Killing Me Softly—Future Challenges in Apoptosis Research


Abstract
:1. Introduction
2. Discussion
2.1. AMARe Mortis
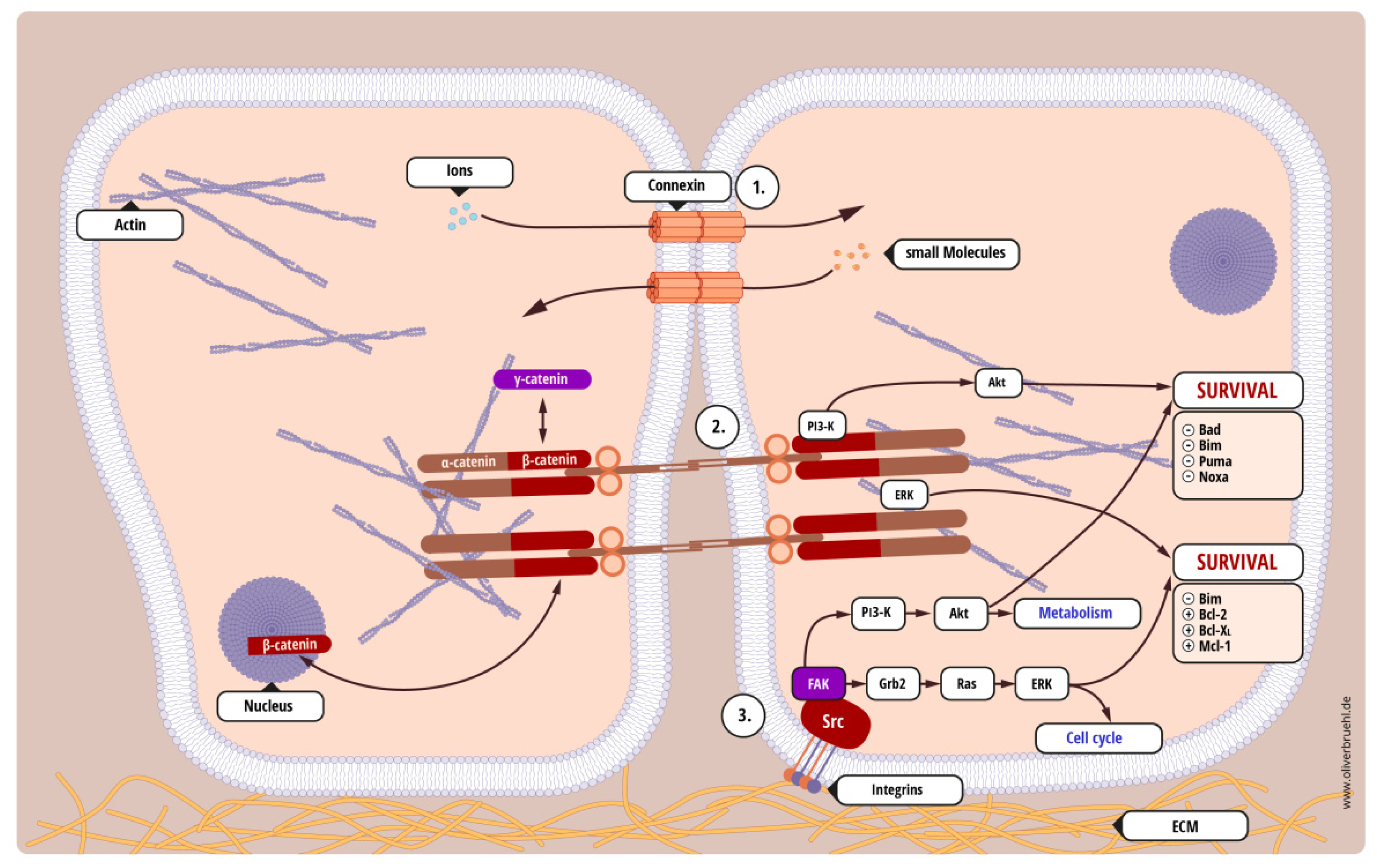
- Homotypic cell-cell interaction—tumor cells interact with other adjacent tumor cells;
- Heterotypic cell-cell interaction—tumor cells interact with other non-malignant cells, for example tumor-associated stroma or healthy tissue that is being invaded;
- Cell-substrate interaction—tumor cells interact with solid structures, such as the extracellular matrix or the basal membrane.
