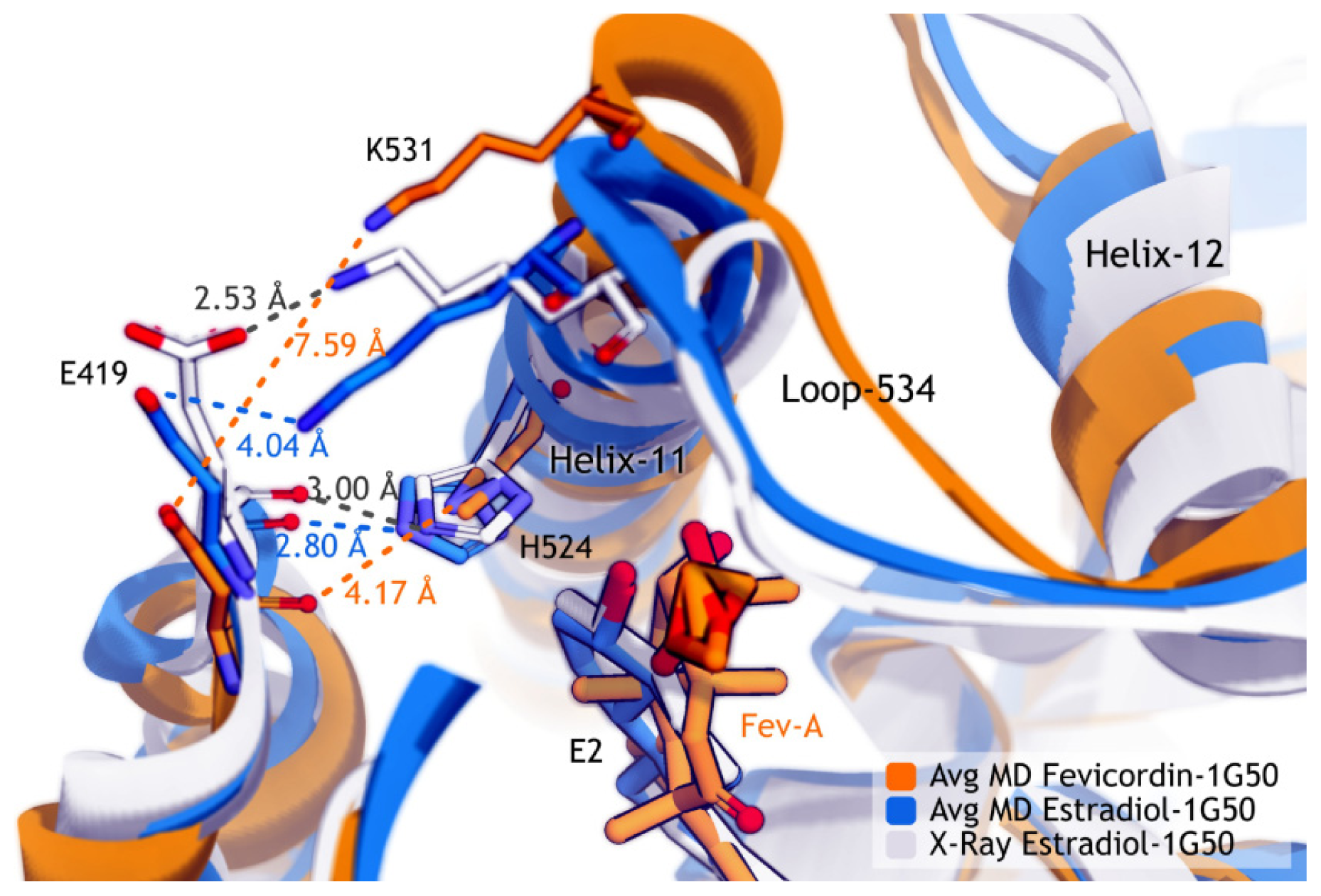
2.3.4. Dihedral Rotation of H524 Side Chain
2.3.5. MM/GBSA Calculation
2.3.6. Pairwise Decomposition of Binding Energy
2.4. Discussion
3. Experimental Section
3.1. Materials
3.1.1. Plant materials
3.1.2. Chemicals
3.1.3. Equipment
3.1.4. Software and Hardware
3.2. Methods
3.2.1. Extraction and Isolation
3.2.2. Assay
3.2.3. Cell Culture
3.2.4. Isolation of Fibroblast Cells from Human Prepuce Skin
3.2.5. Cytotoxycity Tests
3.2.6. Pharmacophore Mapping
3.2.7. Molecular Dynamics Simulation
3.2.8. Binding Energy Calculation
4. Conclusions
Supplementary Information
ijms-15-07225-s001.pdfAcknowledgments
Conflicts of Interest
Abbreviations
| MM/GBSA | Molecular Mechanics/Generalized Born Surface Area |
| RMSF | Root Mean Square Fluctuation |
| RMSD | Root Mean Square Distance |
| PDB | Protein Data Bank |
| hER | Human Estrogen Receptor |
| 4OHT | 4-hydroxy tamoxifen |
| FevA | Fevicordin-A |
| CPI | Cell proliferation inhibition |
| MD | molecular dynamics |
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Irigaray, P.; Newby, J.A.; Clapp, R.; Hardell, L.; Howard, V.; Montagnier, L.; Epstein, S.; Belpomme, D. Lifestyle-related factors and environmental agents causing cancer: An overview. Biomed. Pharmacother 2007, 61, 640–658. [Google Scholar]
- International Agency of Recerarch on Cancer (IARC). Globocan Cancer Incidence, Mortality and Prevalence Worldwide in 2008. Available online: http://www.iarc.fr/en/media-centre/iarcnews/2010/globocan2008.php accessed on 5 January 2013.
- Pudrovska, T.; Anikputa, B. The role of early-life socioeconomic status in breast cancer incidence and mortality: Unraveling life course mechanisms. J. Aging Health 2012, 24, 323–344. [Google Scholar]
- Pudrovska, T.; Anishkin, A.; Shen, Y. Early-life socioeconomic status and the prevalence of breast cancer in later life. Res. Aging 2012, 34, 302–320. [Google Scholar]
- Baquet, C.R.; Commiskey, P. Socioeconomic factors and breast carcinoma in multicultural women. Cancer 2000, 88, 1256–1264. [Google Scholar]
- Robert, S.A.; Strombom, I.; Trentham-Dietz, A.; Hampton, J.M.; McElroy, J.A.; Newcomb, P.A.; Remington, P.L. Socioeconomic risk factors for breast cancer: Distinguishing individual- and community-level effects. Epidemiology 2004, 15, 442–450. [Google Scholar]
- Haryono, S.J. Familial Breast Cancer: Exploring Predisposing Gene Most and Calculation of Risk Genes; Gadjah Mada University: Yogyakarta, Indonesia, 2012. [Google Scholar]
- Nomizu, T.; Matsuzaki, M.; Katagata, N.; Kobayashi, Y.; Sakuma, T.; Monma, T.; Saito, M.; Watanabe, F.; Midorikawa, S.; Yamaguchi, Y. A case of familial breast cancer with double heterozygosity for brca1 and brca2 genes. Breast Cancer 2012. [Google Scholar] [CrossRef]
- Russo, J.; Russo, I.H. The role of estrogen in the initiation of breast cancer. J. Steroid Biochem. Mol. Biol 2006, 102, 89–96. [Google Scholar]
