Comparative Transcriptional Profiling of Three Super-Hybrid Rice Combinations

Abstract
:
1. Introduction
2. Results and Discussion
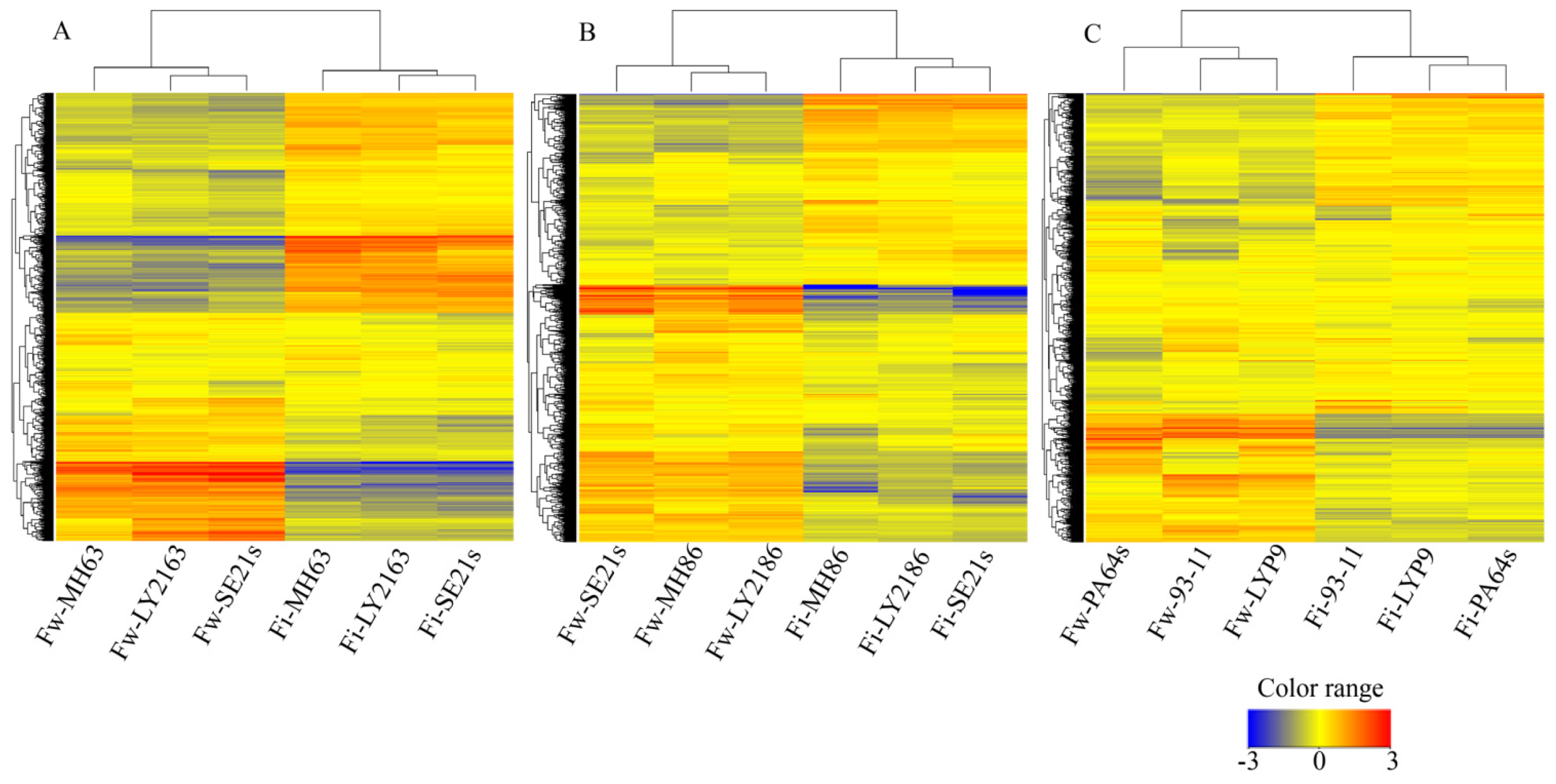
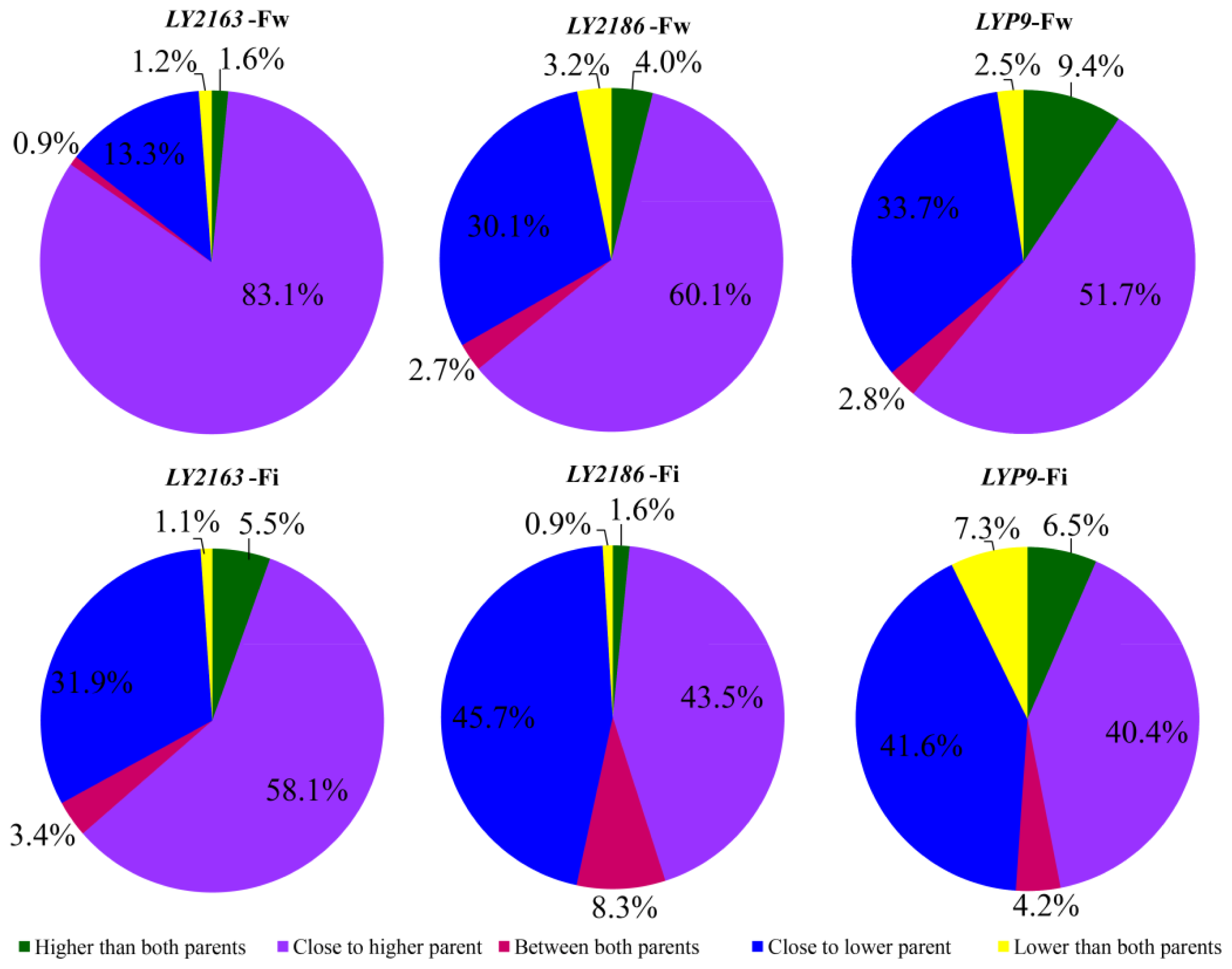
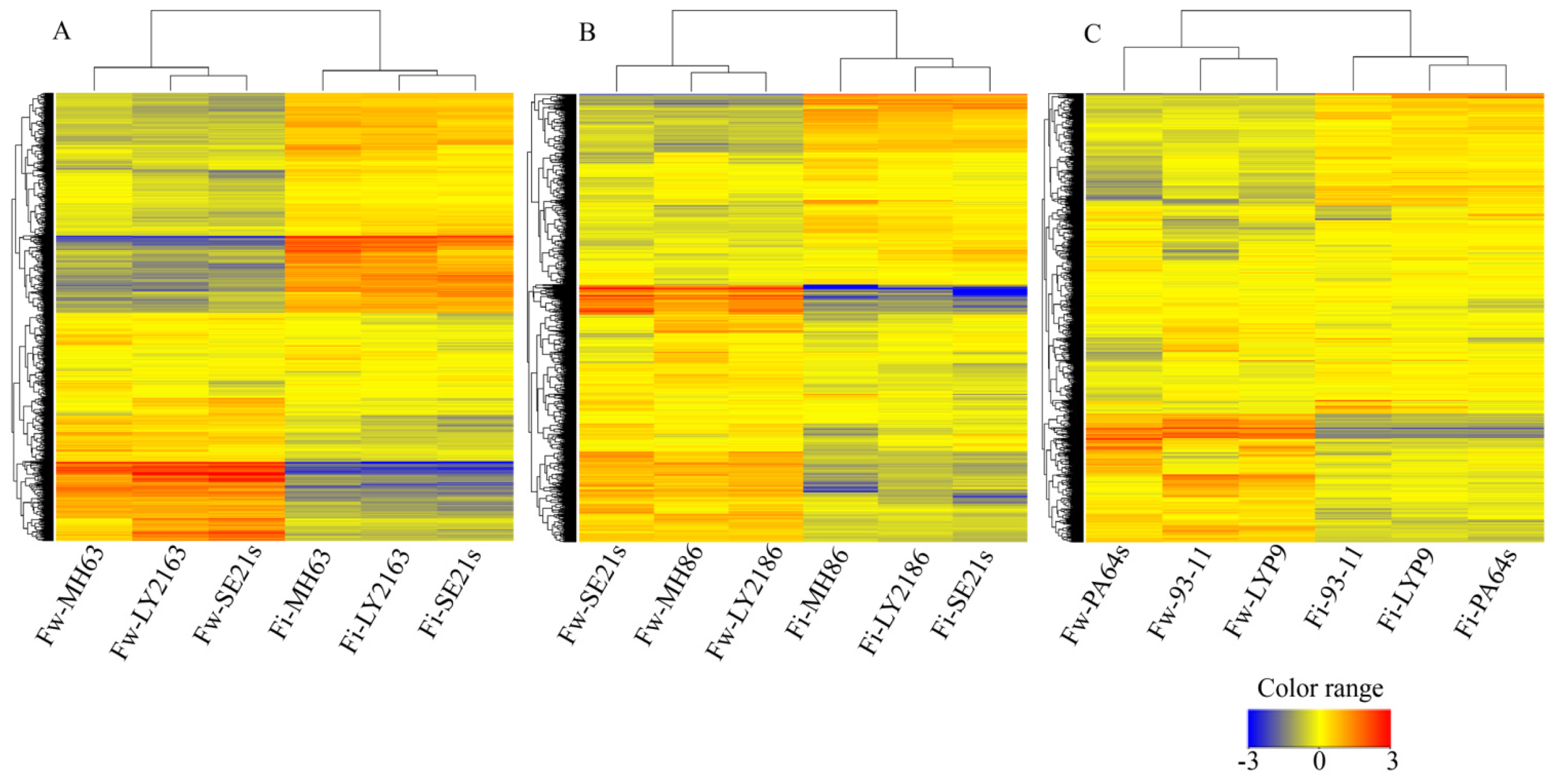
2.1. Transcriptional Profiles of Three Super-Hybrid Rice Combinations
