Towards Personalized Medicine Mediated by in Vitro Virus-Based Interactome Approaches

Abstract
:1. Introduction
2. Comprehensive Protein–Protein Interaction Analysis in Post-Genome Analysis
3. Interactome Analysis Using Next-Generation Sequencing in the Personal-Genome Era
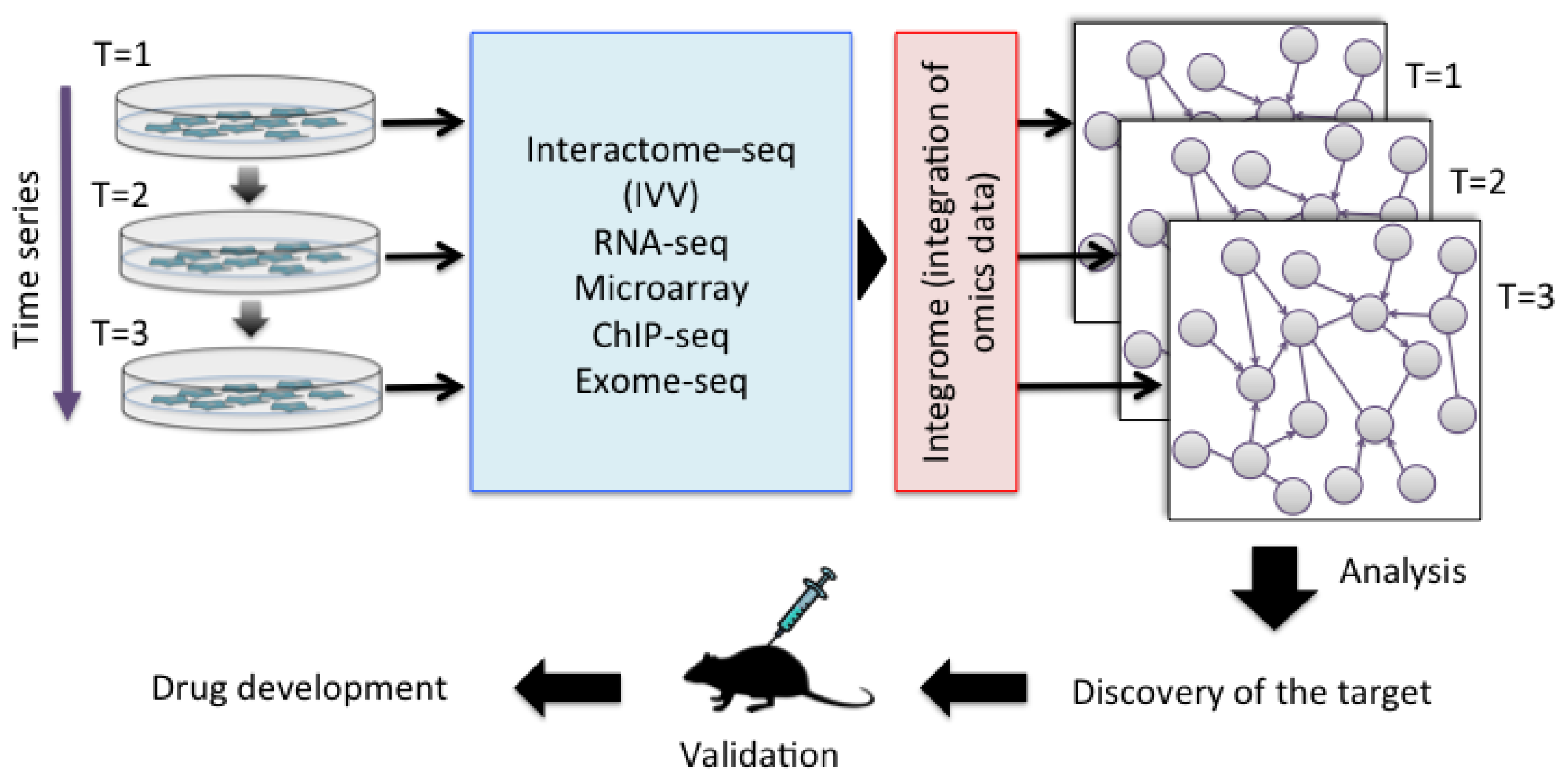
4. Future Prospects for Interactome Analysis in Personal Genomics
Acknowledgments
Conflicts of Interest
- Author ContributionsH.O. and E.M.S. designed the experiments, developed the methods and prepared the manuscript. E.M.S. supervised the projects.