- Platet, N.; Cathiard, A.M.; Gleizes, M.; Garcia, M. Estrogens and their receptors in breast cancer progression: A dual role in cancer proliferation and invasion. Crit. Rev. Oncol. Hematol 2004, 51, 55–67. [Google Scholar]
- Yue, W.; Santen, R.J.; Wang, J.P.; Li, Y.; Verderame, M.F.; Bocchinfuso, W.P.; Korach, K.S.; Devanesan, P.; Todorovic, R.; Rogan, E.G.; et al. Genotoxic metabolites of estradiol in breast: Potential mechanism of estradiol induced carcinogenesis. J. Steroid Biochem. Mol. Biol 2003, 86, 477–486. [Google Scholar]
- Harvell, D.M.; Strecker, T.E.; Tochacek, M.; Xie, B.; Pennington, K.L.; McComb, R.D.; Roy, S.K.; Shull, J.D. Rat strain-specific actions of 17beta-estradiol in the mammary gland: Correlation between estrogen-induced lobuloalveolar hyperplasia and susceptibility to estrogen-induced mammary cancers. Proc. Natl. Acad. Sci. USA 2000, 97, 2779–2784. [Google Scholar]
- Huetz, P.; Kamarulzaman, E.E.; Wahab, H.A.; Mavri, J. Chemical reactivity as a tool to study carcinogenicity: Reaction between estradiol and estrone 3,4-quinones ultimate carcinogens and guanine. J. Chem. Inf. Comput. Sci 2004, 44, 310–314. [Google Scholar]
- Prall, O.W.; Rogan, E.M.; Sutherland, R.L. Estrogen regulation of cell cycle progression in breast cancer cells. J. Steroid Biochem. Mol. Biol 1998, 65, 169–174. [Google Scholar]
- Calderon-Margalit, R.; Paltiel, O. Prevention of breast cancer in women who carry brca1 or brca2 mutations: A critical review of the literature. Int. J. Cancer 2004, 112, 357–364. [Google Scholar]
- Gadducci, A.; Biglia, N.; Sismondi, P.; Genazzani, A.R. Breast cancer and sex steroids: Critical review of epidemiological, experimental and clinical investigations on etiopathogenesis, chemoprevention and endocrine treatment of breast cancer. Gynecol. Endocrinol 2005, 20, 343–360. [Google Scholar]
- Keinan-Boker, L.; van Der Schouw, Y.T.; Grobbee, D.E.; Peeters, P.H. Dietary phytoestrogens and breast cancer risk. Am. J. Clin. Nutr 2004, 79, 282–288. [Google Scholar]
- Peeters, P.H.; Keinan-Boker, L.; van der Schouw, Y.T.; Grobbee, D.E. Phytoestrogens and breast cancer risk. Review of the epidemiological evidence. Breast Cancer Res. Treat 2003, 77, 171–183. [Google Scholar]
- De Lemos, M.L. Effects of soy phytoestrogens genistein and daidzein on breast cancer growth. Ann. Pharmacother 2001, 35, 1118–1121. [Google Scholar]
- Tamir, S.; Eizenberg, M.; Somjen, D.; Stern, N.; Shelach, R.; Kaye, A.; Vaya, J. Estrogenic and antiproliferative properties of glabridin from licorice in human breast cancer cells. Cancer Res 2000, 60, 5704–5709. [Google Scholar]
- Abdallah, H.H.; Mavri, J.; Repic, M.; Lee, V.S.; Wahab, H.A. Chemical reaction of soybean flavonoids with DNA: A computational study using the implicit solvent model. Int. J. Mol. Sci 2012, 13, 1269–1283. [Google Scholar]