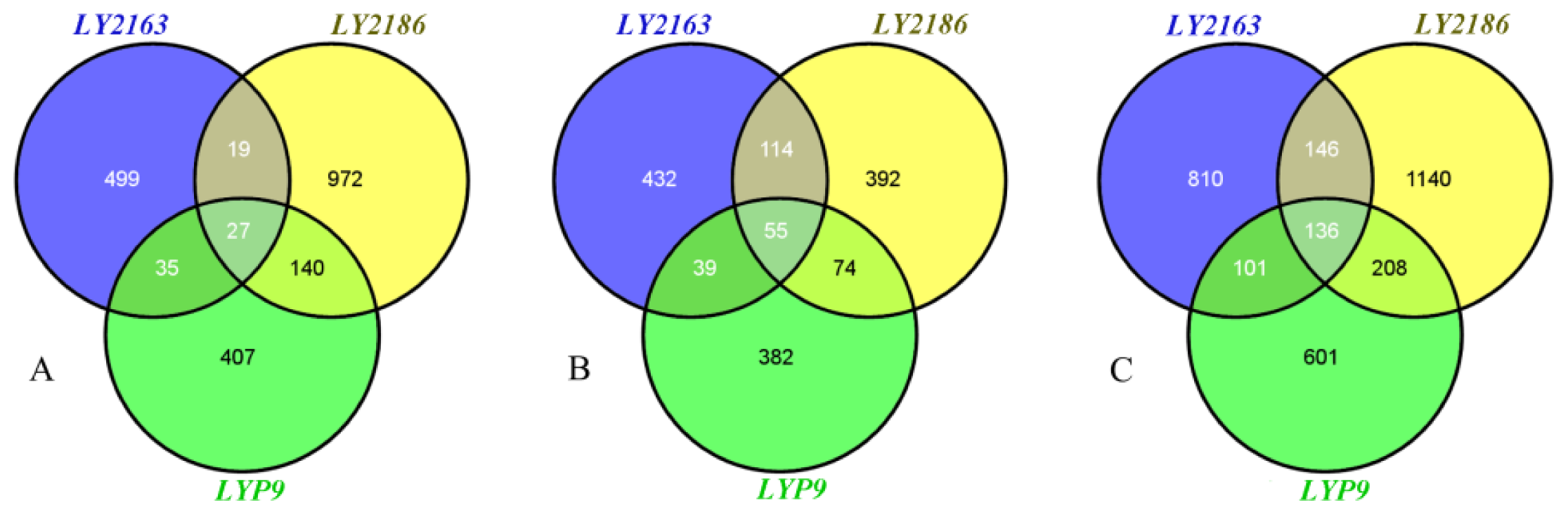
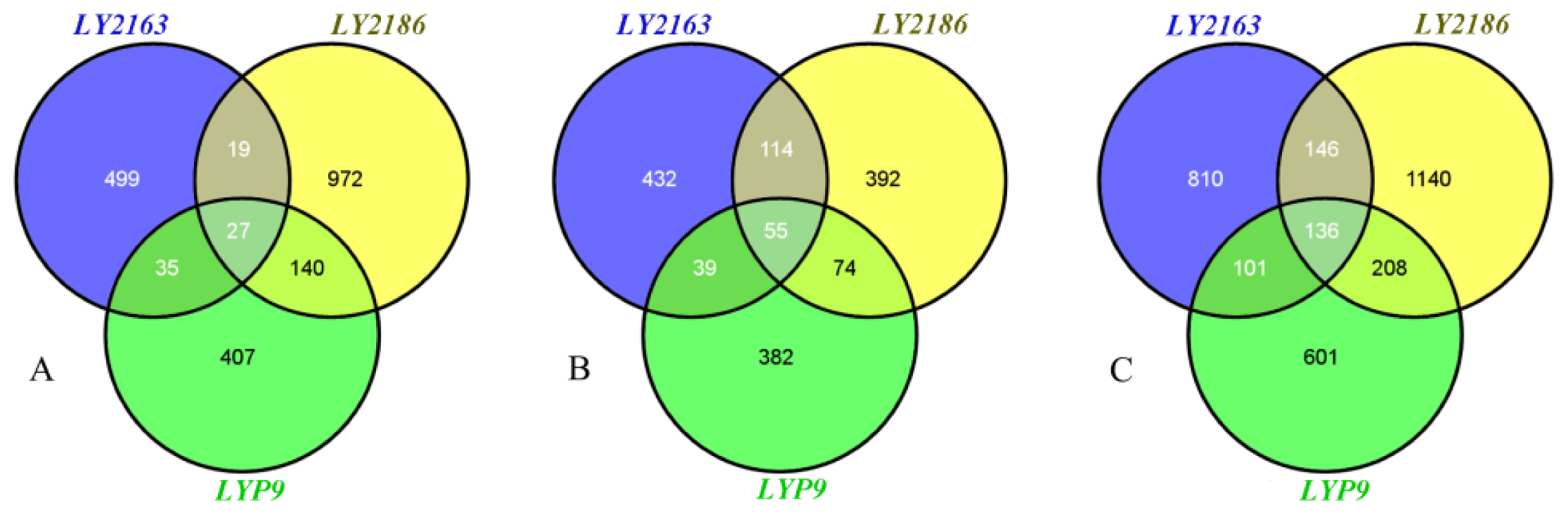
2.2. DGs Functions in the Three Hybrid Combinations
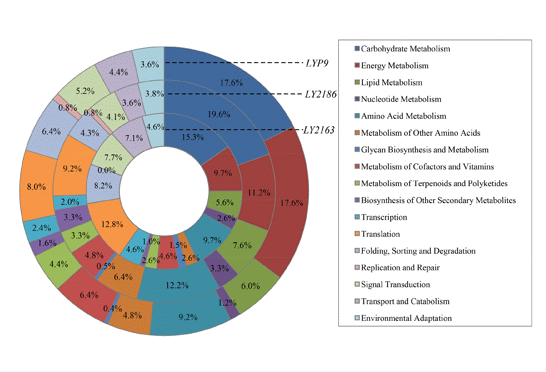
2.3. Metabolic Pathway Analysis of the DGs
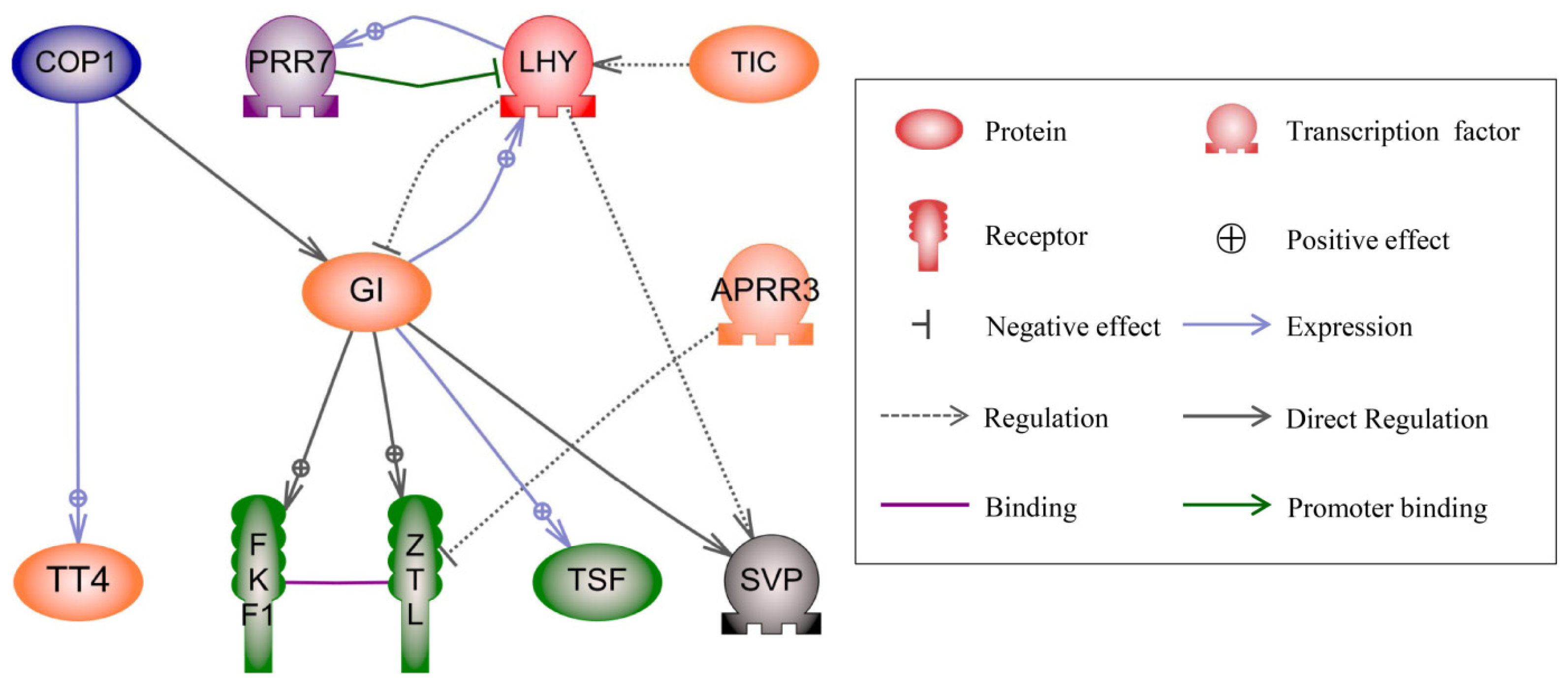