References
- Hamdi, A.; Colas, P. Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol. Sci 2012, 33, 109–118. [Google Scholar]
- Dunham, W.H.; Mullin, M.; Gingras, A.C. Affinity-purification coupled to mass spectrometry: Basic principles and strategies. Proteomics 2012, 12, 1576–1590. [Google Scholar]
- Deane, C.M.; Salwinski, L.; Xenarios, I.; Eisenberg, D. Protein interactions: Two methods for assessment of the reliability of high throughput observations. Mol. Cell. Proteomics 2002, 1, 349–356. [Google Scholar]
- Edwards, A.M.; Kus, B.; Jansen, R.; Greenbaum, D.; Greenblatt, J.; Gerstein, M. Bridging structural biology and genomics: Assessing protein interaction data with known complexes. Trends Genet 2002, 18, 529–536. [Google Scholar]
- Miyamoto-Sato, E.; Nemoto, N.; Kobayashi, K.; Yanagawa, H. Specific bonding of puromycin to full-length protein at the C-terminus. Nucleic Acids Res 2000, 28, 1176–1182. [Google Scholar]
- Miyamoto-Sato, E.; Takashima, H.; Fuse, S.; Sue, K.; Ishizaka, M.; Tateyama, S.; Horisawa, K.; Sawasaki, T.; Endo, Y.; Yanagawa, H. Highly stable and efficient mRNA templates for mRNA-protein fusions and C-terminally labeled proteins. Nucleic Acids Res 2003, 31, e78. [Google Scholar]
- Nemoto, N.; Miyamoto-Sato, E.; Husimi, Y.; Yanagawa, H. In vitro virus: Bonding of mRNA bearing puromycin at the 3′-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro. FEBS Lett. 1997, 414, 405–408. [Google Scholar]
