MALDI Q-TOF CID MS for Diagnostic Ion Screening of Human Milk Oligosaccharide Samples

Abstract
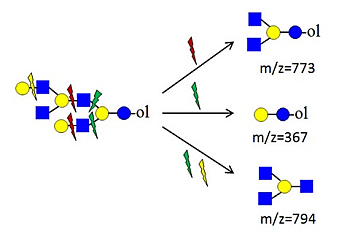
:
1. Introduction
2. Results and Discussion
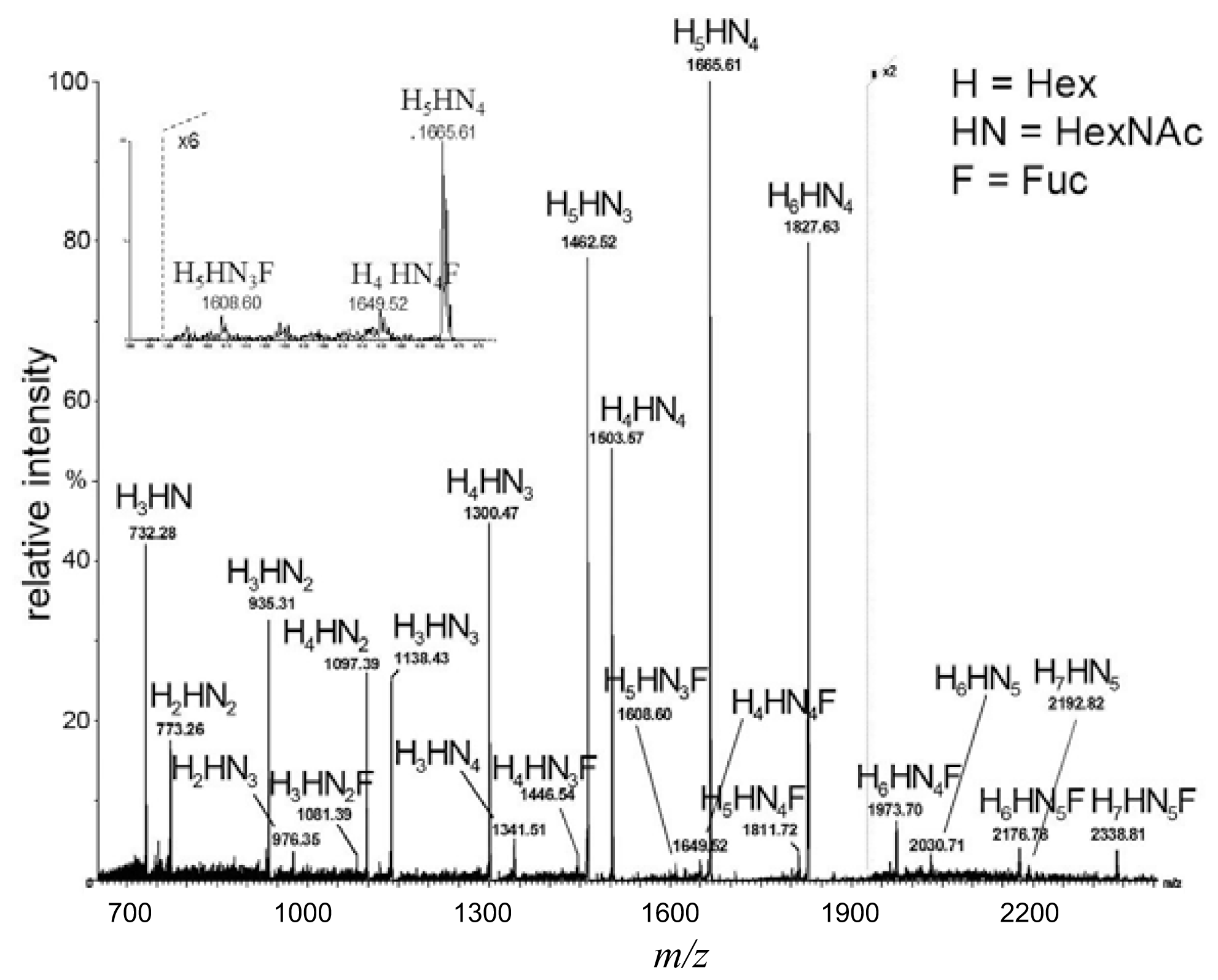
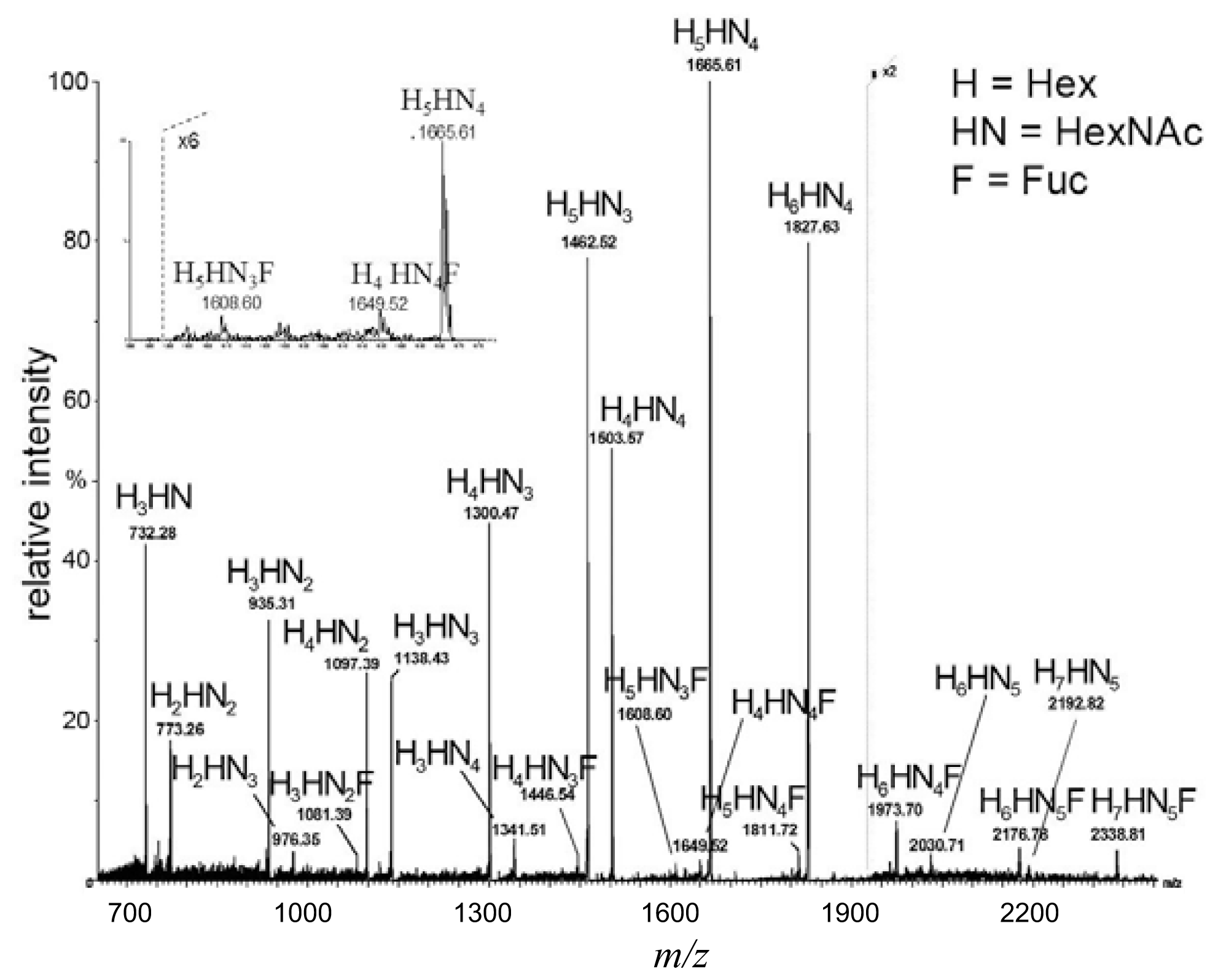
2.1. MALDI TOF (Matrix-Assisted Laser Desorption/Ionization Time-of-Flight) Mapping of Complex Oligosaccharide Mixtures