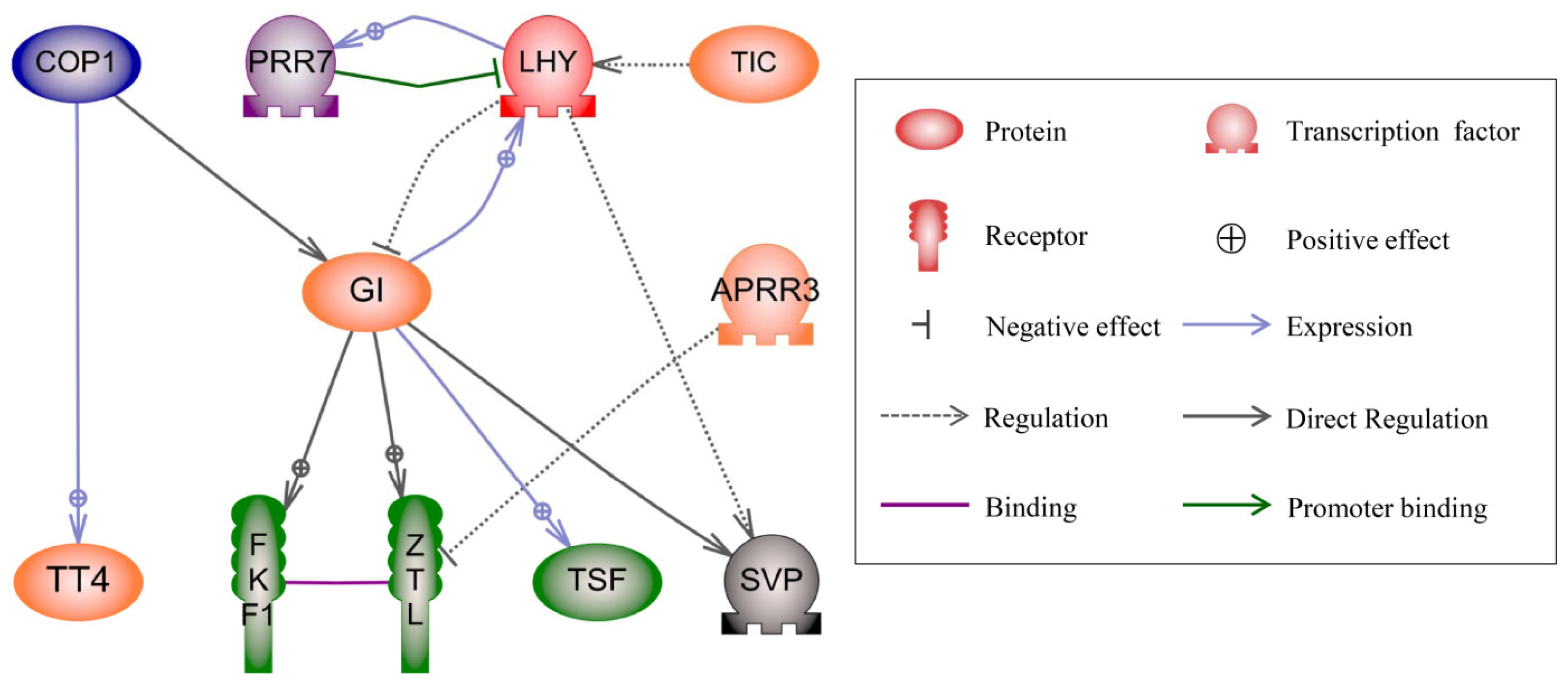
2.4. Regulatory Network Analysis of DGs in the Three Hybrids
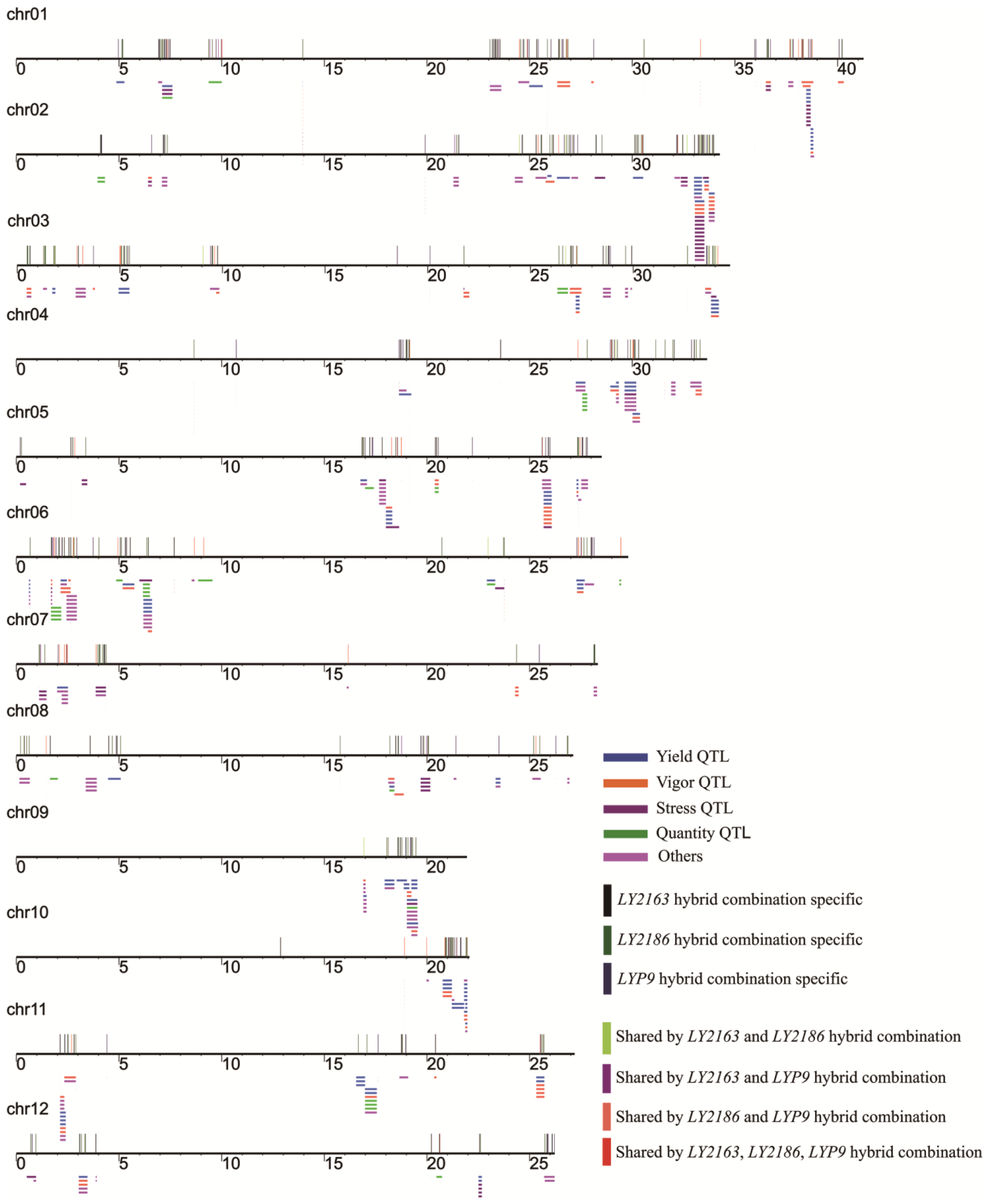
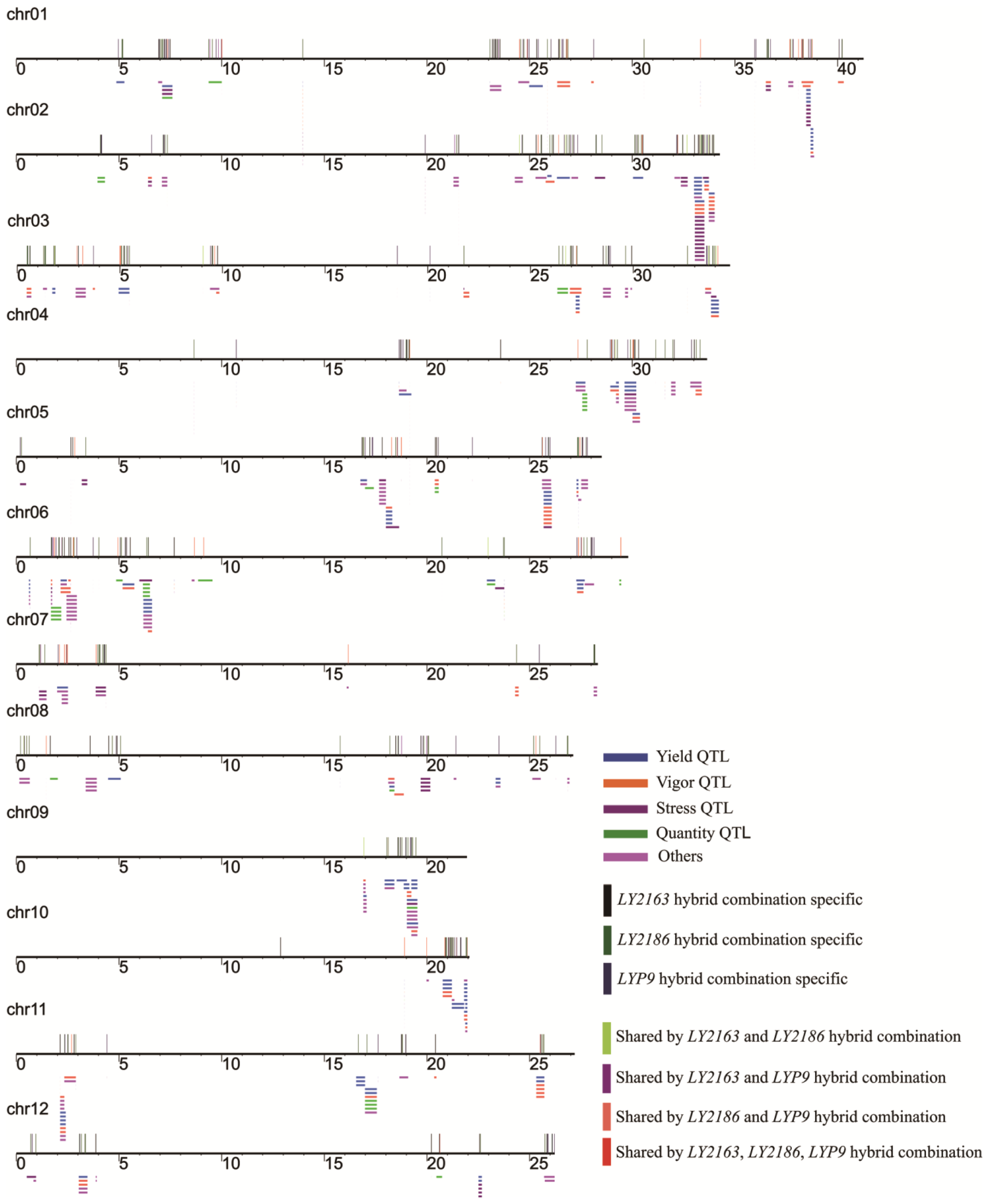
2.5. Mapping of DGs Quantitative Trait Loci (QTLs)
3. Experimental Section
3.1. Plant Materials
3.2. Microarray Experiment and Data Processing
3.3. Functional Category and Metabolic Pathway Analysis
3.4. Regulatory Network Analysis
3.5. Mapping DGs to Rice QTLs
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsY.P., G.W. and L.Z. initiated the research, carried out the biological experiments, performed the computational analyses, co-designed and drafted the manuscript. G.L. co-designed, analyzed the data and revised the manuscript. X.W. participated in the biological experiments and material collections. Z.Z. designed, interpreted the results and finalized the manuscript. All authors read and approved the final manuscript.
