Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction

Abstract
:
{kind=link}
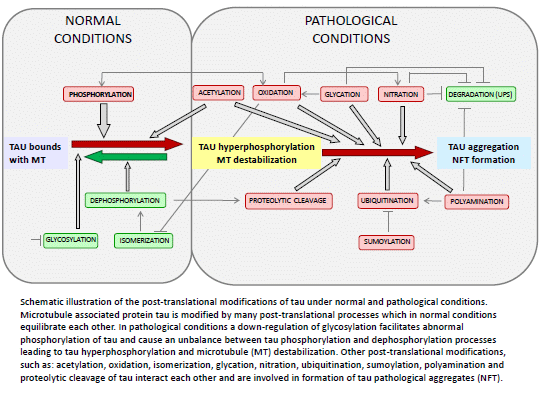{kind=link}
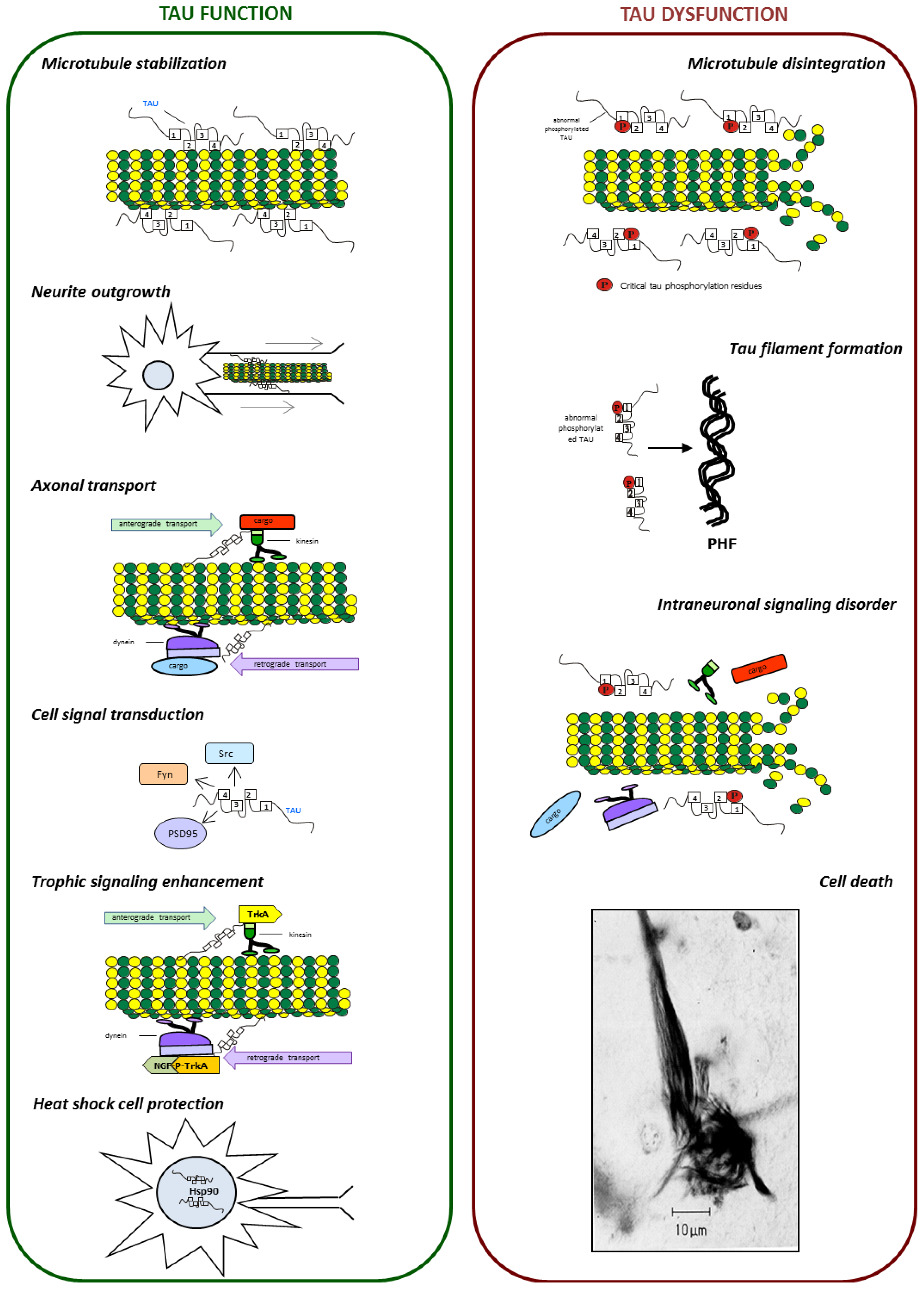
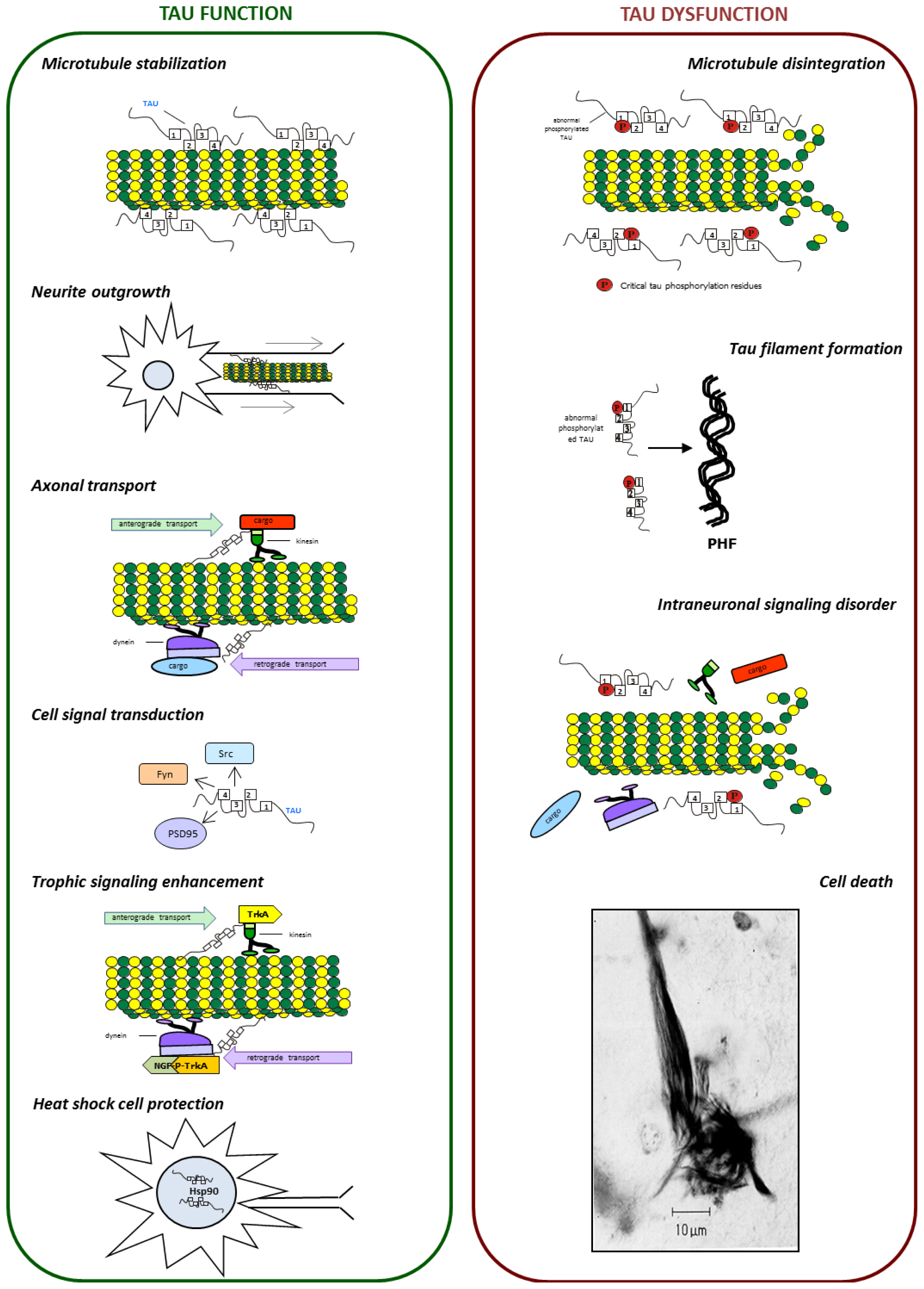{kind=link}
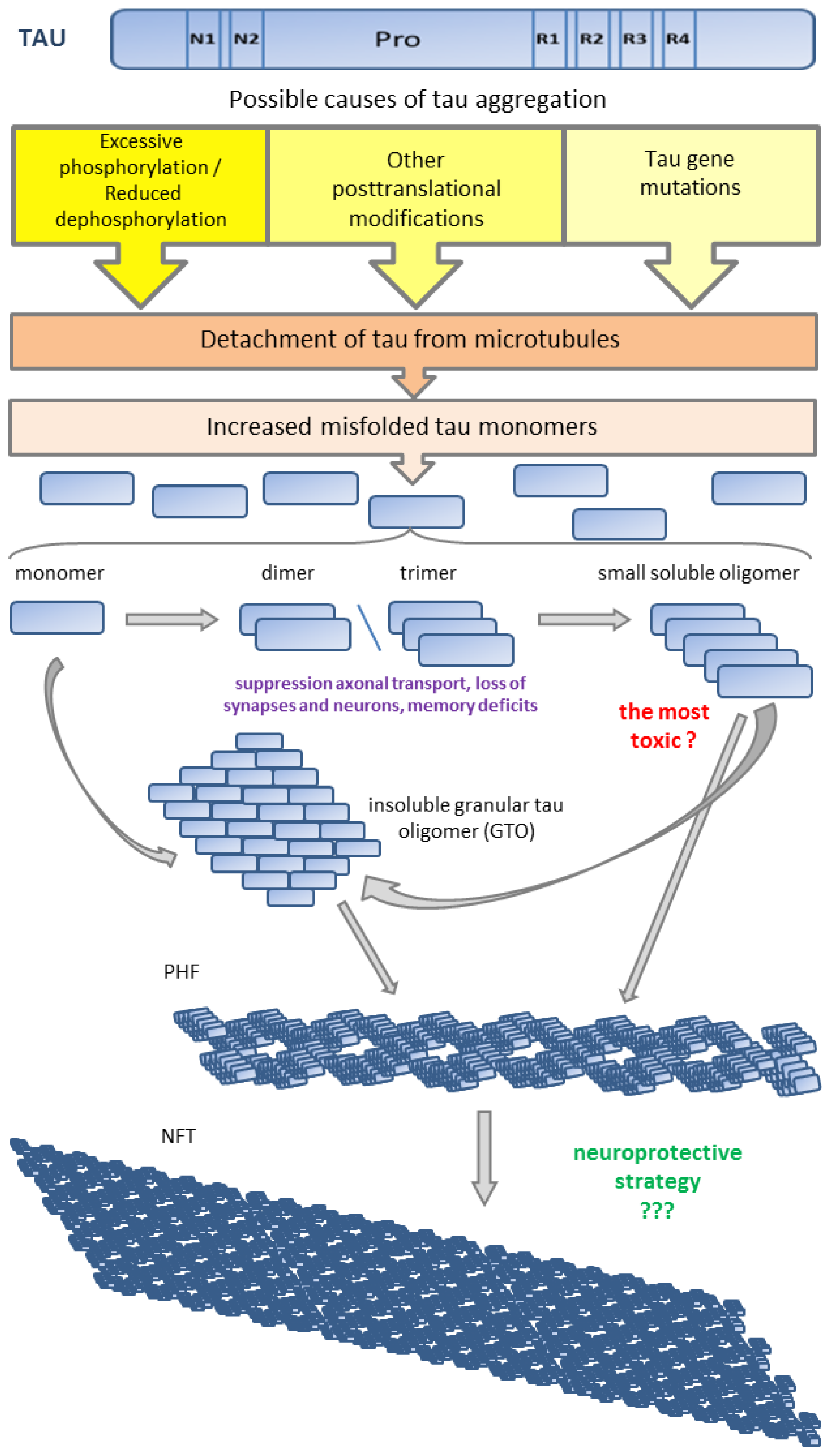{kind=link}
1. Introduction
2. Tau Protein
3. Role of Tau Protein in Neurons
4. Post-Translational Modifications of Tau
4.1. Phosphorylation and Dephosphorylation of Tau Protein
4.1.1. Tau Kinases
4.1.1.1. GSK-3
4.1.1.2. Cdk5
4.1.1.3. JNK
4.1.1.4. CK1
4.1.1.5. Dyrk1A
4.1.1.6. AMPK
4.1.1.7. MARKs
4.1.1.8. PKA
4.1.1.9. TPKI and TPKII
4.1.2. Tau Phosphatases
4.1.2.1. PP2B
4.1.2.2. PP2A
4.1.2.3. PP1
4.1.2.4. PP5
4.1.2.5. CacyBP/SIP
4.1.2.6. TNAP
4.2. Other Post-Translational Modifications
5. Proteins Interacting with Tau
5.1. Amyloid-β
5.2. Pin1
5.3. Fyn Kinase
5.4. Heat Shock Proteins
5.5. FKBP51 and FKBP52 Immunophilins
5.6. α-Synuclein
5.7. PACSIN1
6. Tau Dysfunction
6.1. Tau Aggregates
6.2. Tau and Microtubule Instability
6.3. Tau and Neuronal Transport Defects
6.4. Tau and Neurotrophin Signaling
7. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| 17-AAG | 17-N-allyamino-17-demethoxygeldanamycin |
| ABP | actin-binding proteins |
| AD | Alzheimer’s disease |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| AMPK | adenosine-monophosphate-activated protein kinase |
| APP | amyloid precursor protein |
| a-SN | α-synuclein |
| ATP | adenosine triphosphate |
| Aβ | amyloid beta |
| BDNF | brain-derived neurotrophic factor |
| CacyBP/SIP | calcyclin-binding protein/siah-1-interacting protein |
| cAMP | cyclic adenosine monophosphate |
| CBD | corticobasal degeneration |
| Cdk5 | cyclin-dependent kinase 5 |
| CHIP | chaperone-associated ubiquitin ligase CHIP |
| CK1 | casein kinase 1 |
| CK1 | casein kinase 1 |
| DAPK | death-associated protein kinase |
| DRG | dorsal root ganglia |
| DS | Down syndrome |
| Dyrk1A | dual specificity tyrosine-phosphorylation-regulated kinase 1A |
| E-10 | exon 10 |
| E-2 | exon 2 |
| E-3 | exon 3 |
| EGF | epidermal growth factor |
| EHD | Eps15 homology domain |
| ERK1/2 | kinase extracellular signal-regulated kinase 1/2 |
| F-actin | actin filaments |
| FKBB | FK506-binding protein |
| FTDP-17 | frontotemporal dementia with parkinsonism linked to chromosome 17 |
| Fyn | Proto-oncogene tyrosine-protein kinase Fyn |
| GABA | gamma-aminobutyric acid |
| G-actin | globular actin monomers |
| GCs | growth cones |
| Grb2 | growth factor receptor-bound protein 2 |
| GSK-3 | glycogen synthase kinase-3 |
| GTP | guanosine triphosphate |
| HAP1 | Huntingtin-associated protein 1 |
| Hsc70 | heat shock cognate 70 protein |
| Hsp70 | heat shock protein 70 |
| Hsp90 | Heat shock protein 90 |
| Hsps | heat shock proteins |
| I2 | SET/inhibitor 2 |
| JNK | c-Jun amino-terminal kinase |
| KHC/KIF5 | kinesin-1 |
| KIF1 | kinesin-3 |
| KIF3 | kinesin-2 |