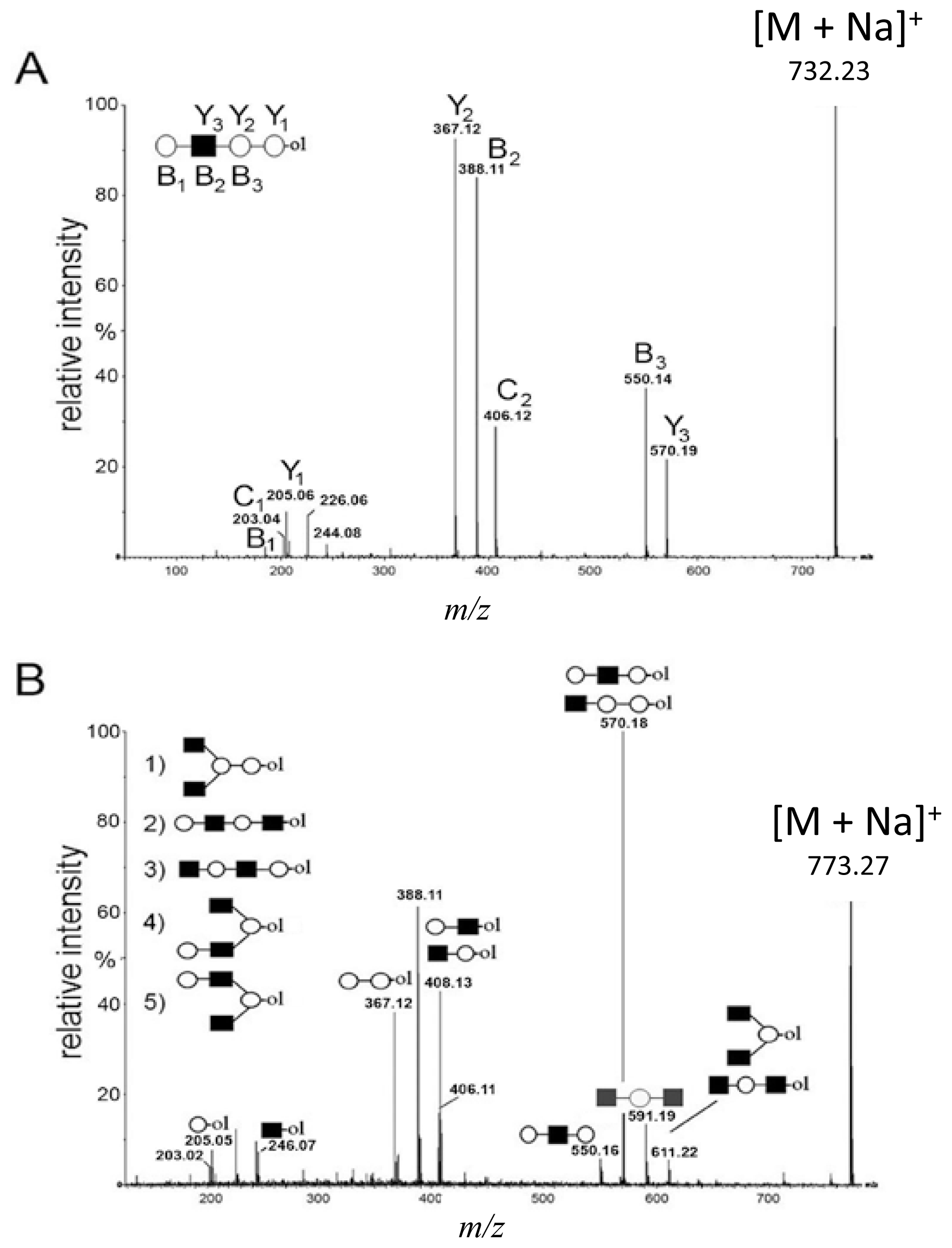
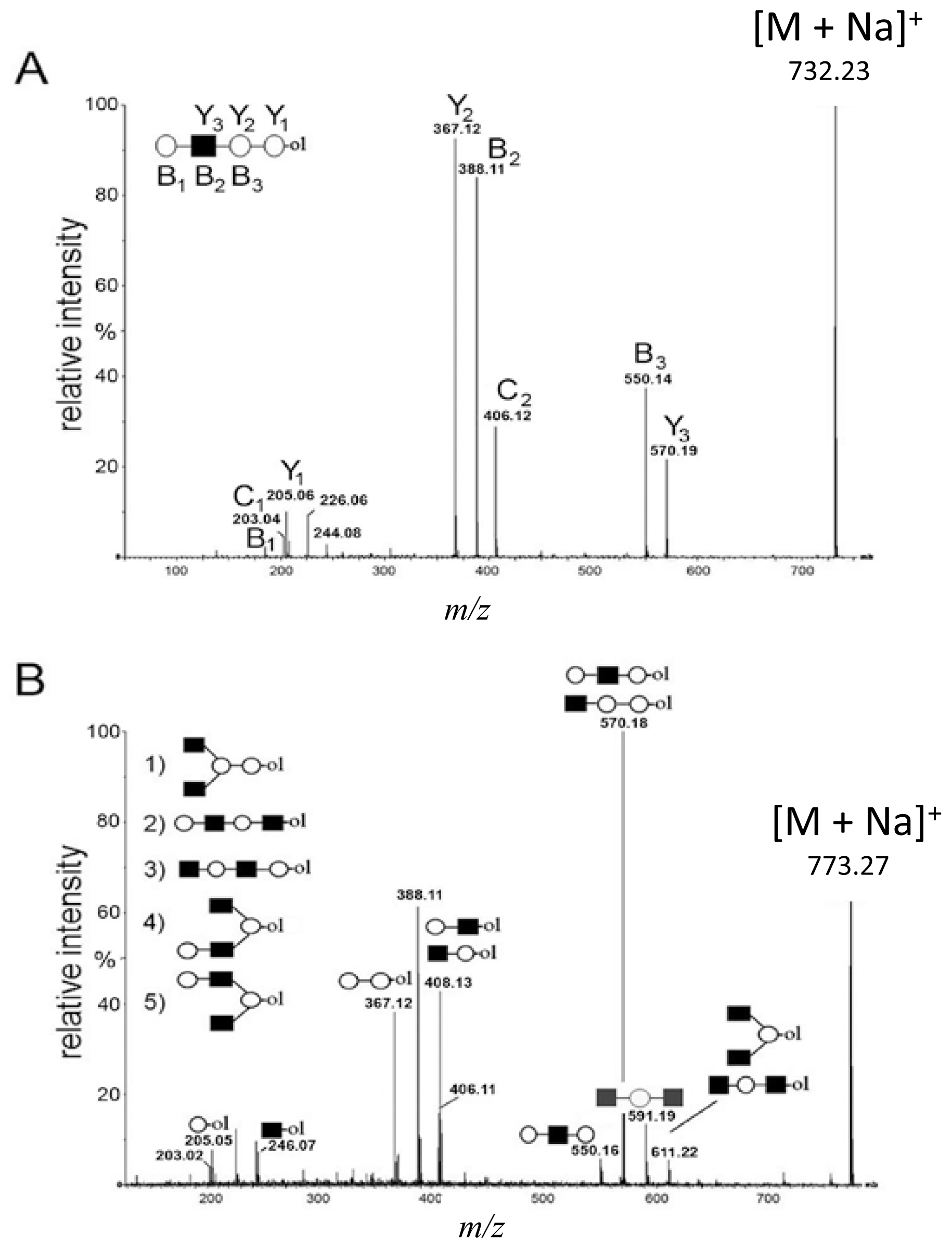
2.2. De Novo MALDI Q-TOF CID Data Analysis Using Diagnostic Ions
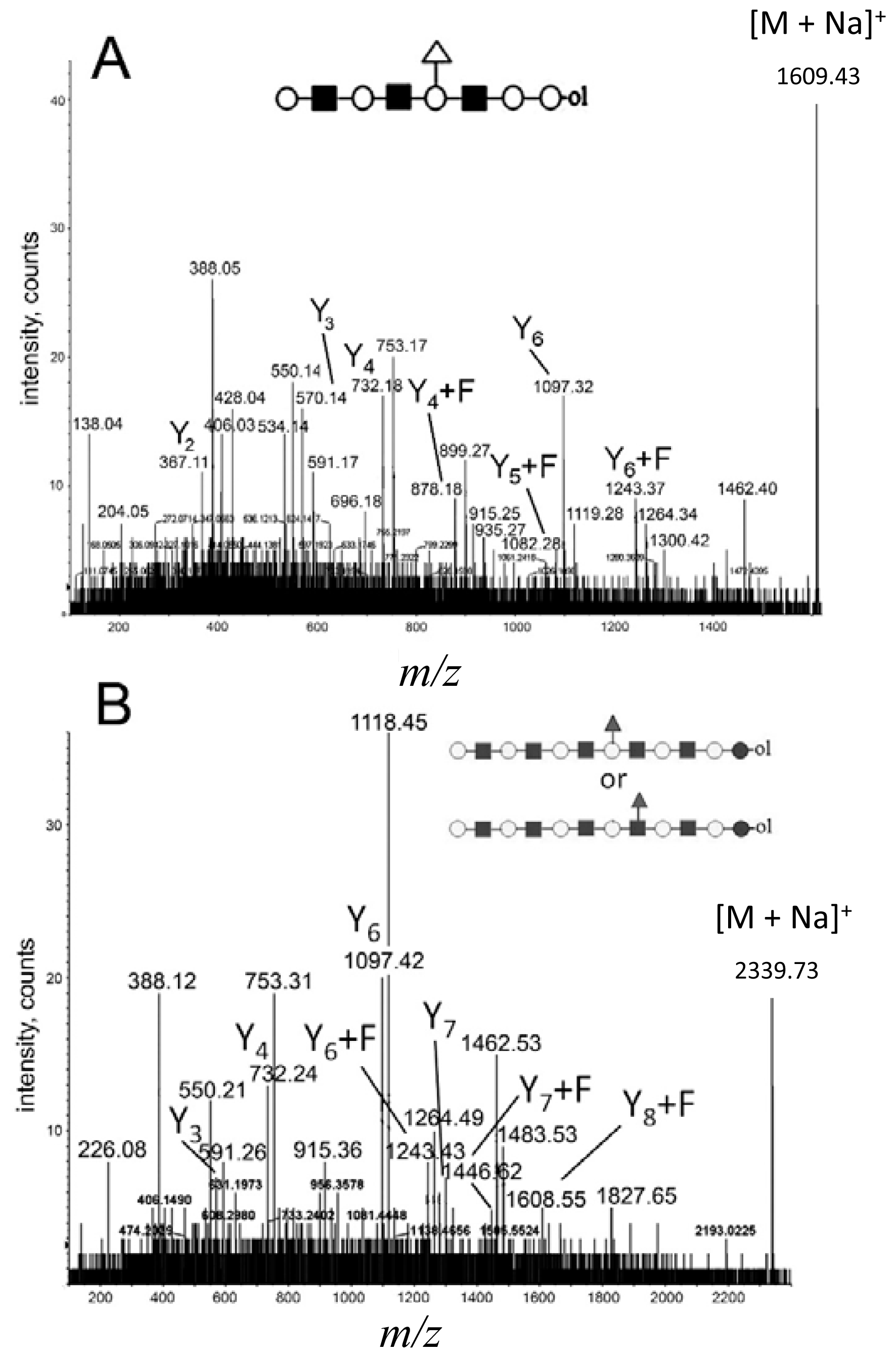
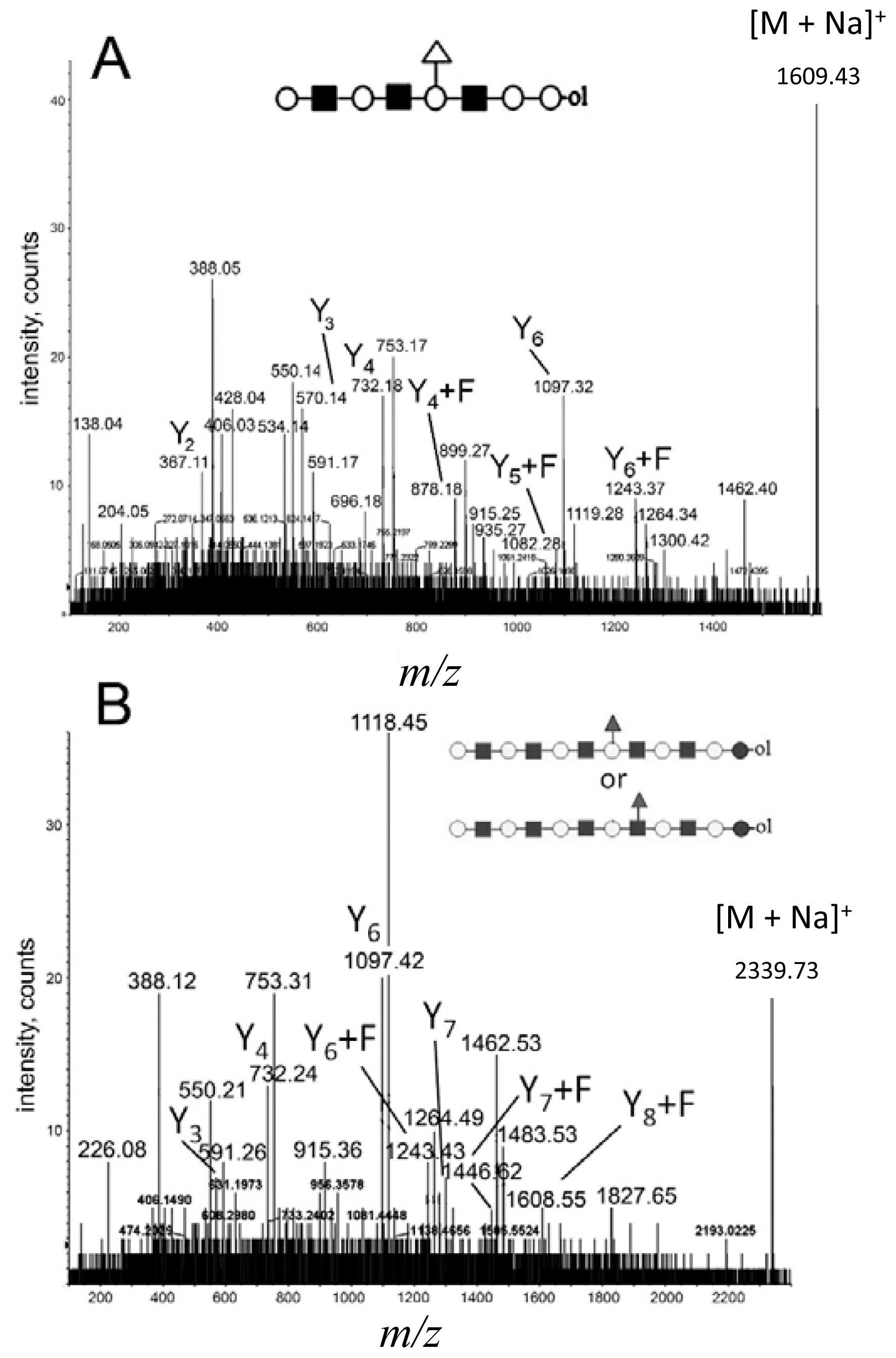
2.3. Analysis of Branched Higher Molecular Weight Oligosaccharides
2.4. MALDI Q-TOF CID of Low-Intensity and High Molecular Weight Parent Ions