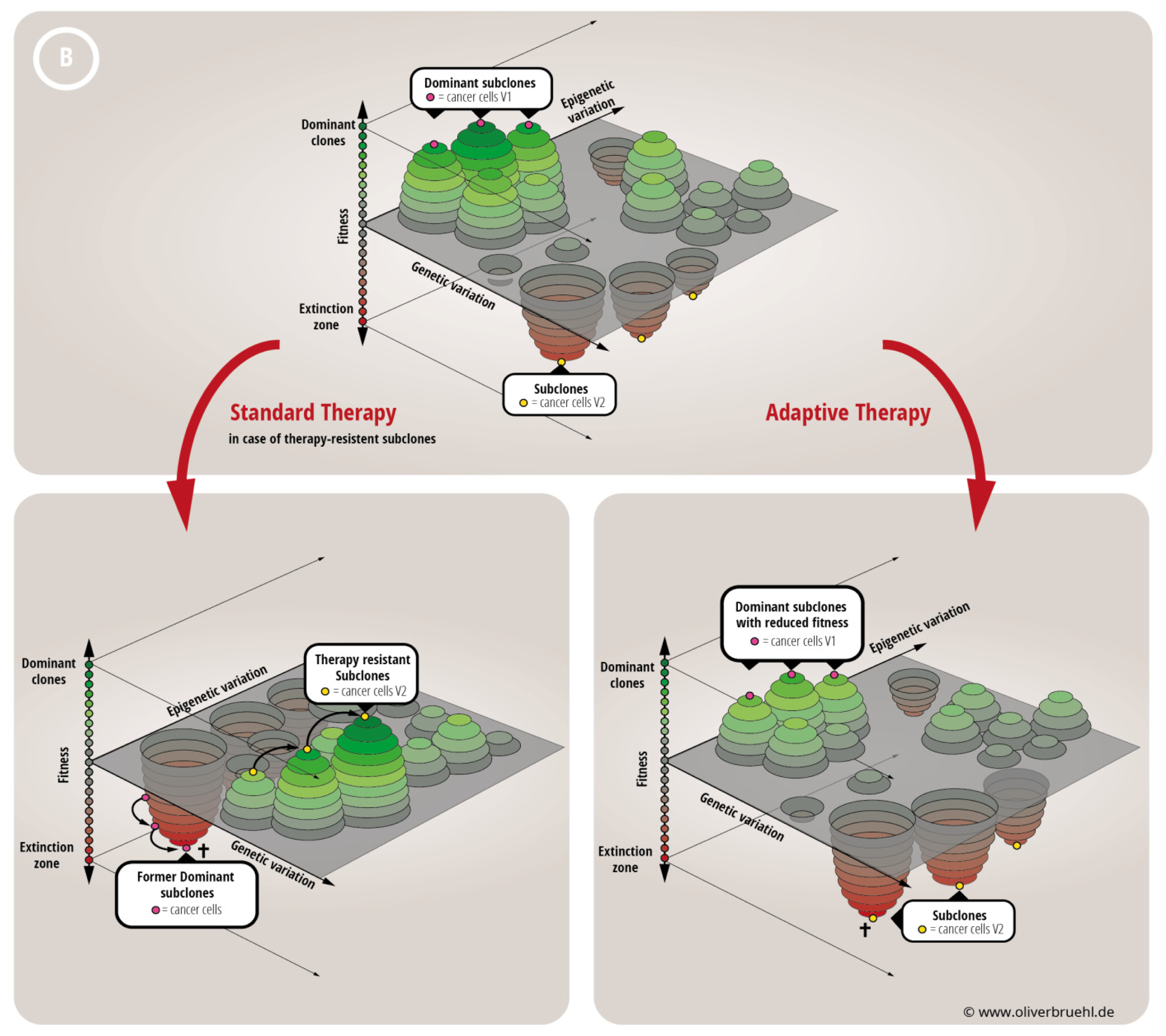