| KIFs | kinesin proteins |
| KO | knockout |
| Kv | voltage-gated potassium channels |
| M1 | muscarinic receptor 1 |
| M3 | muscarinic receptor 3 |
| MAP2 | microtubule associated protein |
| MAPKs | mitogen-activated protein kinases |
| MAPs | microtubule-associated-proteins |
| MAPT | microtubule-associated protein tau |
| MARKs | microtubule-affinity regulating kinases |
| MBD | microtubule binding domain |
| MF | microfilaments |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mRNA | messenger ribonucleic acid |
| mRNP | messenger ribonucleoprotein |
| MT | microtubules |
| MTOC | microtubule organizing center |
| NF | neurofilaments |
| NFH | neurofilament heavy |
| NFM | neurofilament medium |
| NFTs | neurofibrillary tangles |
| NGF | nerve growth factor |
| NHL | neurofilament light |
| NMDAR | N-methyl-d-aspartate receptor |
| NMR | spectroscopy nuclear magnetic resonance spectroscopy |
| non-PDPK | non-proline-directed protein kinase |
| N-WASP | neural Wiskott-Aldrich syndrome protein |
| PACSIN1/SYNDAPIN1 | protein kinase C and casein kinase substrate in neurons protein 1 |
| P-AMPK | activated/phosphorylated AMPK |
| PCH | Pombe Cdc15 homology proteins |
| PD | Parkinson’s disease |
| PDC | parkinsonism dementia complex of Guam |
| PDPK | proline-directed protein kinases |
| PHFs | paired helical filaments |
| PiD | Pick’s disease |
| Pin1 | peptidyl-prolyl cis-trans isomerase 1 |
| PIP3 | phosphatidylinositol (3,4,5)-trisphosphate |
| p-JNK | phospho-c-Jun amino-terminal kinase |
| PKA | protein kinase A |
| PP1 | protein phosphatase 1 |
| PP2A | protein phosphatase 2A |
| PP2B | calcineurin |
| PP5 | protein phosphatase 5 |
| PPI | peptidyl-prolyl cis/trans isomerase |
| PPs | protein phosphatases |
| Pre-mRNA | precursor mRNA |
| PSD-95 | postsynaptic density protein 95 |
| PSP | progressive supranuclear palsy |
| P-tau | phosphorylated tau |
| Rab5 | Ras-related protein Rab-5A |
| SH3 | domain The SRC homology 3 domain |
| Src/cSrc | proto-oncogene tyrosine-protein kinase Src |
| TNAP | tissue-nonspecific alkaline phosphatase |
| TPK | tyrosine protein kinases |
| TPKI | tau protein kinase I |
| TPKII | tau protein kinase II |
| TrkA | neurotrophic tyrosine kinase receptor, type 1 |
| TrkB | neurotrophic tyrosine kinase, receptor, type 2 |
| UPS | ubiquitin proteasome system |
- Author ContributionsEach author has participated sufficiently in the work to take public responsibility for appropriate portions of the article content.
- (1)
- Authors who made substantial contributions to conception and design of the review: G. Niewiadomska, A. Filipek, A. Mietelska-Porowska, M. Goras, and U. Wasik
- (2)
- Authors who participate in drafting the article:U. Wasik—Paragraph 4.1.1A. Filipek—Paragraph 4.1.2M. Goras—Paragraph 5
- (3)
- All Authors gave final approval of the version to be submitted and any revised version.