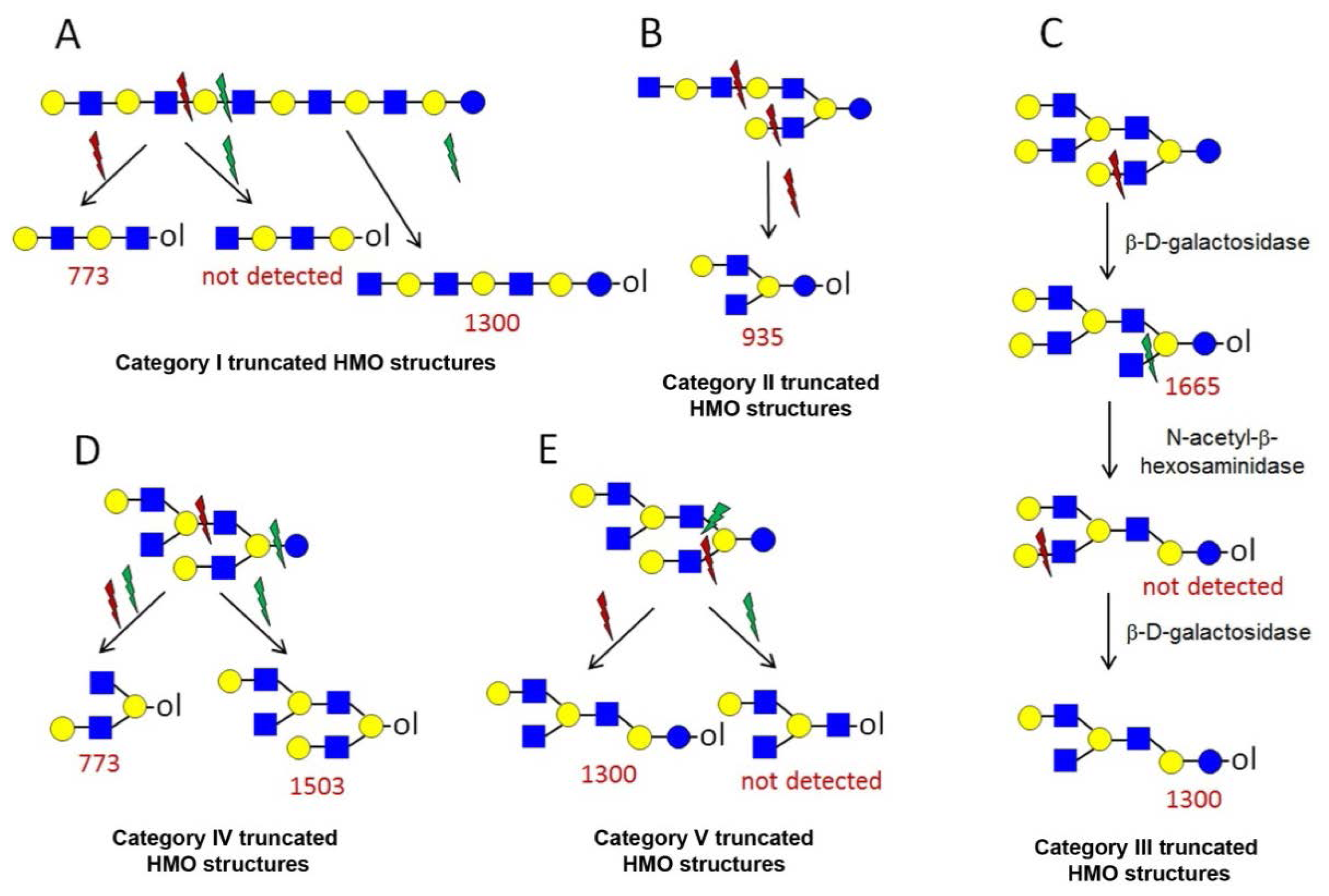
2.5. Analysis of HMO Truncated Structures
2.6. Fragmentation of the Ion at m/z = 1341
2.7. General Conclusions about Identified HMO Structures
3. Experimental Section
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsM.J. performed experiments on the TofSpec 2E and QStar Pulsar i instruments, analysed the data and wrote the manuscript. R.T.W. designed the Micromass prototype MALDI Q-TOF instrument and performed the related experiments in this work. He also contributed to the Experimental Section. G.P. developed the isolation procedure for HMO and prepared the sample for analysis. J.P.K. designed the experiment and wrote the manuscript.