- Roberts, R.W.; Szostak, J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar]
- Keefe, A.D.; Szostak, J.W. Functional proteins from a random-sequence library. Nature 2001, 410, 715–718. [Google Scholar]
- Wilson, D.S.; Keefe, A.D.; Szostak, J.W. The use of mRNA display to select high-affinity protein-binding peptides. Proc. Natl. Acad. Sci. USA 2001, 98, 3750–3755. [Google Scholar]
- Amstutz, P.; Forrer, P.; Zahnd, C.; Pluckthun, A. In vitro display technologies: Novel developments and applications. Curr. Opin. Biotechnol 2001, 12, 400–405. [Google Scholar]
- Miyamoto-Sato, E.; Fujimori, S.; Ishizaka, M.; Hirai, N.; Masuoka, K.; Saito, R.; Ozawa, Y.; Hino, K.; Washio, T.; Tomita, M.; et al. A comprehensive resource of interacting protein regions for refining human transcription factor networks. PLoS One 2010, 5, e9289. [Google Scholar]
- Nemoto, N.; Miyamoto-Sato, E.; Yanagawa, H. Fluorescence labeling of the C-terminus of proteins with a puromycin analogue in cell-free translation systems. FEBS Lett 1999, 462, 43–46. [Google Scholar]
- Doi, N.; Takashima, H.; Kinjo, M.; Sakata, K.; Kawahashi, Y.; Oishi, Y.; Oyama, R.; Miyamoto-Sato, E.; Sawasaki, T.; Endo, Y.; et al. Novel fluorescence labeling and high-throughput assay technologies for in vitro analysis of protein interactions. Genome Res 2002, 12, 487–492. [Google Scholar]
- Tai, M.S.; Mudgett-Hunter, M.; Levinson, D.; Wu, G.M.; Haber, E.; Oppermann, H.; Huston, J.S. A bifunctional fusion protein containing Fc-binding fragment B of staphylococcal protein A amino terminal to antidigoxin single-chain Fv. Biochemistry 1990, 29, 8024–8030. [Google Scholar]