References
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar]
- Binder, L.I.; Frankfurter, A.; Rebhun, K.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol 1985, 101, 1371–1378. [Google Scholar]
- Hirokawa, N.; Funakoshi, T.; Sato-Harada, R.; Kanai, Y. Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J. Cell Biol. 1996, 132, 667–679. [Google Scholar]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar]
- Merino-Serrais, P.; Benavides-Piccione, R.; Blazquez-Llorca, L.; Kastanauskaite, A.; Rábano, A.; Avila, J.; DeFelipe, J. The influence of phospho-τ on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain 2013, 136, 1913–1928. [Google Scholar]
- Zempel, H.; Luedtke, J.; Kumar, Y.; Biernat, J.; Dawson, H.; Mandelkow, E.; Mandelkow, E.M. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013, 32, 2920–2937. [Google Scholar]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J 1990, 9, 4225–4230. [Google Scholar]
- Niblock, M.; Gallo, J.M. Tau alternative splicing in familial and sporadic tauopathies. Biochem. Soc. Trans. 2012, 40, 677–680. [Google Scholar]
- Majounie, E.; Cross, W.; Newsway, V.; Dillman, A.; Vandrovcova, J.; Morris, C.M.; Nalls, M.A.; Ferrucci, L.; Owen, M.J.; O’Donovan, M.C.; et al. Variation in tau isoform expression in different brain regions and disease states. Neurobiol. Aging 2013, 34, 1922.e7–1922.e12. [Google Scholar]
- Rodríguez-Martín, T.; Cuchillo-Ibáñez, I.; Noble, W.; Nyenya, F.; Anderton, B.H.; Hanger, D.P. Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 2013, 34, 2146–2157. [Google Scholar]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar]
- Götz, J.; Probst, A.; Spillantini, M.G.; Schäfer, T.; Jakes, R.; Bürki, K.; Goedert, M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995, 14, 1304–1313. [Google Scholar]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar]
- Müller, W.E.; Eckert, A.; Kurz, C.; Eckert, G.P.; Leuner, K. Mitochondrial dysfunction: Common final pathway in brain aging and Alzheimer’s disease—Therapeutic aspects. Mol. Neurobiol 2010, 41, 159–171. [Google Scholar]
- Mondragón-Rodríguez, S.; Perry, G.; Zhu, X.; Moreira, P.I.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein as the link between oxidative stress mitochondrial dysfunction and connectivity failure: Implications for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 940603:1–9940603:6. [Google Scholar]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar]
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Böhm, K.J.; Winner, B. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem 2013, 288, 21742–21754. [Google Scholar]
- Eckert, A.; Nisbet, R.; Grimm, A.; Götz, J. March separate strike together—Role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2013. [Google Scholar] [CrossRef]
- Andrews-Zwilling, Y.; Bien-Ly, N.; Xu, Q.; Li, G.; Bernardo, A.; Yoon, S.; Zwilling, D.; Yan, T.X.; Chen, L.; Huang, Y. Apolipoprotein E4 causes age- and tau-dependent impairment of GABAergic interneurons leading to learning and memory deficits in mice. J. Neurosci 2010, 30, 13707–13717. [Google Scholar]
- Gu, Y.; Oyama, F.; Ihara, Y. Tau is widely expressed in rat tissues. J. Neurochem 1996, 67, 1235–1244. [Google Scholar]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar]
- Witman, G.B.; Cleveland, D.W.; Weingarten, M.D.; Kirschner, M.W. Tubulin requires tau for growth onto microtubule initiating sites. Proc. Natl. Acad. Sci. USA 1976, 73, 4070–4074. [Google Scholar]
- Andreadis, A. Tau splicing and intricacies of dementia. J. Cell. Physiol 2012, 227, 1120–1225. [Google Scholar]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci 2001, 24, 1121–1159. [Google Scholar]
- Tashiro, K.; Hasegawa, M.; Ihara, Y.; Iwatsubo, T. Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. Neuroreport 1997, 8, 2797–2801. [Google Scholar]
- Klein, C.; Kramer, E.M.; Cardine, A.M.; Schraven, B.; Brandt, R.; Trotter, J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 2002, 22, 698–707. [Google Scholar]
- Jeganathan, S.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. The natively unfolded character of Tau and its aggregation to Alzheimer-like paired helical filaments. Biochemistry 2008, 47, 10526–10539. [Google Scholar]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue Tau at single residue resolution. PLoS Biol. 2009, 7, e1000034. [Google Scholar]
- Sergeant, N.; Bretteville, A.; Hamdane, M.; Caillet-Boudin, M.L.; Grognet, P.; Bombois, S.; Blum, D.; Delacourte, A.; Pasquier, F.; Vanmechelen, E.; et al. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Exp. Rev. Proteomics 2008, 5, 207–224. [Google Scholar]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med 2012, 2, a006247. [Google Scholar]
- Steiner, B.; Mandelkow, E.M.; Biernat, J.; Gustke, N.; Meyer, H.E.; Schmidt, B.; Mieskes, G.; Soling, H.D.; Drechsel, D.; Kirschner, M.W. Phosphorylation of microtubule-associated protein tau: Identification of the site for Ca2(þ)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J 1990, 9, 3539–3544. [Google Scholar]
- Brandt, R.; Lee, G. Functional organization of microtubule-associated protein tau Identification of regions which affect microtubule growth nucleation and bundle formation in vitro. J. Biol. Chem 1993, 268, 3414–3419. [Google Scholar]
- Sontag, E.; Nunbhakdi-Craig, V.; Lee, G.; Brandt, R.; Kamibayashi, C.; Kuret, J.; White, C.L., 3rd; Mumby, M.C.; Bloom, G.S. Molecular interactions among protein phosphatase 2A tau and microtubules Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem. 1999, 274, 25490–25498. [Google Scholar]
- Hirokawa, N.; Shiomura, Y.; Okabe, S. Tau proteins: The molecular structure and mode of binding on microtubules. J. Cell Biol. 1988, 107, 1449–1459. [Google Scholar]
- Jung, D.; Filliol, D.; Miehe, M.; Rendon, A. Interaction of brain mitochondria with microtubules reconstituted from brain tubulin and MAP2 or TAU. Cell Motil. Cytoskeleton 1993, 24, 245–255. [Google Scholar]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar]
- Al-Bassam, J.; Ozer, R.S.; Safer, D.; Halpain, S.; and Milligan, R.A. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J. Cell Biol. 2002, 157, 1187–1196. [Google Scholar]
- Sergeant, N.; Delacourte, A.; Buée, L. Tau protein as a differential biomarker of tauopathies. Biochim. Biophys. Acta 2005, 1739, 17915hi. [Google Scholar]
- Georgieff, I.S.; Liem, R.K.H.; Couchie, D.; Mavilia, C.; Nunez, J.; Shelanski, M.L. Expression of high molecular weight tau in the central and peripheral nervous systems. J. Cell Sci. 1993, 105, 729–737. [Google Scholar]
- Wang, J.Z.; Liu, F. Microtubule-associated protein tau in development degeneration and protection of neurons. Prog. Neurobiol 2008, 85, 148–175. [Google Scholar]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H., Jr; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci 2009, 29, 12776–12786. [Google Scholar]
- Kosik, K.S. The molecular and cellular biology of tau. Brain Pathol. 1993, 3, 39–43. [Google Scholar]
- Jho, Y.S.; Zhulina, E.B.; Kim, M.W.; Pincus, P.A. Monte carlo simulations of tau proteins: Effect of phosphorylation. Biophys. J. 2010, 99, 2387–2397. [Google Scholar]
- Fischer, D.; Mukrasch, M.D.; Biernat, J.; Bibow, S.; Blackledge, M.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry 2009, 48, 10047–10055. [Google Scholar]
- Avila, J. Tau kinases and phosphatases: Commentary. J. Cell. Mol. Med. 2008, 12, 258–259. [Google Scholar]
- Dolan, P.J.; Johnson, G.V. The role of tau kinases in Alzheimer’s disease. Curr. Opin. Drug Discov. Devel. 2010, 13, 595–603. [Google Scholar]
- Gustke, N.; Trinczek, B.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein and interactions with microtubules. Biochemistry 1994, 33, 9511–9522. [Google Scholar]
- Seitz, A.; Kojima, H.; Oiwa, K.; Mandelkow, E.M.; Song, Y.H.; Mandelkow, E. Single-molecule investigation of the interference between kinesin tau and MAP2c. EMBO J 2002, 21, 4896–4905. [Google Scholar]
- Mukrasch, M.D.; von Bergen, M.; Biernat, J.; Fischer, D.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. The “jaws” of the tau-microtubule interaction. J. Biol. Chem. 2007, 282, 12230–12239. [Google Scholar]
- Sillen, A.; Barbier, P.; Landrieu, I.; Lefebvre, S.; Wieruszeski, J.M.; Leroy, A.; Peyrot, V.; Lippens, G. NMR investigation of the interaction between the neuronal protein tau and the microtubules. Biochemistry 2007, 46, 3055–3064. [Google Scholar]
- Panda, D.; Samuel, J.C.; Massie, M.; Feinstein, S.C.; Wilson, L. Differential regulation of microtubule dynamics by three- and four-repeat tau: Implications for the onset of neurodegenerative disease. Proc. Nat. Acad. Sci. USA 2003, 100, 9548–9553. [Google Scholar]