- De Padua, L.S.; Bunyapraphatsara, N.; Lemmens, R.H.M.S. Plant resources of South East Asia. In Medical and Poisonous Plants 1 (Prosea); Backhuys Publishers: Leiden, The Netherlands, 1999; Volume 12, pp. 36–38. [Google Scholar]
- Hendra, R.; Ahmad, S.; Oskoueian, E.; Sukari, A.; Shukor, M.Y. Antioxidant, anti-inflammatory and cytotoxicity of phaleria macrocarpa (boerl.) scheff fruit. BMC Complement. Altern. Med 2011, 11, 110. [Google Scholar]
- Achenbach, H.; Waibel, R.; Bubl, U.H.; Constenla, M.A. Constituents of fevillea cordifolia: New norcucurbitacin and cucurbitacin glycosides. J. Nat. Prod 1993, 56, 1506–1519. [Google Scholar]
- Almeidai, F.R.C.; Rao, V.S.N.; Matos, M.E.O. Antiinflammatory, antitumour and antifertility effects in rodents of two nor-cucurbitacin glucosides from wilbrandia species. Phytother. Res 1992, 6, 189–193. [Google Scholar]
- Zivadinovic, D.; Gametchu, B.; Watson, C.S. Membrane estrogen receptor-alpha levels in mcf-7 breast cancer cells predict camp and proliferation responses. Breast Cancer Res 2005, 7, R101–R112. [Google Scholar]
- Johnson, A.E.; Le, I.P.; Buchwalter, A.; Burnatowska-Hledin, M.A. Estrogen-dependent growth and estrogen receptor (er)-alpha concentration in t47d breast cancer cells are inhibited by vacm-1, a cul 5 gene. Mol. Cell. Biochem 2007, 301, 13–20. [Google Scholar]
- Sorich, M.J.; Miners, J.O.; McKinnon, R.A.; Smith, P.A. Multiple pharmacophores for the investigation of human udp-glucuronosyltransferase isoform substrate selectivity. Mol. Pharmacol 2004, 65, 301–308. [Google Scholar]
- Guner, O.F. The impact of pharmacophore modeling in drug design. IDrugs 2005, 8, 567–572. [Google Scholar]
- Celik, L.; Lund, J.D.D.; Schiøtt, B. Conformational dynamics of the estrogen receptor α: Molecular dynamics simulations of the influence of binding site structure on protein dynamics. Biochemistry 2007, 46, 1743–1758. [Google Scholar]
- Eiler, S.; Gangloff, M.; Duclaud, S.; Moras, D.; Ruff, M. Overexpression, purification, and crystal structure of native erα lbd. Protein Expr. Purif 2001, 22, 165–173. [Google Scholar]
- Diantini, A.; Subarnas, A.; Supriyatna, S.; Levita, J.; Abdullah, R.; Ahmad, T.; Faried, A.; Faried, L.S.; Kunia, D.; Wardhani, S.R.; et al. Cytotoxicity of fevicordin-a from phaleria macrocarpa (boerl.) scheff on p388, hela, caski, te-2, te-8 and prepuce’s fibroblast cells. E3 J. Med. Res 2012, 1, 1–5. [Google Scholar]
- Janocko, L.; Larner, J.M.; Hochberg, R.B. The interaction of c-17 esters of estradiol with the estrogen receptor. Endocrinology 1984, 114, 1180–1186. [Google Scholar]
- Hartmann, R.W. Influence of alkyl chain ramification on estradiol receptor binding affinity and intrinsic activity of 1,2-dialkylated 1,2-bis(4- or 3-hydroxyphenyl)ethane estrogens and antiestrogens. J. Med. Chem 1986, 29, 1668–1674. [Google Scholar]