- Legrain, P.; Selig, L. Genome-wide protein interaction maps using two-hybrid systems. FEBS Lett 2000, 480, 32–36. [Google Scholar]
- Rain, J.C.; Selig, L.; de Reuse, H.; Battaglia, V.; Reverdy, C.; Simon, S.; Lenzen, G.; Petel, F.; Wojcik, J.; Schachter, V.; et al. The protein-protein interaction map ofHelicobacter pylori. Nature 2001, 409, 211–215. [Google Scholar]
- Zhou, X.; Ren, L.; Meng, Q.; Li, Y.; Yu, Y.; Yu, J. The next-generation sequencing technology and application. Protein Cell 2010, 1, 520–536. [Google Scholar]
- Chen, C.; Wan, H.; Zhou, Q. The next generation sequencing technology and its application in cancer research. Chin. J. Lung Cancer 2010, 13, 154–159. (in Chinese). [Google Scholar]
- Woollard, P.M.; Mehta, N.A.; Vamathevan, J.J.; van Horn, S.; Bonde, B.K.; Dow, D.J. The application of next-generation sequencing technologies to drug discovery and development. Drug Discov. Today 2011, 16, 512–519. [Google Scholar]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar]
- Yu, H.; Braun, P.; Yildirim, M.A.; Lemmens, I.; Venkatesan, K.; Sahalie, J.; Hirozane-Kishikawa, T.; Gebreab, F.; Li, N.; Simonis, N.; et al. High-quality binary protein interaction map of the yeast interactome network. Science 2008, 322, 104–110. [Google Scholar]