References
- Coppa, G.V.; Gabrielli, O.; Pierani, P.; Catassi, C.; Carlucci, A.; Giorgi, P.L. Changes in carbohydrate composition in human milk over 4 months of lactation. Pediatrics 1993, 91, 637–641. [Google Scholar]
- Wu, S.; Tao, N.; German, J.B.; Grimm, R.; Lebrilla, C.B. Development of an annotated library of neutral human milk oligosaccharides. J. Proteome Res 2010, 9, 4138–4151. [Google Scholar]
- Wu, S.; Grimm, R.; German, J.B.; Lebrilla, C.B. Annotation and structural analysis of sialylated human milk oligosaccharides. J. Proteome Res 2011, 10, 856–868. [Google Scholar]
- Marino, K.; Lane, J.A.; Abrahams, J.L.; Struwe, W.B.; Harvey, D.J.; Marotta, M.; Hickey, R.M.; Rudd, P.M. Method for milk oligosaccharide profiling by 2-aminobenzamide labeling and hydrophilic interaction chromatography. Glycobiology 2011, 21, 1317–1330. [Google Scholar]
- Zivkovic, A.M.; German, J.B.; Lebrilla, C.B.; Mills, D.A. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc. Natl. Acad. Sci. USA 2011, 108, 4653–4658. [Google Scholar]
- Newburg, D.S.; Ruiz-Palacios, G.M.; Morrow, A.L. Human milk glycans protect infants against enteric pathogens. Annu. Rev. Nutr 2005, 25, 37–58. [Google Scholar]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep 2006, 7, 688–693. [Google Scholar]
- Wang, B.; Bing, Y.; Karim, M.; Sun, Y.; McGreevy, P.; Petocz, P.; Held, S.; Brand-Miller, J. Dietary sialic acid supplementation improves learning and memory in piglets. Am. J. Clin. Nutr 2007, 85, 561–569. [Google Scholar]
- Wang, B. Molecular mechanism underlying sialic acid as an essential nutrient for brain development and cognition. Adv. Nutr 2012, 3, 465S–472S. [Google Scholar]
- De Leoz, M.L.; Gaerlan, S.C.; Strum, J.S.; Dimapasoc, L.M.; Mirmiran, M.; Tancredi, D.J.; Smilowitz, J.T.; Kalanetra, K.M.; Mills, D.A.; German, J.B.; et al. Lacto-N-tetraose, fucosylation, and secretor status are highly variable in human milk oligosaccharides from women delivering preterm. J. Prot. Res 2012, 11, 4662–4672. [Google Scholar]
- Stahl, B.; Thurl, S.; Zeng, J.; Karas, M.; Hillenkamp, F.; Steup, M.; Sawatzki, G. Oligosaccharides from human milk as revealed by matrix-assisted laser desorption/ionization mass spectrometry. Anal. Biochem 1994, 223, 218–226. [Google Scholar]
- Finke, B.; Stahl, B.; Pfenninger, A.; Karas, M.; Daniel, H.; Sawatzki, G. Analysis of high-molecular-weight oligosaccharides from human milk by liquid chromatography and MALDI-MS. Anal. Chem 1999, 71, 3755–3762. [Google Scholar]
- Xie, Y.; Lebrilla, C.B. Infrared multiphoton dissociation of alkali metal-coordinated oligosaccharides. Anal. Chem 2003, 75, 1590–1598. [Google Scholar]
- Mechref, Y.; Novotny, M.V. Structural characterization of oligosaccharides using MALDI-TOF/TOF tandem mass spectrometry. Anal. Chem 2003, 75, 4895–4903. [Google Scholar]
- Lewandrowski, U.; Resemann, A.; Sickmann, A. Laser-induced dissociation/high-energy collision-induced dissociation fragmentation using MALDI-TOF/TOF-MS instrumentation for the analysis of neutral and acidic oligosaccharides. Anal. Chem 2005, 77, 3274–3283. [Google Scholar]
- Albrecht, S.; Schols, H.A.; van den Heuvel, E.G.; Voragen, A.G.; Gruppen, H. CE-LIF-MS n profiling of oligosaccharides in human milk and feces of breast-fed babies. Electrophoresis 2010, 31, 1264–1273. [Google Scholar]