- Zlatovic, M.V.; Sukalovoc, V.V.; Kostic-Rajacic, V.; Andric, D.B.; Roglic, G.M. Influence of n–1 substituent properties on binding affinities of arylpiperazines to the binding site of 5-ht1a receptor. J. Serbian Chem. Soc 2006, 71, 1125–1135. [Google Scholar]
- Anstead, G.M.; Carlson, K.E.; Katzenellenbogen, J.A. The estradiol pharmacophore: Ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids 1997, 62, 268–303. [Google Scholar]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar]
- Gangloff, M.; Ruff, M.; Eiler, S.; Duclaud, S.; Wurtz, J.M.; Moras, D. Crystal structure of a mutant herα ligand-binding domain reveals key structural features for the mechanism of partial agonism. J. Biol. Chem 2001, 276, 15059–15065. [Google Scholar]
- Katzenellenbogen, J.A. The 2010 philip s. Portoghese medicinal chemistry lectureship: Addressing the “core issue” in the design of estrogen receptor ligands. J. Med. Chem 2011, 54, 5271–5282. [Google Scholar]
- Jordan, V.C. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J. Med. Chem 2003, 46, 883–908. [Google Scholar]
- Kieser, K.; Kim, D.W.; Carlson, K.; Katzenellenbogen, B.; Katzenellenbogen, J. Characterization of the pharmacophore properties of novel selective estrogen receptor downregulators (serds). J. Med. Chem 2010, 53, 3320–3329. [Google Scholar]
- Nettles, K.W.; Bruning, J.B.; Gil, G.; O’Neill, E.E.; Nowak, J.; Hughs, A.; Kim, Y.; DeSombre, E.R.; Dilis, R.; Hanson, R.N.; et al. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep 2007, 8, 563–568. [Google Scholar]
- El Garrouj, D.; Aumelas, A.; Borgna, J.L. Steroidal affinity labels of the estrogen receptor. 1. 17.Alpha.-(bromoacetoxy)alkyl/alkynylestradiols. J. Med. Chem 1993, 36, 2973–2983. [Google Scholar]
- Hanson, R.N.; Napolitano, E.; Fiaschi, R. Synthesis and evaluation of 11β-substituted 21-chloro/iodo-(17α,20e/z)-19-norpregna-1,3,5(10),20-tetraene-3,17β-diols: High-affinity ligands for the estrogen receptor. J. Med. Chem 1998, 41, 4686–4692. [Google Scholar]
- El Garrouj, D.; Aliau, S.; Aumelas, A.; Borgna, J.-L. Steroidal affinity labels of the estrogen receptor. 2. 17.Alpha.-[(haloacetamido)alkyl]estradiols. J. Med. Chem 1995, 38, 2339–2348. [Google Scholar]
- Nettles, K.W.; Greene, G.L. Ligand control of coregulator recruitment to nuclear receptors. Annu. Rev. Physiol 2005, 67, 309–333. [Google Scholar]
- Levenson, A.S.; Jordan, V.C. The key to the antiestrogenic mechanism of raloxifene is amino acid 351 (aspartate) in the estrogen receptor. Cancer Res 1998, 58, 1872–1875. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Rob, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Gaussian, Inc: Wallingford, CT, USA, 2003. [Google Scholar]
- Accelrys, Discovery Studio Modeling Environment, Release 2.5.5; Accelrys Software Inc: San Diego, CA, USA, 2007.