References
- Melchinger, A.E.; Gumber, R.K. Overview of Heterosis and Heterotic Groups in Agronomic Crops. In Concepts and Breeding of Heterosis in Crop Plants; Lamkey, K.R., Staub, J.E., Eds.; Crop Science Society of America: Madison, WI, USA, 1998; pp. 29–44. [Google Scholar]
- Cheng, S.H.; Zhuang, J.Y.; Fan, Y.Y.; Du, J.H.; Cao, L.Y. Progress in research and development on hybrid rice: A super-domesticate in China. Ann. Bot. 2007, 100, 959–966. [Google Scholar]
- Zhang, L.; Peng, Y.; Dong, Y.; Li, H.; Wang, W.; Zhu, Z. Progress of Genomics and Heterosis Studies in Hybrid Rice. In Polyploid and Hybrid Genomics; Chen, Z.J., Birchler, J.A., Eds.; John Wiley & Sons, Inc.: Oxford, UK, 2013; pp. 117–135. [Google Scholar]
- Birchler, J.A.; Auger, D.L.; Riddle, N.C. In search of the molecular basis of heterosis. Plant Cell 2003, 15, 2236–2239. [Google Scholar]
- Semel, Y.; Nissenbaum, J.; Menda, N.; Zinder, M.; Krieger, U.; Issman, N.; Pleban, T.; Lippman, Z.; Gur, A.; Zamir, D. Overdominant quantitative trait loci for yield and fitness in tomato. Proc. Natl. Acad. Sci. USA 2006, 103, 12981–12986. [Google Scholar]
- Birchler, J.A. Reflections on studies of gene expression in aneuploids. Biochem. J. 2010, 426, 119–123. [Google Scholar]
- Bruce, A.B. The Mendelian theory of heredity and the augmentation of vigor. Science 1910, 32, 627–628. [Google Scholar]
- Jones, D.F. Dominance of linked factors as a means of accounting for heterosis. Genetics 1917, 2, 466–479. [Google Scholar]
- Shull, G.H. The composition of a field of maize. Am. Breed. Assoc. Rep. 1908, 4, 296–301. [Google Scholar]
- East, E.M. Heterosis. Genetics 1936, 21, 375–397. [Google Scholar]
- Yu, S.B.; Li, J.X.; Xu, C.G.; Tan, Y.F.; Gao, Y.J.; Li, X.H.; Zhang, Q.; Saghai Maroof, M.A. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 1997, 94, 9226–9231. [Google Scholar]
- Hochholdinger, F.; Hoecker, N. Towards the molecular basis of heterosis. Trends Plant Sci. 2007, 12, 427–432. [Google Scholar]
- Schnable, P.S.; Springer, N.M. Progress toward understanding heterosis in crop plants. Annu. Rev. Plant Biol. 2013, 64, 71–88. [Google Scholar]
- Krieger, U.; Lippman, Z.B.; Zamir, D. The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat. Genet. 2010, 42, 459–463. [Google Scholar]
- Bao, J.; Lee, S.; Chen, C.; Zhang, X.; Zhang, Y.; Liu, S.; Clark, T.; Wang, J.; Cao, M.; Yang, H.; et al. Serial analysis of gene expression study of a hybrid rice strain (LYP9) and its parental cultivars. Plant Physiol. 2005, 138, 1216–1231. [Google Scholar]
- Song, G.S.; Zhai, H.L.; Peng, Y.G.; Zhang, L.; Wei, G.; Chen, X.Y.; Xiao, Y.G.; Wang, L.; Chen, Y.J.; Wu, B.; et al. Comparative transcriptional profiling and preliminary study on heterosis mechanism of super-hybrid rice. Mol. Plant 2010, 3, 1012–1025. [Google Scholar]
- Song, S.; Qu, H.; Chen, C.; Hu, S.; Yu, J. Differential gene expression in an elite hybrid rice cultivar (Oryza sativa L) and its parental lines based on SAGE data. BMC Plant Biol. 2007, 7, 49. [Google Scholar]
- Wei, G.; Tao, Y.; Liu, G.; Chen, C.; Luo, R.; Xia, H.; Gan, Q.; Zeng, H.; Lu, Z.; Han, Y.; et al. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc. Natl. Acad. Sci. USA 2009, 106, 7695–7701. [Google Scholar]
- Zhang, H.Y.; He, H.; Chen, L.B.; Li, L.; Liang, M.Z.; Wang, X.F.; Liu, X.G.; He, G.M.; Chen, R.S.; Ma, L.G.; et al. A genome-wide transcription analysis reveals a close correlation of promoter INDEL polymorphism and heterotic gene expression in rice hybrids. Mol. Plant 2008, 1, 720–731. [Google Scholar]
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F.; et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 2010, 22, 17–33. [Google Scholar]
- Swanson-Wagner, R.A.; Jia, Y.; DeCook, R.; Borsuk, L.A.; Nettleton, D.; Schnable, P.S. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc. Natl. Acad. Sci. USA 2006, 103, 6805–6810. [Google Scholar]
- Uzarowska, A.; Keller, B.; Piepho, H.P.; Schwarz, G.; Ingvardsen, C.; Wenzel, G.; Lubberstedt, T. Comparative expression profiling in meristems of inbred-hybrid triplets of maize based on morphological investigations of heterosis for plant height. Plant Mol. Biol. 2007, 63, 21–34. [Google Scholar]
- Ni, Z.; Sun, Q.; Liu, Z.; Wu, L.; Wang, X. Identification of a hybrid-specific expressed gene encoding novel RNA-binding protein in wheat seedling leaves using differential display of mRNA. Mol. Gen. Genet. 2000, 263, 934–938. [Google Scholar]
- Wu, L.M.; Ni, Z.F.; Meng, F.R.; Lin, Z.; Sun, Q.X. Cloning and characterization of leaf cDNAs that are differentially expressed between wheat hybrids and their parents. Mol. Genet. Genomics 2003, 270, 281–286. [Google Scholar]
- Yao, Y.; Ni, Z.; Zhang, Y.; Chen, Y.; Ding, Y.; Han, Z.; Liu, Z.; Sun, Q. Identification of differentially expressed genes in leaf and root between wheat hybrid and its parental inbreds using PCR-based cDNA subtraction. Plant Mol. Biol. 2005, 58, 367–384. [Google Scholar]
- Vuylsteke, M.; van Eeuwijk, F.; van Hummelen, P.; Kuiper, M.; Zabeau, M. Genetic analysis of variation in gene expression in Arabidopsis thaliana. Genetics 2005, 171, 1267–1275. [Google Scholar]
- Hoecker, N.; Keller, B.; Muthreich, N.; Chollet, D.; Descombes, P.; Piepho, H.P.; Hochholdinger, F. Comparison of maize (Zea mays L) F1-hybrid and parental inbred line primary root transcriptomes suggests organ-specific patterns of nonadditive gene expression and conserved expression trends. Genetics 2008, 179, 1275–1283. [Google Scholar]
- Ge, X.; Chen, W.; Song, S.; Wang, W.; Hu, S.; Yu, J. Transcriptomic profiling of mature embryo from an elite super-hybrid rice LYP9 and its parental lines. BMC Plant Biol. 2008, 8, 114. [Google Scholar]
- Flint-Garcia, S.A.; Buckler, E.S.; Tiffin, P.; Ersoz, E.; Springer, N.M. Heterosis is prevalent for multiple traits in diverse maize germplasm. PLoS One 2009, 4, e7433. [Google Scholar]
- Stupar, R.M.; Gardiner, J.M.; Oldre, A.G.; Haun, W.J.; Chandler, V.L.; Springer, N.M. Gene expression analyses in maize inbreds and hybrids with varying levels of heterosis. BMC Plant Biol. 2008, 8, 33. [Google Scholar]
- China Rice Data Center. Available online: http://www.ricedata.cn (accessed on 24 January 2014).