- Ninonuevo, M.; An, H.; Yin, H.; Killeen, K.; Grimm, R.; Ward, R.; German, B.; Lebrilla, C. Nanoliquid chromatography-mass spectrometry of oligosaccharides employing graphitized carbon chromatography on microchip with a high-accuracy mass analyzer. Electrophoresis 2005, 26, 3641–3649. [Google Scholar]
- Wu, S.; Salcedo, J.; Tang, N.; Waddell, K.; Grimm, R.; German, J.B.; Lebrilla, C.B. Employment of tandem mass spectrometry for the accurate and specific identification of oligosaccharide structures. Anal. Chem 2012, 84, 7456–7462. [Google Scholar]
- Amano, J.; Osanai, M.; Orita, T.; Sugahara, D.; Osumi, K. Structural determination by negative-ion MALDI-QIT-TOFMSn after pyrene derivatization of variously fucosylated oligosaccharides with branched decaose cores from human milk. Glycobiology 2009, 19, 601–614. [Google Scholar]
- Bruntz, R.; Dabrowski, U.; Dabrowski, J.; Ebersold, A.; Peter-Katalinić, J.; Egge, H. Fucose-containing oligosaccharides from human milk from a donor of blood group 0 Lea nonsecretor. Biol. Chem. Hoppe Seyler 1988, 369, 257–273. [Google Scholar]
- Wiederschain, G.Y.; Newburg, D.S. Glycosidase activities and sugar release in human milk. Adv. Exp. Med. Biol 2001, 501, 573–577. [Google Scholar]
- Perreault, H.; Lattová, E.; Šagi, D.; Peter-Katalinić, J. MALDI-MS of glycans and glycoconjugates. In MALDI MS: A Practical Guide to Instrumentation, Methods and Applications, 2nd ed.; Hillenkamp, F., Peter-Katalinić, J., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp. 239–272. [Google Scholar]
- Domon, B.; Costello, C.E. A systematic nomenclature for carbohydrate fragmentation in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J 1988, 5, 397–409. [Google Scholar]
- Chai, W.; Piskarev, V.E.; Zhang, Y.; Lawson, A.M.; Kogelberg, H. Structural determination of novel lacto-N-decaose and its monofucosylated analogue from human milk by electrospray tandem mass spectrometry and 1H NMR spectroscopy. Arch. Biochem. Biophys 2005, 434, 116–127. [Google Scholar]
- Kobata, A. Structures and application of oligosaccharides in human milk. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci 2010, 86, 731–747. [Google Scholar]
- Ericson, C.; Phung, Q.T.; Horn, D.M.; Peters, E.C.; Fitchett, J.R.; Ficarro, S.B.; Salomon, A.R.; Brill, L.M.; Brock, A. An automated noncontact deposition interface for liquid chromatography matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem 2003, 75, 2309–2315. [Google Scholar]
- Amano, J.; Sugahara, D.; Osumi, K.; Tanaka, K. Negative-ion MALDI-QIT-TOFMSn for structural determination of fucosylated and sialylated oligosaccharides labeled with a pyrene derivative. Glycobiology 2009, 19, 592–600. [Google Scholar]
- Vakhrushev, S.Y.; Dadimov, D.; Peter-Katalinić, J. Software platform for high-throughput glycomics. Anal. Chem 2009, 81, 3252–3260. [Google Scholar]
 denotes N-acetylglucosamine,
denotes N-acetylglucosamine,
 galactose and
galactose and
 glucose.
glucose.
 ,
,
 and
and
 denote ions relevant to the explanation in the text.
denotes N-acetylglucosamine,
galactose and
glucose.
,
and
denote ions relevant to the explanation in the text.