- Santarella, R.A.; Skiniotis, G.; Goldie, K.N.; Tittmann, P.; Gross, H.; Mandelkow, E.M.; Mandelkow, E.; Hoenger, A. Surface-decoration of microtubules by human tau. J. Mol. Biol. 2004, 339, 539–553. [Google Scholar]
- Mandell, J.W.; Banker, G.A. A spatial gradient of tau protein phosphorylation in nascent axons. J. Neurosci. 1996, 16, 5727–5740. [Google Scholar]
- Yu, J.Z.; Rasenick, M.M. Tau associates with actin in differentiating PC12 cells. FASEB J. 2006, 20, 1452–1461. [Google Scholar]
- He, H.J.; Wang, X.S.; Pan, R.; Wang, D.L.; Liu, M.N.; He, R.Q. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol 2009, 10, 81. [Google Scholar]
- Farias, G.A.; Muñoz, J.P.; Garrido, J.; Maccioni, R.B. Tubulin actin and tau protein interactions and the study of their macromolecular assemblies. J. Cell. Biochem 2002, 85, 315–324. [Google Scholar]
- Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Lee, G. Tau impacts on growth-factor-stimulated actin remodeling. J. Cell Sci. 2007, 120, 748–757. [Google Scholar]
- Niewiadomska, G.; Baksalerska-Pazera, M.; Riedel, G. Altered cellular distribution of phospho-tau proteins coincides with impaired retrograde axonal transport in neurons of aged rats. Ann. N. Y. Acad. Sci. 2005, 1048, 287–295. [Google Scholar]
- Niewiadomska, G.; Baksalerska-Pazera, M.; Lenarcik, I.; Riedel, G.J. Compartmental protein expression of Tau GSK-3beta and TrkA in cholinergic neurons of aged rats. J. Neural Transm. 2006, 113, 1733–1746. [Google Scholar]
- Leugers, C.J.; Lee, G. Tau potentiates nerve growth factor-induced mitogen-activated protein kinase signaling and neurite initiation without a requirement for microtubule binding. J. Biol. Chem. 2010, 285, 19125–19134. [Google Scholar]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase phospholipase Cgamma1 Grb2 and Src family kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar]
- Lee, G.; Thangvel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J. Neurosci. 2004, 4, 2304–2312. [Google Scholar]
- Miyasaka, T.; Watanabe, A.; Saito, Y.; Murayama, S.; Mann, D.M.; Yamazaki, M.; Ravid, R.; Morishima-Kawashima, M.; Nagashima, K.; Ihara, Y. Visualization of newly deposited tau in neurofibrillary tangles and neuropil threads. J. Neuropathol. Exp. Neurol. 2005, 64, 665–674. [Google Scholar]
- Ledesma, M.D.; Bonay, P.; Colaço, C.; Avila, J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J. Biol. Chem. 1994, 269, 21614–21619. [Google Scholar]
- Reyes, J.F.; Reynolds, M.R.; Horowitz, P.M.; Fu, Y.; Guillozet-Bongaarts, A.L.; Berry, R.; Binder, L.I. A possible link between astrocyte activation and tau nitration in Alzheimer’s disease. Neurobiol. Dis. 2008, 31, 198–208. [Google Scholar]
- Arnold, C.S.; Johnson, G.V.; Cole, R.N.; Dong, D.L.; Lee, M.; Hart, G.W. The microtubule-associated protein tau is extensiveely modified with O-linked N-acetylglucosamine. J. Biol. Chem. 1996, 271, 28741–28744. [Google Scholar]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar]
- Landino, L.M.; Skreslet, T.E.; Alston, J.A. Cysteine oxidation of tau and microtubule-associated protein-2 by peroxynitrite: Modulation of microtubule assembly kinetics by the thioredoxin reductase system. J. Biol. Chem. 2004, 279, 35101–35105. [Google Scholar]
- Wilhelmus, M.M.; Grunberg, S.C.; Bol, J.G.; van Dam, A.M.; Hoozemans, J.J.; Rozemuller, A.J.; Drukarch, B. Transglutaminases and transglutaminase-catalyzed cross-links colocalize with the pathological lesions in Alzheimer’s disease brain. Brain Pathol 2009, 19, 612–622. [Google Scholar]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar]
- Cripps, D.; Thomas, S.N.; Jeng, Y.; Yang, F.; Davies, P.; Yang, A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48 Lys-11 and Lys-6 ubiquitin conjugation. J. Biol. Chem. 2006, 281, 10825–10838. [Google Scholar]
- Mondragón-Rodríguez, S.; Basurto-Islas, G.; Binder, L.I.; García-Sierra, F. Conformational changes and cleavage; are these responsible for the tau aggregation in Alzheimer’s disease? Futur. Neurol. 2009, 4, 39–53. [Google Scholar]
- Bhat, R.V.; Shanley, J.; Correll, M.P.; Fieles, W.E.; Keith, R.A.; Scott, C.W.; Lee, C.M. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. USA 2000, 26, 11074–11079. [Google Scholar]
- Wang, Q.M.; Fiol, C.J.; DePaoli-Roach, A.A.; Roach, P.J. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J. Biol. Chem 1994, 269, 14566–14574. [Google Scholar]
- Giese, K.P. GSK-3: A key player in neurodegeneration and memory. IUBMB Life 2009, 61, 516–521. [Google Scholar]
- Ishiguro, K.; Shiratsuchi, A.; Sato, S.; Omori, A.; Arioka, M.; Kobayashi, S.; Uchida, T.; Imahori, K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993, 325, 167–172. [Google Scholar]
- Lovestone, S.; Reynolds, C.H.; Latimer, D.; Davis, D.R.; Anderton, B.H.; Gallo, J.M.; Hanger, D.; Mulot, S.; Marquardt, B.; Stabel, S.; et al. Alzheimer disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 1994, 4, 1077–1086. [Google Scholar]
- Reynolds, C.H.; Betts, J.C.; Blackstock, W.P.; Nebreda, A.R.; Anderton, B.H. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: Differences in vitro between the mitogen-activated protein kinases ERK2 c-Jun N-terminal kinase and P38 and glycogen synthase kinase-3beta. J. Neurochem. 2000, 74, 1587–1595. [Google Scholar]
- Pei, J.J.; Tanaka, T.; Tung, Y.C.; Braak, E.; Iqbal, K.; Grundke-Iqbal, I. Distribution levels and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 1997, 56, 70–78. [Google Scholar]
- Leroy, K.; Boutajangout, A.; Authelet, M.; Woodgett, J.R.; Anderton, B.H.; Brion, J.P. The active form of glycogen synthase kinase-3β is associated with granulovacuolar degeneration in neurons in Alzheimers’s disease. Acta Neuropathol 2002, 103, 91–99. [Google Scholar]
- Yamaguchi, H.; Ishiguro, K.; Uchida, T.; Takashima, A.; Lemere, C.A.; Imahori, K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5 a component of TPK II. Acta Neuropathol. 1996, 92, 232–241. [Google Scholar]
- Alvarez, G.; Munoz-Montano, J.R.; Satrustegui, J.; Avila, J.; Bogonez, E.; Diaz-Nido, J. Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 1999, 453, 260–264. [Google Scholar]
- Tan, W.F.; Cao, X.Z.; Wang, J.K.; Lv, H.W.; Wu, B.Y. Protective effects of lithium treatment for spatial memory deficits induced by tau hyperphosphorylation in splenectomized rats. Clin. Exp. Pharmacol. Physiol. 2010, 37, 1010–1015. [Google Scholar]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E.M.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–156. [Google Scholar]
- Muntané, G.; Dalfó, E.; Martinez, A.; Ferrer, I. Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease and in Parkinson’s disease and related alpha-synucleinopathies. Neuroscience 2008, 152, 913–923. [Google Scholar]
- Gaig, C.; Ezquerra, M.; Marti, M.J.; Valldeoriola, F.; Muñoz, E.; Lladó, A.; Rey, M.J.; Cardozo, A.; Molinuevo, J.L.; Tolosa, E. Screening for the LRRK2 G2019S and codon-1441 mutations in a pathological series of Parkinsonian syndromes and frontotemporal lobar degeneration. J. Neurol. Sci. 2008, 270, 94e8. [Google Scholar]
- Wang, Y.; Liu, W.; He, X.; Zhou, F. Parkinson’s Disease-Associated Dj-1 Mutations increase abnormal phosphorylation of Tau protein through Akt/Gsk-3β pathways. J. Mol. Neurosci. 2013, 51, 911–918. [Google Scholar]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar]
- Köhler, C.; Dinekov, M.; Götz, J. Active glycogen synthase kinase-3 and tau pathology-related tyrosine phosphorylation in pR5 human tau transgenic mice. Neurobiol. Aging 2013, 34, 1369–1379. [Google Scholar]
- Dhavan, R.; Tsai, L.H. A decade of CDK5. Nat. Rev. Mol. Cell. Biol. 2001, 2, 749–759. [Google Scholar]
- Lee, M.S.; Kwon, Y.T.; Li, M.; Peng, J.; Friedlander, R.M.; Tsai, L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 405, 360–364. [Google Scholar]
- Noble, W.; Olm, V.; Takata, K.; Casey, E.; Mary, O.; Meyerson, J.; Gaynor, K.; LaFrancois, J.; Wang, L.; Kondo, T.; et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 2003, 38, 555–565. [Google Scholar]
- Cruz, J.C.; Tseng, H.C.; Goldman, J.A.; Shih, H.; Tsai, L.H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 2003, 30, 471–83. [Google Scholar]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar]
- Piedrahita, D.; Hernández, I.; López-Tobón, A.; Fedorov, D.; Obara, B.; Manjunath, B.S.; Boudreau, R.L.; Davidson, B.; Laferla, F.; Gallego-Gómez, J.C.; et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer mice. J. Neurosci 2010, 30, 13966–13976. [Google Scholar]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 2006, 45, 3134–3145. [Google Scholar]