- Accelrys, Discovery Studio Modeling Environment, Release 3.5; Accelrys Software Inc: San Diego, CA, USA, 2007.
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. Amber 11; University of California: San Francisco, CA, USA, 2010. [Google Scholar]
- American Type Culture Collection (ATCC), Mtt Cell Proliferation Assay Instructions; ATCC: Manassas, VA, USA, 2001; pp. 1–6.
- Hida, T.; Ueda, R.; Takahashi, T.; Watanabe, H.; Kato, T.; Suyama, M.; Sugiura, T.; Ariyoshi, Y. Chemosensitivity and radiosensitivity of small cell lung cancer cell lines studied by a newly developed 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (mtt) hybrid assay. Cancer Res 1989, 49, 4785–4790. [Google Scholar]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. Charmm: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- Qin, Z.; Kastrati, I.; Chandrasena, R.E.; Liu, H.; Yao, P.; Petukhov, P.A.; Bolton, J.L.; Thatcher, G.R. Benzothiophene selective estrogen receptor modulators with modulated oxidative activity and receptor affinity. J. Med. Chem 2007, 50, 2682–2692. [Google Scholar]
- Gauthier, S.; Caron, B.; Cloutier, J.; Dory, Y.L.; Favre, A.; Larouche, D.; Mailhot, J.; Ouellet, C.; Schwerdtfeger, A.; Leblanc, G.; et al. (s)-(+)-4-[7-(2,2-dimethyl-1-oxopropoxy)-4-methyl-2-[4- [2-(1-piperidinyl)- ethoxy]phenyl]-2h-1-benzopyran-3-yl]-phenyl 2,2-dimethylpropanoate (em- 800): A highly potent, specific, and orally active nonsteroidal antiestrogen. J. Med. Chem 1997, 40, 2117–2122. [Google Scholar]
- Chao, E.Y.; Collins, J.L.; Gaillard, S.; Miller, A.B.; Wang, L.; Orband-Miller, L.A.; Nolte, R.T.; McDonnell, D.P.; Willson, T.M.; Zuercher, W.J. Structure-guided synthesis of tamoxifen analogs with improved selectivity for the orphan errgamma. Bioorg. Med. Chem. Lett 2006, 16, 821–824. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem 2003, 24, 1999–2012. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model 2006, 25, 247–260. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [Google Scholar]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. Mmpbsa.Py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput 2012, 8, 3314–3321. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | H-Bond Acceptor (atom@res) | H-Bond Donor (H···atom@res) | Percentage Occupancy (%) | Average Distance (Å) | Average Angle (°) |
|---|---|---|---|---|---|
| E2-1G50 | OE2@E353 | H3···O1@E2 | 91.88 | 2.58 | 102.86 |
| ND1@H524 | H21···O2@E2 | 54.41 | 2.87 | 103.06 | |
| OE1@E353 | H3···O1@E2 | 0.25 | 2.89 | 67.97 | |
| FevA-1G50 | OE2@E353 | H2···O1@FEV | 99.88 | 2.60 | 106.21 |
| OE2@E353 | H17···O2@FEV | 99.01 | 2.65 | 107.55 | |
| O@G521 | H27···O4@FEV | 56.23 | 2.77 | 100.46 | |
| ND1@H524 | H31···O5@FEV | 12.28 | 2.85 | 103.73 | |