denote ions relevant to the explanation in the text.
denotes N-acetylglucosamine,
galactose and
glucose.
,
and
denote ions relevant to the explanation in the text.

 and
and
 depict various possible hydrolysis events, each of which, alone (A–D right, E) or combined (D left), yield different oligosaccharide structure types shown in the figure.
denotes N-acetylglucosamine,
galactose and
glucose.
and
depict various possible hydrolysis events, each of which, alone (A–D right, E) or combined (D left), yield different oligosaccharide structure types shown in the figure.
denotes N-acetylglucosamine,
galactose and
glucose.
depict various possible hydrolysis events, each of which, alone (A–D right, E) or combined (D left), yield different oligosaccharide structure types shown in the figure.
denotes N-acetylglucosamine,
galactose and
glucose.
and
depict various possible hydrolysis events, each of which, alone (A–D right, E) or combined (D left), yield different oligosaccharide structure types shown in the figure.
denotes N-acetylglucosamine,
galactose and
glucose.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of ions | m/z | Type | Structure | |
|---|---|---|---|---|
| non-diagnostic ions | 753 | B4 or internal B/Y fragment ion |  | |
| 771 | C4 or internal C/Z fragment ion | 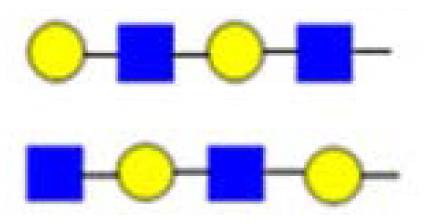 | ||
| diagnostic ions | 205 | hexose at reducing end |  | |
| 246 | HexNAc at reducing end (non-biosynthetic) |  | ||
| 367 | contains biosynthetic core lactose unit |  | ||
| 794 | branching at Hexn≥4 | 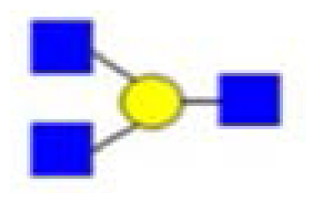 | ||
| conditional diagnostic ions | 408 | non-biosynthetic reducing end structure | no 246 |  |
| no 205 | 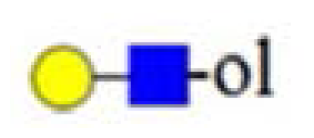 | |||
| 611 | non-biosynthetic reducing end structure | no 246 |  | |
| no 205 | 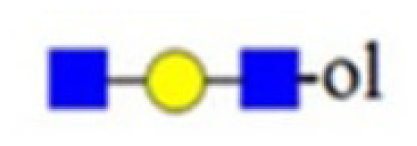 | |||
| 732 | Y4 with core lactose—linear oligosaccharide | no 773 and no 794 |  | |
| 773 | branching at Hex2 (biosynthetic structure) | yes 367 and no 408 |  | |
denotes N-acetylglucosamine,
galactose and
glucose.| Monosaccharide composition | Biosynthetic | Truncated with lactose core | Truncated without lactose core | Diagnostic ions |
|---|---|---|---|---|
| H3HN (732) | 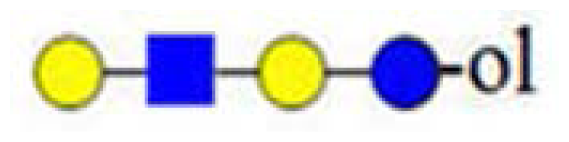 | - | - | 367 |
| H2HN2 (773) | - | 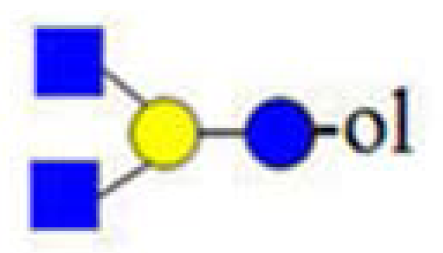 |  | 246, 367, 408 |
| H3HN2 (935) | - | 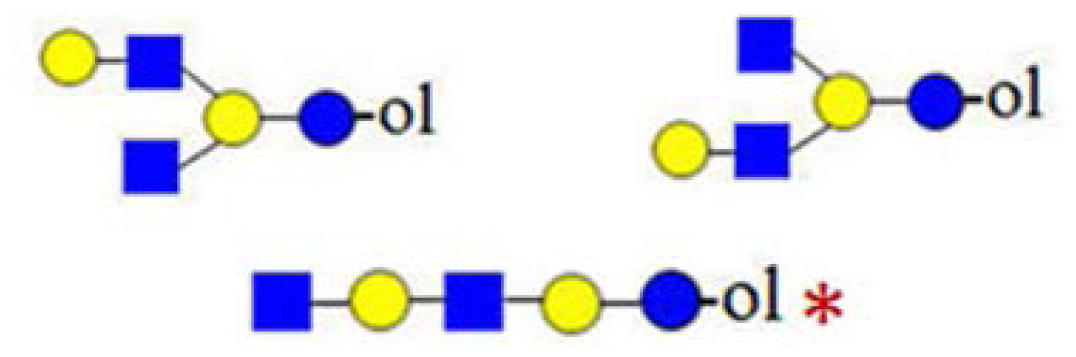 | - | 367, 773 |
| H2HN3 (976) | - | - | 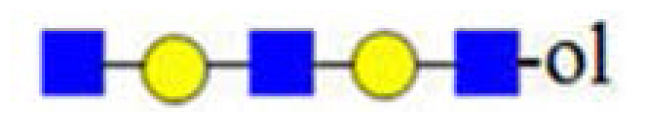 | - |
| H4HN2 (1097) |  | - | - | 367 |
| H3HN3 (1138) | - |  |  | 246, 367, 408, 773, 794 |
| H4HN3 (1300) | - |  | - | 367, 773, 794 |
| H3HN4 (1341) | - |  | - | 773, 794 |
| H5HN3 (1462) |  | - | - | 367 |
| H4HN4 (1503) | - |  | 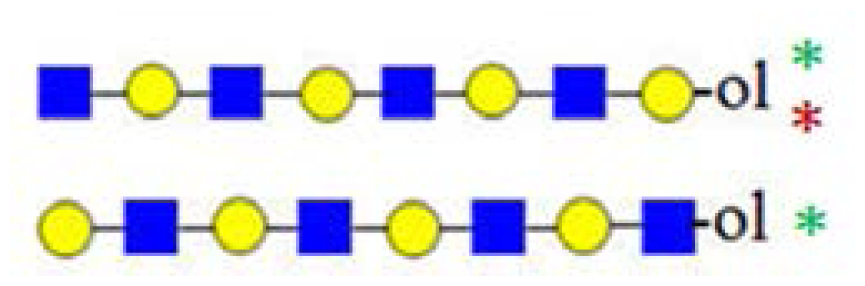 | 367, 408, 773, 794 |
| H5HN4 (1665) | - | 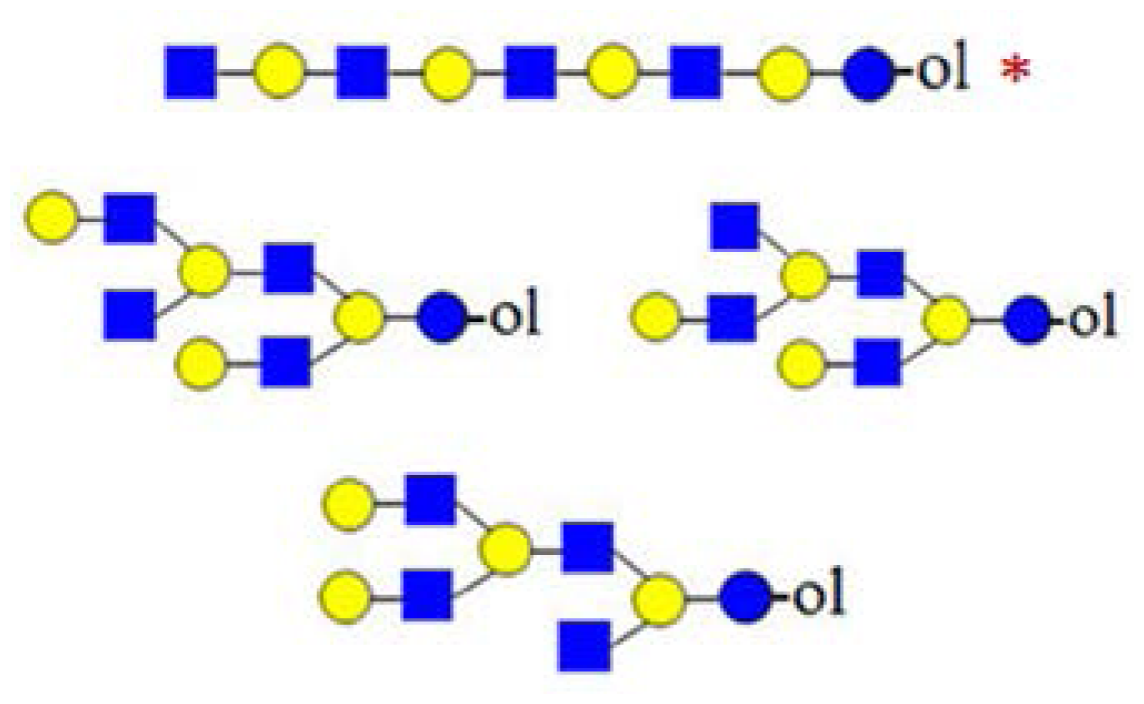 | - | 367, 773, 794 |
| H6HN4 (1827) | 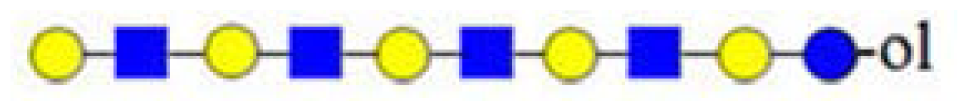 | - | - | 367 |
| H5HN3F (1609) |  | - | - | 878 |
| H6HN4F (1973) | 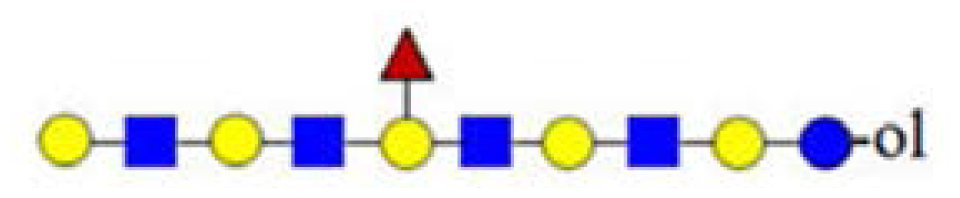 | - | - | 1243 |
| H7HN5F (2338) |  | - | - | 1243 ? |
denotes N-acetylglucosamine,
galactose,
glucose and
 fucose. structures that could be neither confirmed nor excluded. present in very small amounts.