- Liou, Y.C.; Sun, A.; Ryo, A.; Zhou, X.Z.; Yu, Z.X.; Huang, H.K.; Uchida, T.; Bronson, R.; Bing, G.; Li, X.; et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 2003, 424, 556–61. [Google Scholar]
- Yotsumoto, K.; Saito, T.; Asada, A.; Oikawa, T.; Kimura, T.; Uchida, C.; Ishiguro, K.; Uchida, T.; Hasegawa, M.; Hisanaga, S. Effect of Pin1 or microtubule binding on dephosphorylation of FTDP-17 mutant Tau. J. Biol. Chem 2009, 284, 16840–16847. [Google Scholar]
- Kimura, T.; Tsutsumi, K.; Taoka, M.; Saito, T.; Masuda-Suzukake, M.; Ishiguro, K.; Plattner, F.; Uchida, T.; Isobe, T.; Hasegawa, M.; et al. Isomerase Pin1 stimulates dephosphorylation of tau protein at cyclin-dependent kinase (Cdk5)-dependent Alzheimer phosphorylation sites. J. Biol. Chem 2013, 288, 7968–7977. [Google Scholar]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B. Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P) protein kinase of 38 kDa (p38-P) stress-activated protein kinase (SAPK/JNK-P) and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J. Neural Transm 2001, 108, 1397–1415. [Google Scholar]
- Mohit, A.A.; Martin, J.H.; Miller, C.A. p493F12 kinase: A novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron 1995, 14, 67–78. [Google Scholar]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar]
- Knippschild, U.; Gocht, A.; Wolff, S.; Huber, N.; Löhler, J.; Stöter, M. The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell. Signal. 2005, 17, 675–689. [Google Scholar]
- Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of tau protein by casein kinase-1 converts it to an abnormal Alzheimer-like state. J. Neurochem 1995, 64, 1420–1423. [Google Scholar]
- Kuret, J.; Johnson, G.S.; Cha, D.; Christenson, E.R.; DeMaggio, A.J.; Hoekstra, M.F. Casein kinase 1 is tightly associated with paired-helical filaments isolated from Alzheimer disease brain. J. Neurochem. 1997, 69, 2506–2515. [Google Scholar]
- Li, G.; Yin, H.; Kuret, J. Casein kinase 1 delta phosphorylates tau and disrupts its binding to microtubules. J. Biol. Chem. 2004, 279, 15938–15945. [Google Scholar]
- Schwab, C.; DeMaggio, A.J.; Ghoshal, N.; Binder, L.I.; Kuret, J.; McGeer, P.L. Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol. Aging 2000, 21, 503–510. [Google Scholar]
- Yasojima, K.; Kuret, J.; DeMaggio, A.J.; McGeer, E.; McGeer, P.L. Casein kinase 1 delta mRNA is upregulated in Alzheimer disease brain. Brain Res 2000, 865, 116–120. [Google Scholar]
- Ferrer, I.; Barrachina, M.; Puig, B.; Martínez de Lagrán, M.; Martí, E.; Avila, J.; Dierssen, M. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease Down syndrome Pick disease and related transgenic models. Neurobiol. Dis. 2005, 20, 392–400. [Google Scholar]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.X. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar]
- Ryoo, S.R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.J.; Cho, H.J.; Lee, H.W.; Kim, I.S.; Cheon, Y.H.; Ahn, Y.S.; Chung, S.H.; et al. DYRK1A-mediated hyperphosphorylation of Tau A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem 2007, 282, 34850–34857. [Google Scholar]
- Wegiel, J.; Kaczmarski, W.; Barua, M.; Kuchna, I.; Nowicki, K.; Wang, K.C.; Wegiel, J.; Yang, S.M.; Frackowiak, J.; Mazur-Kolecka, B.; et al. Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J. Neuropathol. Exp. Neurol 2011, 70, 36–50. [Google Scholar]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer disease and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar]
- Thornton, C.; Bright, N.J.; Sastre, M.; Muckett, P.J.; Carling, D. AMP-activated protein kinase (AMPK) is a tau kinase activated in response to amyloid β-peptide exposure. Biochem. J. 2011, 434, 503–512. [Google Scholar]
- Timm, T.; Marx, A.; Panneerselvam, S.; Mandelkow, E.; Mandelkow, E.M. Structure and regulation of MARK a kinase involved in abnormal phosphorylation of Tau protein. BMC Neurosci 2008, 2, S9. [Google Scholar]
- Chin, J.Y.; Knowles, R.B.; Schneider, A.; Drewes, G.; Mandelkow, E.M.; Hyman, B.T. Microtubule-affinity regulating kinase (MARK) is tightly associated with neurofibrillary tangles in Alzheimer brain: A fluorescence resonance energy transfer study. J. Neuropathol. Exp. Neurol. 2000, 59, 966–971. [Google Scholar]
- Gustke, N.; Steiner, B.; Mandelkow, E.M.; Biernat, J.; Meyer, H.E.; Goedert, M.; Mandelkow, E. The Alzheimer-like phosphorylation of tau protein reduces microtubule binding and involves Ser-Pro and Thr-Pro motifs. FEBS Lett. 1992, 307, 199–205. [Google Scholar]
- Augustinack, J.C.; Sanders, J.L.; Tsai, L.H.; Hyman, B.T. Colocalization and fluorescence resonance energy transfer between cdk5 and AT8 suggests a close association in pre-neurofibrillary tangles and neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 2002, 61, 557–564. [Google Scholar]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar]
- Mocanu, M.M.; Nissen, A.; Eckermann, K.; Khlistunova, I.; Biernat, J.; Drexler, D.; Petrova, O.; Schönig, K.; Bujard, H.; Mandelkow, E.; et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation synaptic decay neuronal loss and coassembly with endogenous Tau in inducible mouse models of tauopathy. J. Neurosci. 2008, 28, 737–748. [Google Scholar]
- Gu, G.J.; Lund, H.; Wu, D.; Blokzijl, A.; Classon, C.; von Euler, G.; Landegren, U.; Sunnemark, D.; Kamali-Moghaddam, M. Role of individual MARK isoforms in phosphorylation of tau at Ser262 in Alzheimer disease. Neuromol. Med 2013, 15, 458–469. [Google Scholar]
- Wu, P.R.; Tsai, P.I.; Chen, G.C.; Chou, H.J.; Huang, Y.P.; Chen, Y.H.; Lin, M.Y.; Kimchi, A.; Chien, C.T.; Chen, R.H. DAPK activates MARK1/2 to regulate microtubule assembly neuronal differentiation and tau toxicity. Cell Death Differ 2011, 18, 1507–1520. [Google Scholar]
- Sun, L.; Wang, X.; Liu, S.; Wang, Q.; Wang, J.; Bennecib, M.; Gong, C.X.; Sengupta, A.; Grundke-Iqbal, I.; Iqbal, K. Bilateral injection of isoproterenol into hippocampus induces Alzheimer-like hyperphosphorylation of tau and spatial memory deficit in rat. FEBS Lett. 2005, 579, 251–258. [Google Scholar]
- Liu, F.; Liang, Z.; Shi, J.; Yin, D.; El-Akkad, E.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. PKA modulates GSK-3beta- and cdk5-catalyzed phosphorylation of tau in site- and kinase-specific manners. FEBS Lett 2006, 580, 6269–6274. [Google Scholar]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar]
- Liu, S.J.; Zhang, J.Y.; Li, H.L.; Fang, Z.Y.; Wang, Q.; Deng, H.M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K.; Wang, J.Z. Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J. Biol. Chem 2004, 279, 50078–50088. [Google Scholar]
- Arioka, M.; Tsukamoto, M.; Ishiguro, K.; Kato, R.; Sato, K.; Imahori, K.; Uchida, T. Tau protein kinase II is involved in the regulation of the normal phosphorylation state of tau protein. J. Neurochem. 1993, 60, 461–468. [Google Scholar]
- Imahori, K.; Hoshi, M.; Ishiguro, K.; Sato, K.; Takahashi, M.; Shiurba, R.; Yamaguchi, H.; Takashima, A.; Uchida, T. Possible role of tau protein kinases in pathogenesis of Alzheimer disease. Neurobiol. Aging 1998, 19, S93–S98. [Google Scholar]
- Rank, K.B.; Pauley, A.M.; Bhattacharya, K.; Wang, Z.; Evans, D.B.; Fleck, T.J.; Johnston, J.A.; Sharma, S.K. Direct interaction of soluble human recombinant tau protein with Abeta 1–42 results in tau aggregation and hyperphosphorylation by tau protein kinase II. FEBS Lett. 2002, 514, 263–268. [Google Scholar]
- Takashima, A.; Honda, T.; Yasutake, K.; Michel, G.; Murayama, O.; Murayama, M.; Ishiguro, K.; Yamaguchi, H. Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25–35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res. 1998, 31, 317–323. [Google Scholar]
- Braithwaite, S.P.; Stock, J.B.; Lombroso, P.J.; Nairn, A.C. Protein phosphatases and Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2012, 106, 343–379. [Google Scholar]
- Garver, T.D.; Kincaid, R.L.; Conn, R.A.; Billingsley, M.L. Reduction of calcineurin activity in brain by antisense oligonucleotides leads to persistent phosphorylation of tau protein at Thr181 and Thr231. Mol. Pharmacol. 1999, 55, 632–641. [Google Scholar]
- Rahman, A.; Grundke-Iqbal, I.; Iqbal, K. PP2B isolated from human brain preferentially dephosphorylates Ser-262 and Ser-396 of the Alzheimer disease abnormally hyperphosphorylated tau. J. Neural Transm. 2006, 113, 219–230. [Google Scholar]
- Qian, W.; Yin, X.; Hu, W.; Sh, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X.; Liu, F. Activation of protein phosphatase 2B and hyperphosphorylation of Tau in Alzheimer’s disease. J. Alzheimers Dis. 2011, 23, 617–627. [Google Scholar]
- Kim, Y.; Lee, Y.I.; Seo, M.; Kim, S.Y.; Lee, J.E.; Youn, H.D.; Kim, Y.S.; Juhnn, Y.S. Calcineurin dephosphorylates glycogen synthase kinase-3 beta at serine-9 in neuroblast-derived cells. J. Neurochem. 2009, 111, 344–354. [Google Scholar]