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 20: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar]
- Law, P.J.; Claudel-Renard, C.; Joubert, F.; Louw, A.I.; Berger, D.K. MADIBA: A web server toolkit for biological interpretation of Plasmodium and plant gene clusters. BMC Genomics 2008, 9, 105. [Google Scholar]
- Bar-Even, A.; Noor, E.; Lewis, N.E.; Milo, R. Design and analysis of synthetic carbon fixation pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 8889–8894. [Google Scholar]
- Fujimoto, R.; Taylor, J.M.; Shirasawa, S.; Peacock, W.J.; Dennis, E.S. Heterosis of Arabidopsis hybrids between C24 and Col is associated with increased photosynthesis capacity. Proc. Natl. Acad. Sci. USA 2012, 109, 7109–7114. [Google Scholar]
- Li, J.; Ou-Lee, T.M.; Raba, R.; Amundson, R.G.; Last, R.L. Arabidopsis flavonoid mutants are hypersensitive to UV-B irradiation. Plant cell 1993, 5, 171–179. [Google Scholar]
- Bieza, K.; Lois, R. An Arabidopsis mutant tolerant to lethal ultraviolet-B levels shows constitutively elevated accumulation of flavonoids and other phenolics. Plant Physiol. 2001, 126, 1105–1115. [Google Scholar]
- Peer, W.A.; Murphy, A.S. Flavonoids and auxin transport: Modulators or regulators? Trends Plant Sci. 2007, 12, 556–563. [Google Scholar]
- Korn, M.; Peterek, S.; Mock, H.P.; Heyer, A.G.; Hincha, D.K. Heterosis in the freezing tolerance and sugar and flavonoid contents of crosses between Arabidopsis thaliana accessions of widely varying freezing tolerance. Plant Cell Environ. 2008, 31, 813–827. [Google Scholar]
- Shen, H.; He, H.; Li, J.; Chen, W.; Wang, X.; Guo, L.; Peng, Z.; He, G.; Zhong, S.; Qi, Y.; et al. Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 2012, 24, 875–892. [Google Scholar]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar]
- Nikitin, A.; Egorov, S.; Daraselia, N.; Mazo, I. Pathway studio—The analysis and navigation of molecular networks. Bioinformatics 2003, 19, 2155–2157. [Google Scholar]
- Farre, E.M.; Harmer, S.L.; Harmon, F.G.; Yanovsky, M.J.; Kay, S.A. Overlapping and distinct roles of PRR7 and PRR9 in the Arabidopsis circadian clock. Curr. Biol. 2005, 15, 47–54. [Google Scholar]
- Nakamichi, N.; Kiba, T.; Henriques, R.; Mizuno, T.; Chua, N.H.; Sakakibara, H. Pseudo-response regulators 9 7 and 5 are transcriptional repressors in the Arabidopsis circadian clock. Plant Cell 2010, 22, 594–605. [Google Scholar]
- Dalchau, N.; Baek, S.J.; Briggs, H.M.; Robertson, F.C.; Dodd, A.N.; Gardner, M.J.; Stancombe, M.A.; Haydon, M.J.; Stan, G.B.; Goncalves, J.M.; et al. The circadian oscillator gene GIGANTEA mediates a long-term response of the Arabidopsis thaliana circadian clock to sucrose. Proc. Natl. Acad. Sci. USA 2011, 108, 5104–5109. [Google Scholar]
- Mizoguchi, T.; Wheatley, K.; Hanzawa, Y.; Wright, L.; Mizoguchi, M.; Song, H.R.; Carre, I.A.; Coupland, G. LHY and CCA1 are partially redundant genes required to maintain circadian rhythms in Arabidopsis. Dev. Cell 2002, 2, 629–641. [Google Scholar]
- Murakami, M.; Tago, Y.; Yamashino, T.; Mizuno, T. Comparative overviews of clock-associated genes of Arabidopsis thaliana and Oryza sativa. Plant Cell Physiol. 2007, 48, 110–121. [Google Scholar]
- Yu, J.W.; Rubio, V.; Lee, N.Y.; Bai, S.; Lee, S.Y.; Kim, S.S.; Liu, L.; Zhang, Y.; Irigoyen, M.L.; Sullivan, J.A.; et al. COP1 and ELF3 control circadian function and photoperiodic flowering by regulating GI stability. Mol. Cell 2008, 32, 617–630. [Google Scholar]
- Fornara, F.; Panigrahi, K.C.; Gissot, L.; Sauerbrunn, N.; Ruhl, M.; Jarillo, J.A.; Coupland, G. Arabidopsis DOF transcription factors act redundantly to reduce CONSTANS expression and are essential for a photoperiodic flowering response. Dev. Cell 2009, 17, 75–86. [Google Scholar]
- Kim, W.Y.; Fujiwara, S.; Suh, S.S.; Kim, J.; Kim, Y.; Han, L.; David, K.; Putterill, J.; Nam, H.G.; Somers, D.E. ZEITLUPE is a circadian photoreceptor stabilized by GIGANTEA in blue light. Nature 2007, 449, 356–360. [Google Scholar]
- Harmer, S.L. The circadian system in higher plants. Ann. Rev. Plant Biol. 2009, 60, 357–377. [Google Scholar]
- Yakir, E.; Hilman, D.; Harir, Y.; Green, R.M. Regulation of output from the plant circadian clock. FEBS J. 2007, 274, 335–345. [Google Scholar]
- Graf, A.; Schlereth, A.; Stitt, M.; Smith, A.M. Circadian control of carbohydrate availability for growth in Arabidopsis plants at night. Proc. Natl. Acad. Sci. USA 2010, 107, 9458–9463. [Google Scholar]
- Ni, Z.; Kim, E.D.; Ha, M.; Lackey, E.; Liu, J.; Zhang, Y.; Sun, Q.; Chen, Z.J. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 2009, 457, 327–331. [Google Scholar]
- Youens-Clark, K.; Buckler, E.; Casstevens, T.; Chen, C.; DeClerck, G.; Derwent, P.; Dharmawardhana, P.; Jaiswal, P.; Kersey, P.; Karthikeyan, A.S. Gramene database in 2010: Updates and extensions. Nucleic Acids Res. 2011, 39, D1085–D1094. [Google Scholar]