- Yu, H.; Tardivo, L.; Tam, S.; Weiner, E.; Gebreab, F.; Fan, C.; Svrzikapa, N.; Hirozane-Kishikawa, T.; Rietman, E.; Yang, X.; et al. Next-generation sequencing to generate interactome datasets. Nat. Methods 2011, 8, 478–480. [Google Scholar]
- Weimann, M.; Grossmann, A.; Woodsmith, J.; Ozkan, Z.; Birth, P.; Meierhofer, D.; Benlasfer, N.; Valovka, T.; Timmermann, B.; Wanker, E.E.; et al. A Y2H-seq approach defines the human protein methyltransferase interactome. Nat. Methods 2013, 10, 339–342. [Google Scholar]
- Fujimori, S.; Hirai, N.; Ohashi, H.; Masuoka, K.; Nishikimi, A.; Fukui, Y.; Washio, T.; Oshikubo, T.; Yamashita, T.; Miyamoto-Sato, E. Next-generation sequencing coupled with a cell-free display technology for high-throughput production of reliable interactome data. Sci. Rep 2012, 2, 691. [Google Scholar]
- Roberts, R.W. Totally in vitro protein selection using mRNA-protein fusions and ribosome display. Curr. Opin. Chem. Biol 1999, 3, 268–273. [Google Scholar]
- Wang, H.; Liu, R. Advantages of mRNA display selections over other selection techniques for investigation of protein-protein interactions. Expert Rev. Proteomics 2011, 8, 335–346. [Google Scholar]
- Takahashi, T.T.; Austin, R.J.; Roberts, R.W. mRNA display: Ligand discovery, interaction analysis and beyond. Trends Biochem. Sci 2003, 28, 159–165. [Google Scholar]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar]
- Baker, M. Big biology: The ‘omes puzzle. Nature 2013, 494, 416–419. [Google Scholar]