- Iqbal, K.; Alonso, A.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar]
- Landrieu, I.; Smet-Nocca, C.; Amniai, L.; Louis, J.V.; Wieruszeski, J.M.; Goris, J.; Janssens, V.; Lippens, G. Molecular implication of PP2A and Pin1 in the Alzheimer’s disease specific hyperphosphorylation of Tau. PLoS One 2011, 6, e21521. [Google Scholar]
- Merrick, S.E.; Demoise, D.C.; Lee, V.M. Site-specific dephosphorylation of tau protein at Ser202/Thr205 in response to microtubule depolymerization in cultured human neurons involves protein phosphatase 2A. J. Biol. Chem 1996, 271, 5589–5594. [Google Scholar]
- Sontag, J.M.; Nunbhakdi-Craig, V.; White, C.L., 3rd; Halpain, S.; Sontag, E. The protein phosphatase PP2A/Balpha binds to the microtubule-associated proteins Tau and MAP2 at a motif also recognized by the kinase Fyn: Implications for tauopathies. J. Biol. Chem. 2012, 287, 14984–14993. [Google Scholar]
- Tanimukai, H.; Grundke-Iqbal, I.; Iqbal, K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am. J. Pathol. 2005, 166, 1761–1771. [Google Scholar]
- Gharbi-Ayachi, A.; Labbe, J.C.; Burgess, A.; Vigneron, S.; Strub, J.M.; Brioudes, E.; van-Dorsselaer, A.; Castro, A.; Lorca, T. The substrate of Greatwall kinase Arpp 19 controls mitosis by inhibiting protein phosphatase 2A. Science 2010, 330, 1673–1677. [Google Scholar]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010, 330, 1670–1673. [Google Scholar]
- Bollen, M. Combinatorial control of protein phosphatase-1. Trends Biochem. Sci. 2001, 26, 426–431. [Google Scholar]
- Rahman, A.; Grundke-Iqbal, I.; Iqbal, K. Phosphothreonine-212 of Alzheimer abnormally hyperphosphorylated tau is a preferred substrate of protein phosphatase-1. Neurochem. Res. 2005, 30, 277–287. [Google Scholar]
- Lapointe, N.E.; Morfini, G.; Pigino, G.; Gaisina, I.N.; Kozikowski, A.P.; Binder, L.I.; Brady, S.T. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J. Neurosci. Res. 2009, 87, 440–451. [Google Scholar]
- Kanaan, N.M.; Morfini, G.A.; Lapointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Rossie, S.; Gong, C.X. Dephosphorylation of tau by protein phosphatase 5: Impairment in Alzheimer’s disease. J. Biol. Chem. 2005, 280, 1790–1796. [Google Scholar]
- Yamaguchi, F.; Umeda, Y.; Shimamoto, S.; Tsuchiya, M.; Tokumitsu, H.; Tokuda, M.; Kobayashi, R. S100 Proteins Modulate Protein Phosphatase 5 Function: A link between Ca2+ signal transduction and protein dephosphorylation. J. Biol. Chem 2012, 287, 13787–13798. [Google Scholar]
- Fedrizzi, L.; Carafoli, E. Ca2+ dysfunction in neurodegenerative disorders: Alzheimer’s disease. Biofactors 2011, 37, 189–196. [Google Scholar]
- Wasik, U.; Schneider, G.; Mietelska-Porowska, A.; Mazurkiewicz, M.; Fabczak, H.; Weis, S.; Zabke, C.; Harrington, C.R.; Filipek, A.; Niewiadomska, G. Calcyclin binding protein and Siah-1 interacting protein in Alzheimer’s disease pathology: Neuronal localization and possible function. Neurobiol. Aging 2013, 34, 1380–1388. [Google Scholar]
- Kilanczyk, E.; Filipek, S.; Filipek, A. ERK1/2 is dephosphorylated by a novel phosphatase— CacyBP/SIP. Biochem. Biophys. Res. Commun 2011, 404, 179–183. [Google Scholar]
- Diaz-Hernandez, M.; Gomez-Ramos, A.; Rubio, A.; Gomez-Villafuertes, R.; Naranjo, J.R.; Miras-Portugal, M.T.; Avila, J. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J. Biol. Chem 2010, 285, 32539–32548. [Google Scholar]
- Necula, M.; Kuret, J. Pseudophosphorylation and glycation of tau protein enhance but do not trigger fibrillization in vitro. J. Biol. Chem 2004, 279, 49694– 49703. [Google Scholar]
- Smet-Nocca, C.; Broncel, M.; Wieruszeski, J.M.; Tokarski, C.; Hanoulle, X.; Leroy, A.; Landrieu, I.; Rolando, C.; Lippensa, G.; Hackenberger, C.P.R. Identification of O-GlcNAc sites within peptides of the Tau protein and their impact on phosphorylation. Mol. BioSyst. 2011, 7, 1420–1429. [Google Scholar]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar]
- Borghgraef, P.; Menuet, C.; Theunis, C.; Louis, J.V.; Devijver, H.; Maurin, H.; Smet-Nocca, C.; Lippens, G.; Hilaire, G.; Gijsen, H.; et al. Increasing brain protein O-GlcNAc-ylation mitigates breathing defects and mortality of TauP301L mice. PLoS One 2013. [Google Scholar] [CrossRef]
- Liu, F.; Shi, J.; Tanimukai, H.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 2009, 132, 1820–1832. [Google Scholar]
- Takahashi, M.; Tsujioka, Y.; Yamada, T.; Tsuboi, Y.; Okada, H.; Yamamoto, T.; Liposits, Z. Glycosylation of microtubule-associated protein tau in Alzheimer’s disease brain. Acta Neuropathol. 1999, 7, 635–641. [Google Scholar]
- Ledesma, M.D.; Medina, M.; Avila, J. The in vitro formation of recombinant tau polymers: Effect of phosphorylation and glycation. Mol. Chem. Neuropathol 1996, 27, 249–258. [Google Scholar]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar]
- Zhang, Y.J.; Xu, Y.F.; Liu, Y.H.; Yin, J.; Li, H.L.; Wang, Q.; Wang, J.Z. Peroxynitrite induces Alzheimer-like tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB J 2006, 20, 1431–1442. [Google Scholar]
- Singer, S.M.; Zainelli, G.M.; Norlund, M.A.; Lee, J.M.; Muma, N.A. Transglutaminase bonds in neurofibrillary tangles and paired helical filament tau early in Alzheimer’s disease. Neurochem. Int. 2002, 40, 17–30. [Google Scholar]
- Bancher, C.; Grundke-Iqbal, I.; Iqbal, K.; Fried, V.A.; Smith, H.T.; Wisniewski, H.M. Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res. 1991, 539, 11–18. [Google Scholar]
- Riederer, I.M.; Schiffrin, M.; Kovari, E.; Bouras, C.; Riederer, B.M. Ubiquitination and cysteine nitrosylation during aging and Alzheimer’s disease. Brain Res. Bull. 2009, 80, 233–241. [Google Scholar]
- Takahashi, K.; Ishida, M.; Komano, H.; Takahashi, H. SUMO-1 immunoreactivity co-localizes with phospho-Tau in APP transgenic mice but not in mutant Tau transgenic mice. Neurosci. Lett. 2008, 441, 90–93. [Google Scholar]
- Newman, M.; Musgrave, I.F.; Lardelli, M. Alzheimer disease: Amyloidogenesis the presenilins and animal models. Biochim. Biophys. Acta 2007, 1772, 285–297. [Google Scholar]
- Amadoro, G.; Corsetti, V.; Ciotti, M.T.; Florenzano, F.; Capsoni, S.; Amato, G.; Calissano, P. Endogenous Aβ causes cell death via early tau phosphorylation. Neurobiol. Aging 2011, 32, 969–990. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci 2011, 12, 67–72. [Google Scholar]
- Vossel, K.A.; Zhang, K.; Brodbeck, J.; Daub, A.C.; Sharma, P.; Finkbeiner, S.; Cui, B.; Mucke, L. Tau reduction prevents Aβ-induced defects in axonal transport. Science 2010, 330, 198. [Google Scholar]
- Fein, J.A.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Yang, F.; Cole, G.M.; Gylys, K.H. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am. J. Pathol. 2008, 172, 1683–1692. [Google Scholar]
- Takahashi, R.H.; Capetillo-Zarate, E.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Co-occurrence of Alzheimer’s disease beta-amyloid and tau pathologies at synapses. Neurobiol. Aging 2010, 31, 1145–1152. [Google Scholar]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar]
- Hamdane, M.; Dourlen, P.; Bretteville, A.; Sambo, A.V.; Ferreira, S.; Ando, K.; Kerdraon, O.; Bégard, S.; Geay, L.; Lippens, G.; et al. Pin1 allows for differential Tau dephosphorylation in neuronal cells. Mol. Cell. Neurosci 2006, 1–2, 155–160. [Google Scholar]
- Lim, J.; Balastik, M.; Lee, T.H.; Nakamura, K.; Liou, Y.C.; Sun, A.; Finn, G.; Pastorino, L.; Lee, V.M.; Lu, K.P. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J. Clin. Investig 2008, 118, 1877–1889. [Google Scholar]
- Bulbarelli, A.; Lonati, E.; Cazzaniga, E.; Gregori, M.; Masserini, M. Pin1 affects Tau phosphorylation in response to Aβ-oligomers. Mol. Cell. Neurosci 2009, 42, 75–80. [Google Scholar]
- Ma, S.L.; Pastorino, L.; Zhou, X.Z.; Lu, K.P. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3β (GSK3β) activity: Novel mechanism for Pin1 to protect against Alzheimer disease. J. Biol. Chem. 2012, 287, 6969–6973. [Google Scholar]
- Tamayev, R.; Zhou, D.; D’Adamio, L. The interactome of the amyloid-β precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol. Neurodegener. 2009, 4, 28. [Google Scholar]
- Williamson, R.; Usardi, A.; Hanger, D.P.; Anderton, B.H. Membrane-bound beta-amyloid oligomers are recruited into lipid rafts by a fyn-dependent mechanism. FEBS J. 2008, 22, 1552–1559. [Google Scholar]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic network and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci 2011, 31, 700–711. [Google Scholar]
- Hernandez, P.; Lee, G.; Sjoberg, M.; Maccioni, R.B. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25–35): Involvement of lipid rafts. J. Alzheimers. Dis 2009, 16, 149–156. [Google Scholar]