- Wang, E.; Wang, J.; Zhu, X.; Hao, W.; Wang, L.; Li, Q.; Zhang, L.; He, W.; Lu, B.; Lin, H.; et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 2008, 40, 1370–1374. [Google Scholar]
- Xing, Y.; Zhang, Q. Genetic and molecular bases of rice yield. Ann. Rev. Plant Biol. 2010, 61, 421–442. [Google Scholar]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L ssp indica). Science 2002, 296, 79–92. [Google Scholar]
- Yu, J.; Wang, J.; Lin, W.; Li, S.; Li, H.; Zhou, J.; Ni, P.; Dong, W.; Hu, S.; Zeng, C.; et al. The genomes of Oryza sativa: A history of duplications. PLoS Biol. 2005, 3, e38. [Google Scholar]
- Ma, L.; Chen, C.; Liu, X.; Jiao, Y.; Su, N.; Li, L.; Wang, X.; Cao, M.; Sun, N.; Zhang, X.; et al. A microarray analysis of the rice transcriptome and its comparison to Arabidopsis. Genome Res. 2005, 15, 1274–1283. [Google Scholar]
- Eisen, M.B.; Brown, P.O. DNA arrays for analysis of gene expression. Methods Enzymol. 1999, 303, 179–205. [Google Scholar]
- Bachem, C.W.B.; Oomen, R.J.F.J.; Visser, R.G.F. Transcript imaging with cDNA-AFLP: A step-by-step protocol. Plant Mol. Biol. Rep. 1998, 16, 157–157. [Google Scholar]
- Ma, L.; Gao, Y.; Qu, L.; Chen, Z.; Li, J.; Zhao, H.; Deng, X.W. Genomic evidence for COP1 as a repressor of light-regulated gene expression and development in Arabidopsis. Plant Cell 2002, 14, 2383–2398. [Google Scholar]
- Ma, L.; Li, J.; Qu, L.; Hager, J.; Chen, Z.; Zhao, H.; Deng, X.W. Light control of Arabidopsis development entails coordinated regulation of genome expression and cellular pathways. Plant Cell 2001, 13, 2589–2607. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez gene: Gene-centered information at NCBI. Nucleic Acids. Res. 2011, 39, D52–D57. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Developmental stage | LY2163 | LY2186 | LYP9 | Three hybrid combinations |
|---|---|---|---|---|
| Flowering (Fw) | 580 (1.6%) | 1,158 (3.1%) | 609 (1.6%) | 2,099 (5.7%) |
| Filling (Fi) | 640 (1.7%) | 635 (1.7%) | 550 (1.5%) | 1,488 (4.0%) |
| Fw + Fi | 1,193 (3.2%) | 1,630 (4.4%) | 1,046 (2.8%) | 3,142 (8.5%) |
| Overall rank a | Metabolic pathway b | LY2163 | LY2186 | LYP9 | |||
|---|---|---|---|---|---|---|---|
| p-value c | Rank d | p-value c | Rank d | p-value c | Rank d | ||
| 1 | Carbon fixation | 5.37 × 10−3 | 1 | 2.36 × 10−6 | 1 | 1.03 × 10−4 | 1 |
| 2 | Flavonoid biosynthesis | 0.185 | 3 | 0.192 | 4 | 0.116 | 3 |
| 3 | Metabolism of xenobiotics by cytochrome P450 | 0.224 | 4 | 0.151 | 3 | 0.266 | 8 |
| 4 | Photosynthesis | 0.045 | 2 | 0.493 | 13 | 0.056 | 2 |
| 5 | Reductive carboxylate cycle (CO2 fixation) | 0.514 | 6 | 0.258 | 6 | 0.284 | 10 |
| 6 | Oxidative phosphorylation | 0.595 | 10 | 0.062 | 2 | 0.659 | 26 |
| 7 | Glycolysis/Gluconeogenesis | 0.779 | 17 | 0.414 | 10 | 0.345 | 11 |
| 8 | Tricarboxylic acid (TCA) cycle | 0.341 | 5 | 0.312 | 9 | 0.665 | 28 |
| 9 | Starch and sucrose metabolism | 0.755 | 16 | 0.297 | 8 | 0.632 | 23 |
| 10 | Streptomycin biosynthesis | 0.857 | 26 | 0.561 | 17 | 0.129 | 4 |
| Category | LY2163 | LY2186 | LYP9 | Shared by three hybrid combinations |
|---|---|---|---|---|
| Number of DGs | 1,193 | 1,630 | 1,046 | 136 |
| DGs located to QTL | 1,004 | 1,411 | 894 | 120 |
| DGs located to yield-related QTL | 989 | 1385 | 884 | 119 |
| Number of QTL located by DGs | 4,057 | 4,326 | 4,114 | 3,884 |
| QTL of yield category | 909 | 1,006 | 969 | 882 |
| QTL of vigor category | 939 | 995 | 953 | 911 |
| QTL of Sterility or fertility category | 137 | 156 | 137 | 124 |
| QTL of quality category | 326 | 341 | 318 | 310 |
| QTL of development category | 427 | 450 | 421 | 412 |
| QTL of abiotic stress category | 346 | 365 | 339 | 315 |
| QTL of biotic stress category | 211 | 220 | 212 | 208 |
| QTL of biochemical category | 108 | 112 | 110 | 107 |
| QTL of anatomy category | 654 | 681 | 655 | 615 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peng, Y.; Wei, G.; Zhang, L.; Liu, G.; Wei, X.; Zhu, Z. Comparative Transcriptional Profiling of Three Super-Hybrid Rice Combinations. Int. J. Mol. Sci. 2014, 15, 3799-3815. https://doi.org/10.3390/ijms15033799
Peng Y, Wei G, Zhang L, Liu G, Wei X, Zhu Z. Comparative Transcriptional Profiling of Three Super-Hybrid Rice Combinations. International Journal of Molecular Sciences. 2014; 15(3):3799-3815. https://doi.org/10.3390/ijms15033799
Chicago/Turabian StylePeng, Yonggang, Gang Wei, Lei Zhang, Guozhen Liu, Xiaoli Wei, and Zhen Zhu. 2014. "Comparative Transcriptional Profiling of Three Super-Hybrid Rice Combinations" International Journal of Molecular Sciences 15, no. 3: 3799-3815. https://doi.org/10.3390/ijms15033799
APA StylePeng, Y., Wei, G., Zhang, L., Liu, G., Wei, X., & Zhu, Z. (2014). Comparative Transcriptional Profiling of Three Super-Hybrid Rice Combinations. International Journal of Molecular Sciences, 15(3), 3799-3815. https://doi.org/10.3390/ijms15033799