fucose. structures that could be neither confirmed nor excluded. present in very small amounts.| Monosaccharide composition | HMO structure types identified |
|---|---|
| Hn+2HNn |  |
| Hn+1HNn | 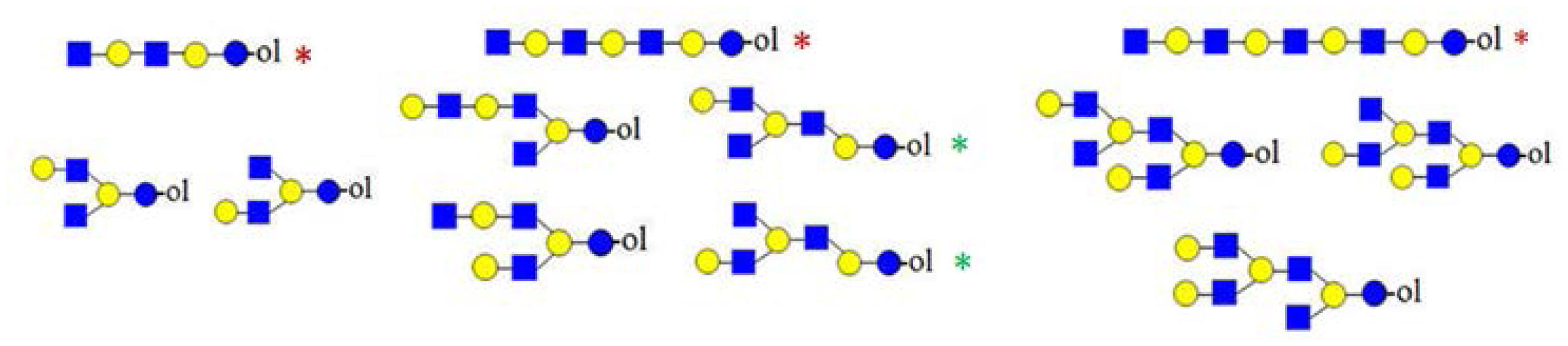 |
| HnHNn | 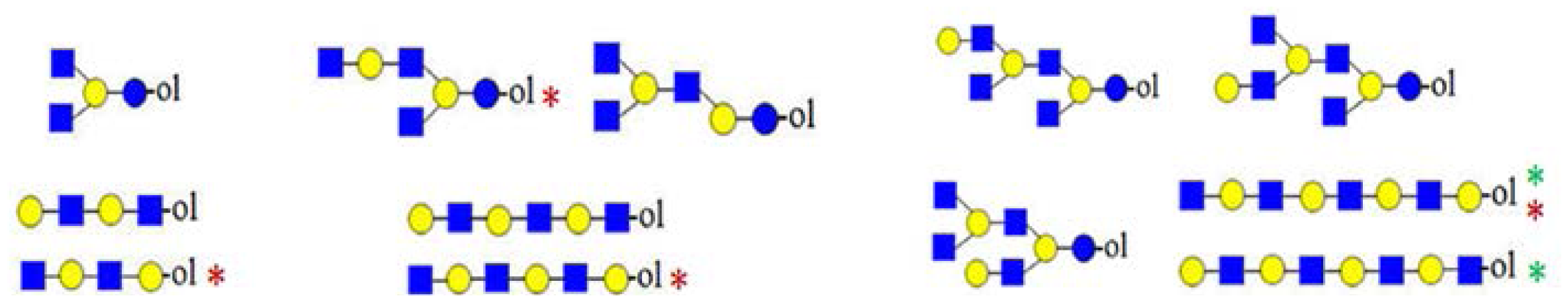 |
| Hn−1HNn | 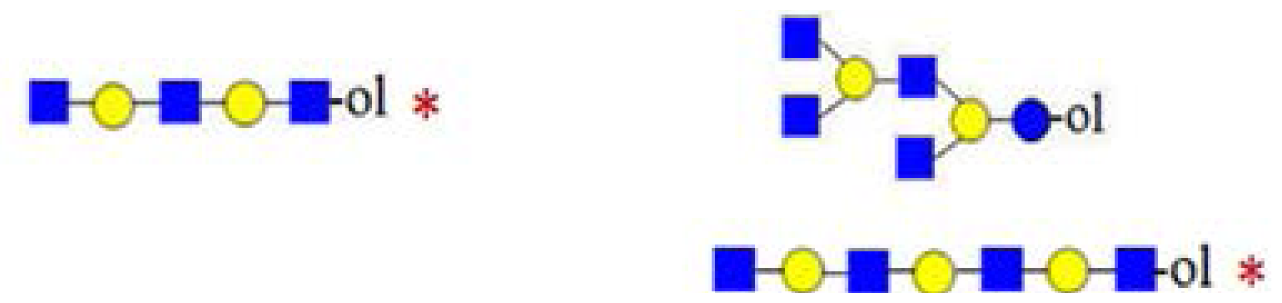 |
denotes N-acetylglucosamine,
galactose and
glucose. structures that could be neither confirmed nor excluded. present in very small amounts.© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jovanović, M.; Tyldesley-Worster, R.; Pohlentz, G.; Peter-Katalinić, J. MALDI Q-TOF CID MS for Diagnostic Ion Screening of Human Milk Oligosaccharide Samples. Int. J. Mol. Sci. 2014, 15, 6527-6543. https://doi.org/10.3390/ijms15046527
Jovanović M, Tyldesley-Worster R, Pohlentz G, Peter-Katalinić J. MALDI Q-TOF CID MS for Diagnostic Ion Screening of Human Milk Oligosaccharide Samples. International Journal of Molecular Sciences. 2014; 15(4):6527-6543. https://doi.org/10.3390/ijms15046527
Chicago/Turabian StyleJovanović, Marko, Richard Tyldesley-Worster, Gottfried Pohlentz, and Jasna Peter-Katalinić. 2014. "MALDI Q-TOF CID MS for Diagnostic Ion Screening of Human Milk Oligosaccharide Samples" International Journal of Molecular Sciences 15, no. 4: 6527-6543. https://doi.org/10.3390/ijms15046527