- Scales, T.M.; Derkinderen, P.; Leung, K.Y.; Byers, H.L.; Ward, M.A.; Price, C.; Bird, I.N.; Perera, T.; Kellie, S.; Williamson, R.; et al. Tyrosine phosphorylation of tau by the SRC family kinases lck and fyn. Mol. Neurodegener 2011, 6, 12. [Google Scholar]
- Bhaskar, K.; Hobbs, G.A.; Yen, S.H.; Lee, G. Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol. Appl. Neurobiol 2010, 36, 462–477. [Google Scholar]
- Usardi, A.; Pooler, A.M.; Seereeram, A.; Reynolds, C.H.; Derkinderen, P.; Anderton, B.; Hanger, D.P.; Noble, W.; Williamson, R. Tyrosine phosphorylation of tau regulates its interactions with Fyn SH2 domains but not SH3 domains altering the cellular localization of tau. FEBS J. 2011, 278, 2927–2937. [Google Scholar]
- Pritchard, S.M.; Dolan, P.J.; Vitkus, A.; Johnson, G.W.V. The toxicity of tau in Alzheimer disease: Turnover targets and potential therapeutics. J. Cell. Mol. Med 2011, 15, 1621–1635. [Google Scholar]
- Dou, F.; Netzer, W.J.; Tanemura, K.; Li, F.; Hartl, F.U.; Takashima, A.; Gouras, G.K.; Greengard, P.; Xu, H. Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. USA 2003, 100, 721–726. [Google Scholar]
- Miyata, Y.; Koren, J., III; Kiray, J.; Dickey, C.A.; Gestwicki, J.E. Molecular chaperones and regulation of tau quality control:strategies for drug discovery in tauopathies. Futur. Med. Chem 2011, 3, 1523–1537. [Google Scholar]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; de Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination degradation and aggregation. Hum. Mol. Genet 2004, 13, 703–714. [Google Scholar]
- Jinwal, U.K.; O’Leary, J.C., III; Borysov, S.I.; Jones, J.R.; Li, Q.; Koren, J., III; Abisambra, J.F.; Vestal, G.D.; Lawson, L.Y.; Johnson, A.G.; et al. Hsc70 rapidly engages tau after microtubule destabilization. J. Biol. Chem 2010, 285, 16798–16805. [Google Scholar]
- Jinwal, U.K.; Koren, J., III; Borysov, S.I.; Schmid, A.B.; Abisambra, J.F.; Blair, L.J.; Johnson, A.G.; Jones, J.R.; Shults, C.L.; O’Leary, J.C., III; et al. The Hsp90 cochaperone FKBP51 increases tau stability and polymerizes microtubules. J. Neurosci 2010, 30, 591–599. [Google Scholar]
- Tortosa, E.; Santa-Maria, I.; Moreno, F.; Lim, F.; Perez, M.; Avila, J. Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alzheimers Dis. 2009, 17, 319–325. [Google Scholar]
- Dickey, C.A.; Dunmore, J.; Lu, B.; Wang, J.W.; Lee, W.C.; Kamal, A.; Burrows, F.; Eckman, C.; Hutton, M.; Petrucelli, L. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 2006, 20, 753–765. [Google Scholar]
- Dickey, C.A.; Koren, J.; Zhang, Y.J.; Xu, Y.F.; Jinwal, U.K.; Birnbaum, M.J.; Monks, B.; Sun, M.; Cheng, J.Q.; Patterson, C.; et al. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 3622–3627. [Google Scholar]
- Cao, W.; Konsolaki, M. FKBP immunophilins and Alzheimer’s disease: A chaperoned affair. J. Biosci 2011, 36, 493–498. [Google Scholar]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C., III; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig 2013, 123, 4158–4169. [Google Scholar]
- Chambraud, B.; Sardin, E.; Giustiniani, J.; Dounane, O.; Schumacher, M.; Goedert, M.; Baulieu, E. A role for FKBP52 in Tau protein function. Proc. Natl. Acad. Sci. USA 2010, 107, 2658–2663. [Google Scholar]
- Korff, A.; Liu, C.; Ginghina, C.; Shi, M.; Zhang, J. α-Synuclein in cerebrospinal fluid of Alzheimer’s disease and mild cognitive impairment. J. Alzheimers Dis 2013, 36, 679–688. [Google Scholar]
- Hashiguchi, M.; Hashiguchi, T. Kinase-Kinase Interaction and Modulation of Tau Phosphorylation. Int. Rev. Cell. Mol. Biol 2013, 300, 121–160. [Google Scholar]
- Kawakami, F.; Suzuki, M.; Shimada, N.; Kagiya, G.; Ohta, E.; Tamura, K.; Maruyama, H.; Ichikawa, T. Stimulatory effect of α-synuclein on the tau-phosphorylation by GSK-3β. FEBS J 2011, 278, 4895–4904. [Google Scholar]
- Qureshi, H.Y.; Paudel, H.K. Parkinsonian neurotoxin 1-methyl-4phenyl-1236- tetrahydropyridine (MPTP) and α-synuclein mutations promote Tau protein phosphorylation at Ser 262 and destabilize microtubule cytoskeleton in vitro. J. Biol. Chem 2011, 286, 5055–5068. [Google Scholar]
- Whiteman, I.T.; Gervasio, O.L.; Cullen, K.M.; Guillemin, G.J.; Jeong, E.V.; Witting, P.K.; Antao, S.T.; Minamide, L.S.; Bamburg, J.R.; Goldsbury, C. Activated ADF/cofilin sequesters phosphorylated microtubuleassociated-protein during the assembly of Alzheimer-like neuritic cytoskeletal striations. J. Neurosci 2009, 29, 12994. [Google Scholar]
- Poulain, F.E.; Sobel, A. The microtubule network and neuronal morphogenesis: Dynamic and coordinated orchestration through multiple players. Mol. Cell. Neurosci. 2010, 43, 15–32. [Google Scholar]
- Grimm-Günter, E.S.; Milbrandt, M.; Merkl, B.; Paulsson, M.; Plomann, M. PACSIN proteins bind tubulin andpromotemicrotubule assembly. Exp. Cell Res 2008, 314, 1991–2003. [Google Scholar]
- Liu, Y.; Lv, K.; Li, Z.; Yu, A.C.H.; Chen, J.; Teng, J. PACSIN1 a Tau-interacting protein regulates axonal elongation and branching by facilitating microtubule instability. J. Biol. Chem 2012, 287, 39911–39924. [Google Scholar]
- Crowther, R.A.; Goedert, M. Abnormal tau-containing filaments in neurodegenerative diseases. J. Struct. Biol. 2000, 130, 271–279. [Google Scholar]
- Kosik, K.S.; Shimura, H. Phosphorylated tau and the neurodegenerative foldopathies. Biochim. Biophys. Acta 2005, 1739, 298–310. [Google Scholar]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of Tau in both physiological and pathological conditions. Physiol. Rev 2004, 84, 361–384. [Google Scholar]
- Jeganathan, S.; Hascher, A.; Chinnathambi, S.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 2008, 283, 32066–32076. [Google Scholar]
- Hyman, B.T.; Augustinack, J.C.; Ingelsson, M. Transcriptional and conformational changes of the tau molecule in Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1739, 150–157. [Google Scholar]
- Delacourte, A.; Buee, L. Normal and pathological Tau proteins as factors for microtubule assembly. Int. Rev. Cytol 1997, 171, 167–224. [Google Scholar]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci 2004, 117, 5721–5729. [Google Scholar]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Gertz, H.N.; Xuereb, J.H.; Paykel, E.S.; Brayne, C.; Huppert, F.A.; Mukaetova-Ladinska, E.B.; Mena, R.; et al. Quantitative analysis of tau protein in paired helical filament preparations: Implications for the role of tau protein phosphorylation in PHF assembly in Alzheimer’s disease. Neurobiol. Aging 1995, 16, 409–417. [Google Scholar]
- Feuillette, S.; Miguel, L.; Frébourg, T.; Campion, D.; Lecourtois, M. Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J. Neurochem. 2010, 113, 895–903. [Google Scholar]
- Rocher, A.B.; Crimins, J.L.; Amatrudo, J.M.; Kinson, M.S.; Todd-Brown, M.A.; Lewis, J.; Luebke, J.I. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp. Neurol. 2010, 223, 385–393. [Google Scholar]
- Cowan, C.M.; Bossing, T.; Page, A.; Shepherd, D.; Mudher, A. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 2010, 120, 593–604. [Google Scholar]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol 2013, 13, 4–114. [Google Scholar]
- Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimers Dis. 2008, 14, 393–9. [Google Scholar]
- Kayed, R. Anti-tau oligomers passive vaccination for the treatment of Alzheimer disease. Hum. Vaccin. 2010, 6, 931–5. [Google Scholar]
- Spires-Jones, T.L.; Kopeikina, K.J.; Koffie, R.M.; de Calignon, A.; Hyman, B.T. Are tangles as toxic as they look? J. Mol. Neurosci. 2011, 45, 438–44. [Google Scholar]
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 2011, 50, 10300–10310. [Google Scholar]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-typemice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J. Neurosci 2007, 27, 3650–3662. [Google Scholar]
- Sahara, N.; Maeda, S.; Murayama, M.; Suzuki, T.; Dohmae, N.; Yen, S.H.; Takashima, A. Assembly of two distinct dimers and higher-orderoligomers from full-length tau. Eur. J. Neurosci. 2007, 25, 3020–3029. [Google Scholar]
- Spires-Jones, T.L.; Stoothoff, W.H.; de Calignon, A.; Jones, P.B.; Hyman, B.T. Tau pathophysiology in neurodegeneration: A tangled issue. Trends Neurosci. 2009, 32, 150–159. [Google Scholar]
- Haase, C.; Stieler, J.T.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar]
- Cho, J.H.; Johnson, G.V. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J. Neurochem. 2004, 88, 349–358. [Google Scholar]
- Sun, Q.; Gamblin, T.C. Pseudohyperphosphorylation causing AD-like changes in tau has significant effects on its polymerization. Biochemistry 2009, 48, 6002–6011. [Google Scholar]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; Xie, S.X.; Lee, V.M.; Trojanowski, J.Q. Acetylated tau a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 2012, 135, 807–818. [Google Scholar]