- Dimitrakopoulou, K.; Dimitrakopoulos, G.N.; Sgarbas, K.N.; Bezerianos, A. Tamoxifen integromics and personalized medicine: Dynamic modular transformations underpinning response to tamoxifen in breast cancer treatment. OMICS 2014, 18, 15–33. [Google Scholar]
- Sumida, T.; Yanagawa, H.; Doi, N. In vitro selection of fab fragments by mRNA display and gene-linking emulsion PCR. J. Nucleic Acids 2012, 2012, 371379. [Google Scholar]
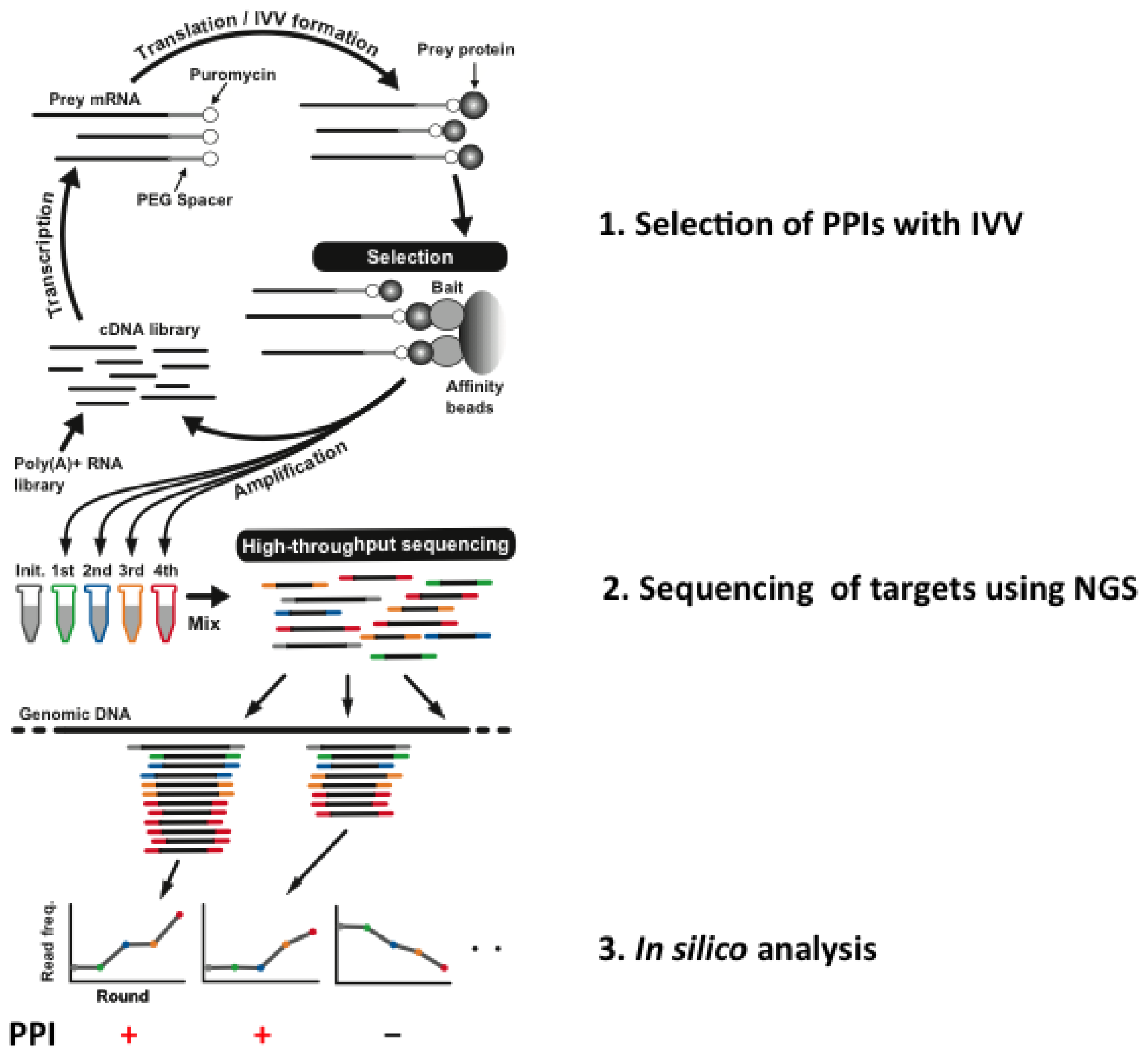

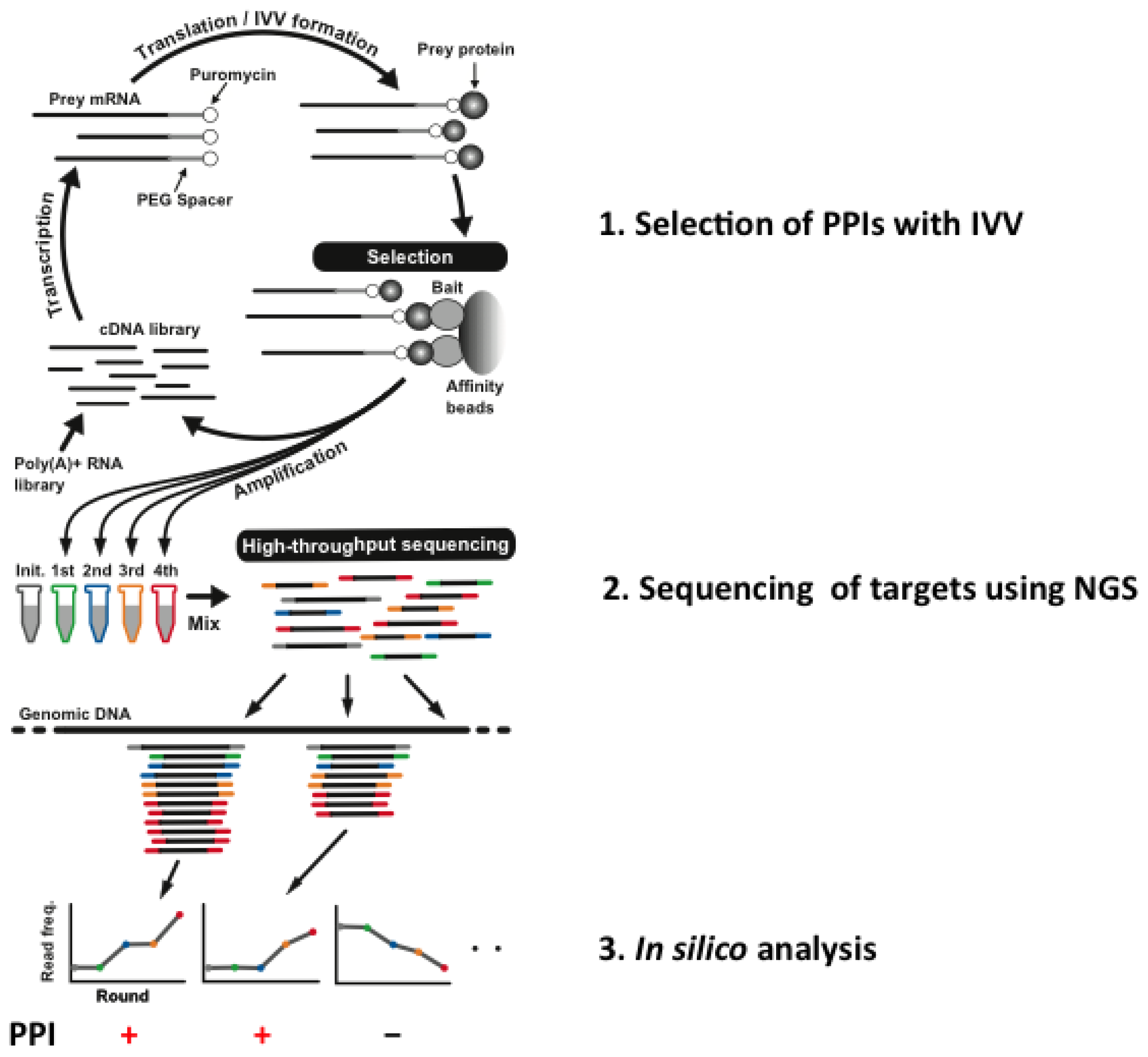{kind=link}
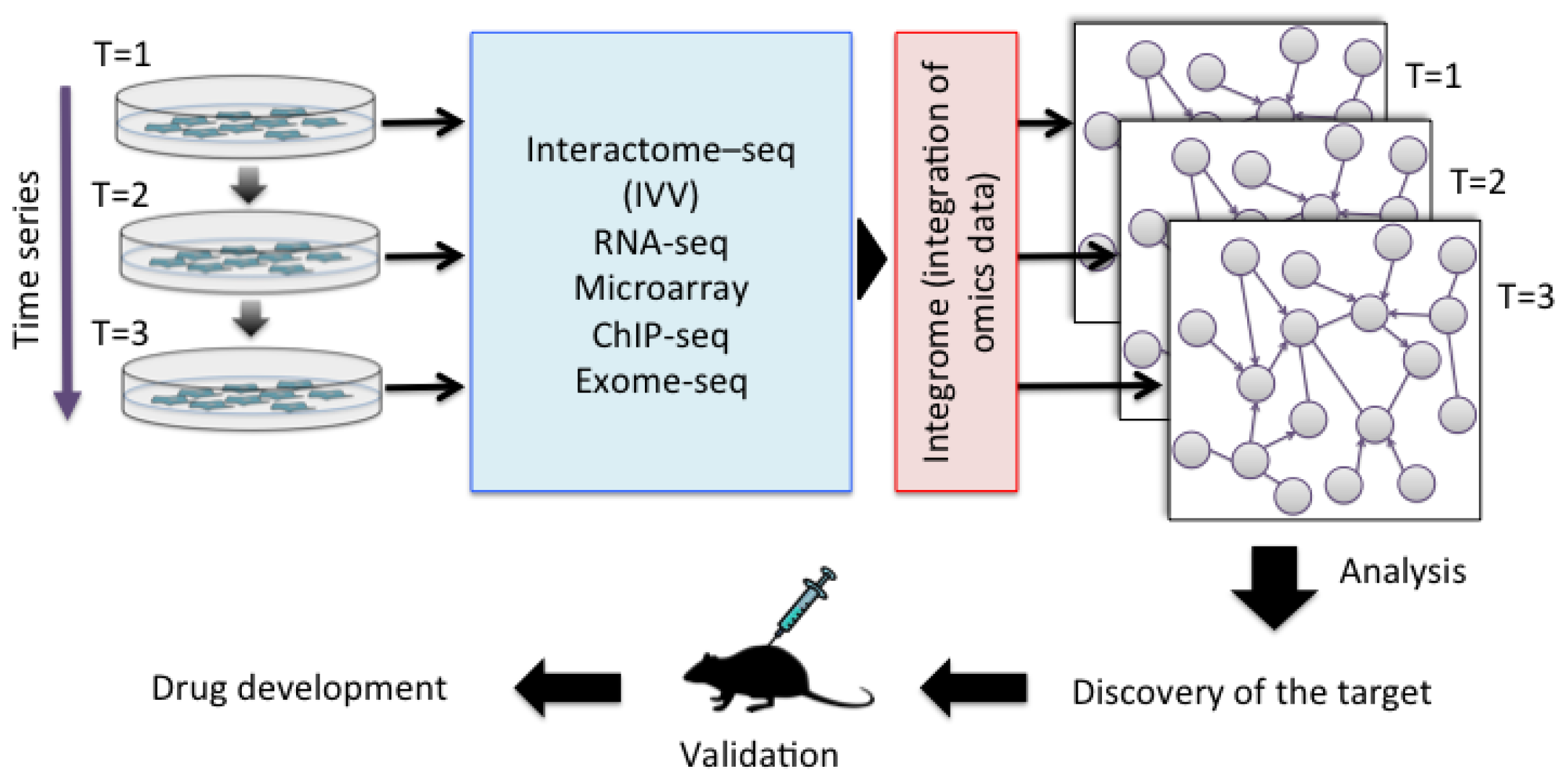{kind=link}
| Method | Experimental system | Library size | Cell cloning required | Next generation sequencing |
|---|---|---|---|---|
| Y2Ha | In vivo | 106 | Yes | Applicable, but limited |
| AP-MSb | In vivo | - | Yes | Inapplicable |
| IVV | In vitro | 1012 | No | Applicable and effective |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ohashi, H.; Miyamoto-Sato, E. Towards Personalized Medicine Mediated by in Vitro Virus-Based Interactome Approaches. Int. J. Mol. Sci. 2014, 15, 6717-6724. https://doi.org/10.3390/ijms15046717
Ohashi H, Miyamoto-Sato E. Towards Personalized Medicine Mediated by in Vitro Virus-Based Interactome Approaches. International Journal of Molecular Sciences. 2014; 15(4):6717-6724. https://doi.org/10.3390/ijms15046717
Chicago/Turabian StyleOhashi, Hiroyuki, and Etsuko Miyamoto-Sato. 2014. "Towards Personalized Medicine Mediated by in Vitro Virus-Based Interactome Approaches" International Journal of Molecular Sciences 15, no. 4: 6717-6724. https://doi.org/10.3390/ijms15046717