| OE1@E353 | H2···O1@FEV | 0.28 | 2.93 | 65.59 | |
| ND1@H524 | H27···O4@FEV | 0.03 | 2.89 | 82.24 | |
| 4OHT-3ERT | OE2@E353 | H13···O1@4OHT | 99.34 | 2.64 | 98.74 |
| OE1@E353 | H13···O1@4OHT | 0.02 | 2.90 | 73.48 | |
| FevA-3ERT | OE2@E353 | H17···O2@FEV | 98.55 | 2.67 | 107.82 |
| ND1@H524 | H31···O5@FEV | 59.2 | 2.85 | 103.49 | |
| O@G521 | H27···O4@FEV | 13.81 | 2.80 | 97.73 | |
| Energy Component * | System | |||
|---|---|---|---|---|
| E2-1G50 | FevA-1G50 | 4OHT-3ERT | FevA-3ERT | |
| Vdw | −39.49 | −62.73 | −49.20 | −56.63 |
| EEL | −25.81 | −43.72 | −19.07 | −42.80 |
| ΔGgas (vdw + EEL) | −65.30 | −106.45 | −68.26 | −99.43 |
| EGB | 35.58 | 68.00 | 25.46 | 59.22 |
| ESURF | −5.08 | −8.39 | −7.37 | −7.88 |
| ΔGsolv (EGB + ESURF) | 30.50 | 59.61 | 18.10 | 51.34 |
| ΔGMMGBSA (ΔGgas + ΔGsolv) | −34.80 | −46.84 | −50.16 | −48.09 |
| Entropy | −15.93 | −34.35 | −28.21 | −30.27 |
| ΔGbinding (ΔGMMGBSA − Entropy) | −18.87 | −12.49 | −21.96 | −17.82 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Muchtaridi, M.; Yusuf, M.; Diantini, A.; Choi, S.B.; Al-Najjar, B.O.; Manurung, J.V.; Subarnas, A.; Achmad, T.H.; Wardhani, S.R.; Wahab, H.A. Potential Activity of Fevicordin-A from Phaleria macrocarpa (Scheff) Boerl. Seeds as Estrogen Receptor Antagonist Based on Cytotoxicity and Molecular Modelling Studies. Int. J. Mol. Sci. 2014, 15, 7225-7249. https://doi.org/10.3390/ijms15057225
Muchtaridi M, Yusuf M, Diantini A, Choi SB, Al-Najjar BO, Manurung JV, Subarnas A, Achmad TH, Wardhani SR, Wahab HA. Potential Activity of Fevicordin-A from Phaleria macrocarpa (Scheff) Boerl. Seeds as Estrogen Receptor Antagonist Based on Cytotoxicity and Molecular Modelling Studies. International Journal of Molecular Sciences. 2014; 15(5):7225-7249. https://doi.org/10.3390/ijms15057225
Chicago/Turabian StyleMuchtaridi, Muchtaridi, Muhammad Yusuf, Ajeng Diantini, Sy Bing Choi, Belal O. Al-Najjar, Jerry V. Manurung, Anas Subarnas, Tri H. Achmad, Savitri R. Wardhani, and Habibah A. Wahab. 2014. "Potential Activity of Fevicordin-A from Phaleria macrocarpa (Scheff) Boerl. Seeds as Estrogen Receptor Antagonist Based on Cytotoxicity and Molecular Modelling Studies" International Journal of Molecular Sciences 15, no. 5: 7225-7249. https://doi.org/10.3390/ijms15057225
APA StyleMuchtaridi, M., Yusuf, M., Diantini, A., Choi, S. B., Al-Najjar, B. O., Manurung, J. V., Subarnas, A., Achmad, T. H., Wardhani, S. R., & Wahab, H. A. (2014). Potential Activity of Fevicordin-A from Phaleria macrocarpa (Scheff) Boerl. Seeds as Estrogen Receptor Antagonist Based on Cytotoxicity and Molecular Modelling Studies. International Journal of Molecular Sciences, 15(5), 7225-7249. https://doi.org/10.3390/ijms15057225