- Alonso, A.D.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar]
- Alonso, A.D.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar]
- Duan, A.R.; Goodson, H.V. Taxol-stabilized microtubules promote the formation of filaments from unmodified full-length Tau in vitro. Mol. Biol. Cell 2012, 23, 4796–4806,. [Google Scholar]
- Masliah, E.; Iimoto, D.S.; Saitoh, T.; Hansen, L.A.; Terry, R.D. Increased immunoreactivity of brain spectrin in Alzheimer disease: A marker for synapse loss? Brain Res. 1990, 531, 36–44. [Google Scholar]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar]
- Billingsley, M.L.; Kincaid, R.L. Regulated phosphorylation and dephosphorylation of tau protein: Effects on microtubule interaction intracellular trafficking and neurodegeneration. Biochem. J. 1997, 323, 577–591. [Google Scholar]
- Bunker, J.M.; Kamath, K.; Wilson, L.; Jordan, M.A.; Feinstein, S.C. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J. Biol. Chem 2006, 281, 11856–11863. [Google Scholar]
- Díaz-Nido, J.; Wandosell, F.; Avila, J. Glycosaminoglycans and beta-amyloid prion and tau peptides in neurodegenerative diseases. Peptides 2002, 23, 1323–1332. [Google Scholar]
- Stokin, G.B.; Goldstein, L.S. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 2006, 75, 607–627. [Google Scholar]
- Kanaan, N.M.; Pigino, G.F.; Brady, S.T.; Lazarov, O.; Binder, L.I.; Morfini, G.A. Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp. Neurol 2013, 246, 44–53. [Google Scholar]
- Chevalier-Larsen, E.; Holzbaur, E.L. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 2006, 1762, 1094–1108. [Google Scholar]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar]
- Van Spronsen, M.; Hoogenraad, C.C. Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 2010, 10, 207–214. [Google Scholar]
- Kapitein, L.C.; Hoogenraad, C.C. Which way to go? Cytoskeletal organization and polarized transport in neurons. Mol. Cell. Neurosci. 2011, 46, 9–20. [Google Scholar]
- Hirokawa, N.; Noda, Y. Intracellular transport and kinesin superfamily proteins KIFs: Structure function and dynamics. Physiol. Rev. 2008, 88, 1089–1118. [Google Scholar]
- Falzone, T.L.; Gunawardena, S.; McCleary, D.; Reis, G.F.; Goldstein, L.S. Kinesin-1 transport reductions enhance human tau hyperphosphorylation aggregation and neurodegeneration in animal models of tauopathies. Hum. Mol. Genet 2010, 19, 4399–4408. [Google Scholar]
- Terada, S.; Kinjo, M.; Aihara, M.; Takei, Y.; Hirokawa, N. Kinesin-1/Hsc70-dependent mechanism of slow axonal transport and its relation to fast axonal transport. EMBO J. 2010, 29, 843–854. [Google Scholar]
- Gunawardena, S.; Goldstein, L.S. Cargo-carrying motor vehicles on the neuronal highway: Transport pathways and neurodegenerative disease. J. Neurobiol 2004, 58, 258–271. [Google Scholar]
- Perlson, E.; Maday, S.; Fu, M.M.; Moughamian, A.J.; Holzbaur, E.L. Retrograde axonal transport: Pathways to cell death? Trends Neurosci 2010, 33, 335–344. [Google Scholar]
- Bendiske, J.; Caba, E.; Brown, Q.B.; Bahr, B.A. Intracellular deposition microtubule destabilization and transport failure: An ‘early’ pathogenic cascade leading to synaptic decline. J. Neuropathol. Exp. Neurol. 2002, 61, 640–650. [Google Scholar]
- Kamal, A.; Stokin, G.B.; Yang, Z.; Xia, C.H.; Goldstein, L.S. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 2000, 28, 449–459. [Google Scholar]
- Morel, M.; Héraud, C.; Nicaise, C.; Suain, V.; Brion, J.P. Levels of kinesin light chain and dynein intermediate chain are reduced in the frontal cortex in Alzheimer’s disease: Implications for axoplasmic transport. Acta Neuropathol 2012, 123, 71–84. [Google Scholar]
- Hashimoto, Y.; Tsuji, O.; Niikura, T.; Yamagishi, Y.; Ishizaka, M.; Kawasumi, M.; Chiba, T.; Kanekura, K.; Yamada, M.; Tsukamoto, E.; et al. Involvement of c-Jun N-terminal kinase in amyloid precursor protein-mediated neuronal cell death. J. Neurochem 2003, 84, 864–877. [Google Scholar]
- Futerman, A.H.; Banker, G.A. The economics of neurite outgrowth—The addition of new membrane to growing axons. Trends Neurosci. 1996, 19, 144–149. [Google Scholar]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles neurofilaments and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 18, 1051–1063. [Google Scholar]
- Mandelkow, E.M.; Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E. Clogging of axons by tau inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 2003, 24, 1079–1085. [Google Scholar]
- Calissano, P.; Matrone, C.; Amadoro, G. Nerve growth factor as a paradigm of neurotrophins related to Alzheimer’s disease. Dev. Neurobiol 2010, 70, 372–383. [Google Scholar]
- Thies, E.; Mandelkow, E.M. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 2007, 27, 2896–2907. [Google Scholar]
- Tatebayashi, Y.; Haque, N.; Tung, Y.C.; Iqbal, K.; Grundke-Iqbal, I. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J. Cell Sci. 2004, 117, 1653–1663. [Google Scholar]
- Watson, F.L.; Heerssen, H.M.; Moheban, D.B.; Lin, M.Z.; Sauvageot, C.M.; Bhattacharyya, A.; Pomeroy, S.L.; Segal, R.A. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J. Neurosci 1999, 15, 7889–7900. [Google Scholar]
- Sandow, S.L.; Heydon, K.; Weible, M.W., II; Reynolds, A.J.; Bartlett, S.E.; Hendry, I.A. Signaling organelle for retrograde axonal transport of internalized neurotrophins from the nerve terminal. Immunol. Cell Biol. 2000, 78, 430–435. [Google Scholar]
- Bhattacharyya, A.; Watson, F.L.; Pomeroy, S.L.; Zhang, Y.Z.; Stiles, C.D.; Segal, R.A. High-resolution imaging demonstrates dynein-based vesicular transport of activated Trk receptors. J. Neurobiol 2002, 51, 302–312. [Google Scholar]
- Heerssen, H.M.; Pazyra, M.F.; Segal, R.A. Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat. Neurosci 2004, 7, 596–604. [Google Scholar]
- Yano, H.; Lee, F.S.; Kong, H.; Chuang, J.; Arevalo, J.; Perez, P.; Sung, C.; Chao, M.V. Association of Trk neurotrophin receptors with components of the cytoplasmic dynein motor. J. Neurosci 2001, 21, RC125. [Google Scholar]
- Yano, H.; Chao, M.V. Mechanisms of neurotrophin receptor vesicular transport. J. Neurobiol 2004, 58, 244–257. [Google Scholar]
- Riccio, A.; Pierchal, B.A.; Ciarallo, C.L.; Ginty, D.D. A NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science 1997, 277, 1097–1100. [Google Scholar]
- Yano, H.; Chao, M.V. Biochemical characterization of intracellular membranes bearing Trk neurotrophin receptors. Neurochem. Res 2005, 30, 767–777. [Google Scholar]
- Wu, C.; Ramirez, A.; Cui, B.; Ding, J.; Delcroix, J.D.; Valletta, J.S.; Liu, J.J.; Yang, Y.; Chu, S.; Mobley, W.C. A functional dynein-microtubule network is required for NGF signaling through the Rap1/MAPK pathway. Traffic 2007, 8, 1503–1520. [Google Scholar]
- Salehi, A.; Delcroix, J.D.; Swaab, D.F. Alzheimer’s disease and NGF signaling. J. Neural Transm 2004, 111, 323–345. [Google Scholar]
- Schindowski, K.; Belarbi, K.; Buée, L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav 2008, 1, 43–56. [Google Scholar]
- Lazarov, O.; Morfini, G.A.; Pigino, G.; Gadadhar, A.; Chen, X.; Robinson, J.; Ho, H.; Brady, S.T.; Sisodia, S.S. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer’s disease-linked mutant presenilin 1. J. Neurosci 2007, 27, 7011–7020. [Google Scholar]
- Corsetti, V.; Amadoro, G.; Gentile, A.; Capsoni, S.; Ciotti, M.T.; Cencioni, M.T.; Atlante, A.; Canu, N.; Rohn, T.T.; Cattaneo, A.; et al. Identification of a caspase-derived N-terminal tau fragment in cellular and animal Alzheimer’s disease models. Mol. Cell. Neurosci. 2008, 38, 381–392. [Google Scholar]
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of tau protein with the dynactin complex. EMBO J 2007, 31, 4546–4554. [Google Scholar]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF signaling control amyloidogenic route and Ab production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar]
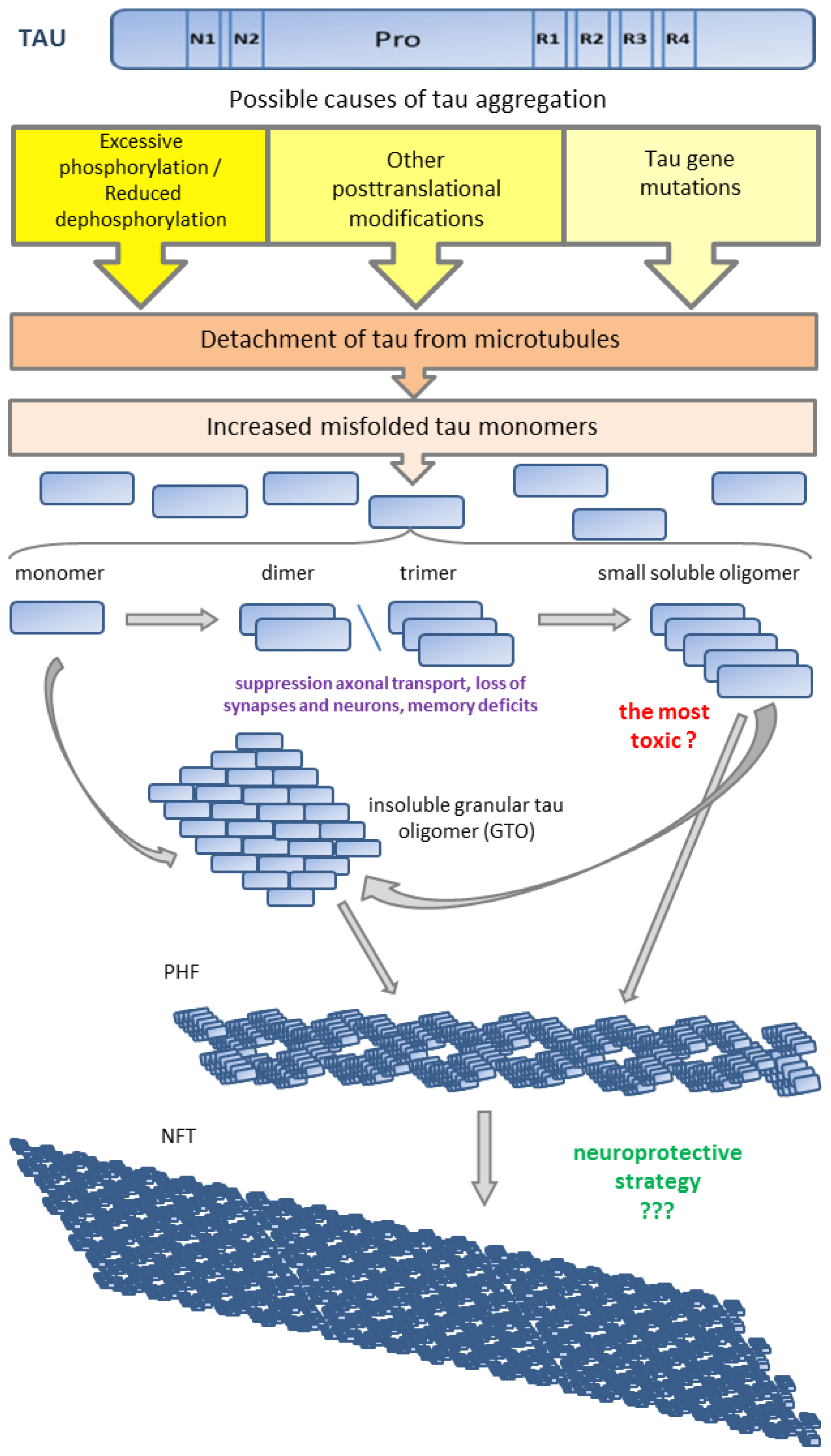
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction. Int. J. Mol. Sci. 2014, 15, 4671-4713. https://doi.org/10.3390/ijms15034671
Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G. Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction. International Journal of Molecular Sciences. 2014; 15(3):4671-4713. https://doi.org/10.3390/ijms15034671
Chicago/Turabian StyleMietelska-Porowska, Anna, Urszula Wasik, Marcelina Goras, Anna Filipek, and Grazyna Niewiadomska. 2014. "Tau Protein Modifications and Interactions: Their Role in Function and Dysfunction" International Journal of Molecular Sciences 15, no. 3: 4671-4713. https://doi.org/10.3390/ijms15034671