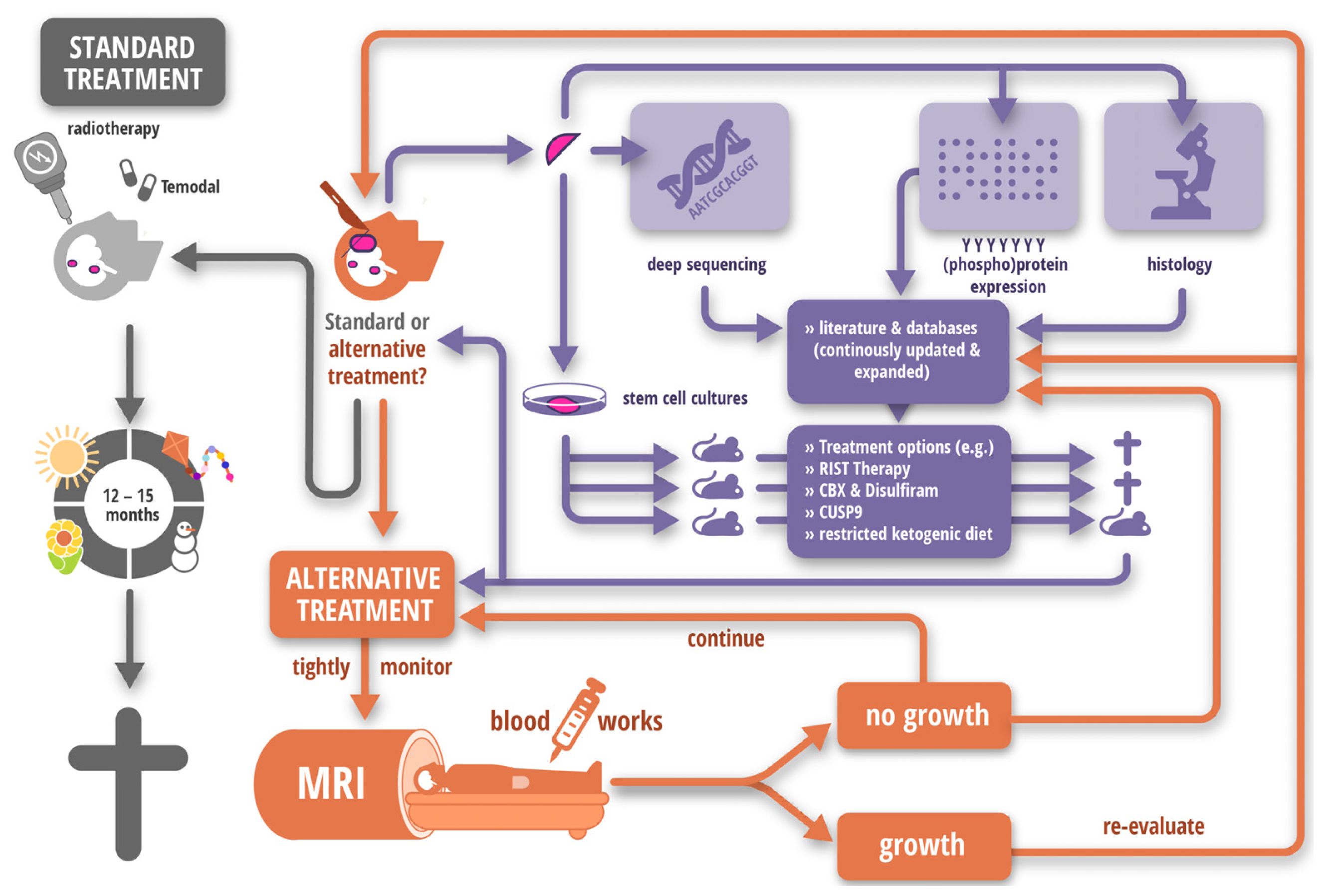
2.2. Tempus Occidendi et Tempus Sanandi
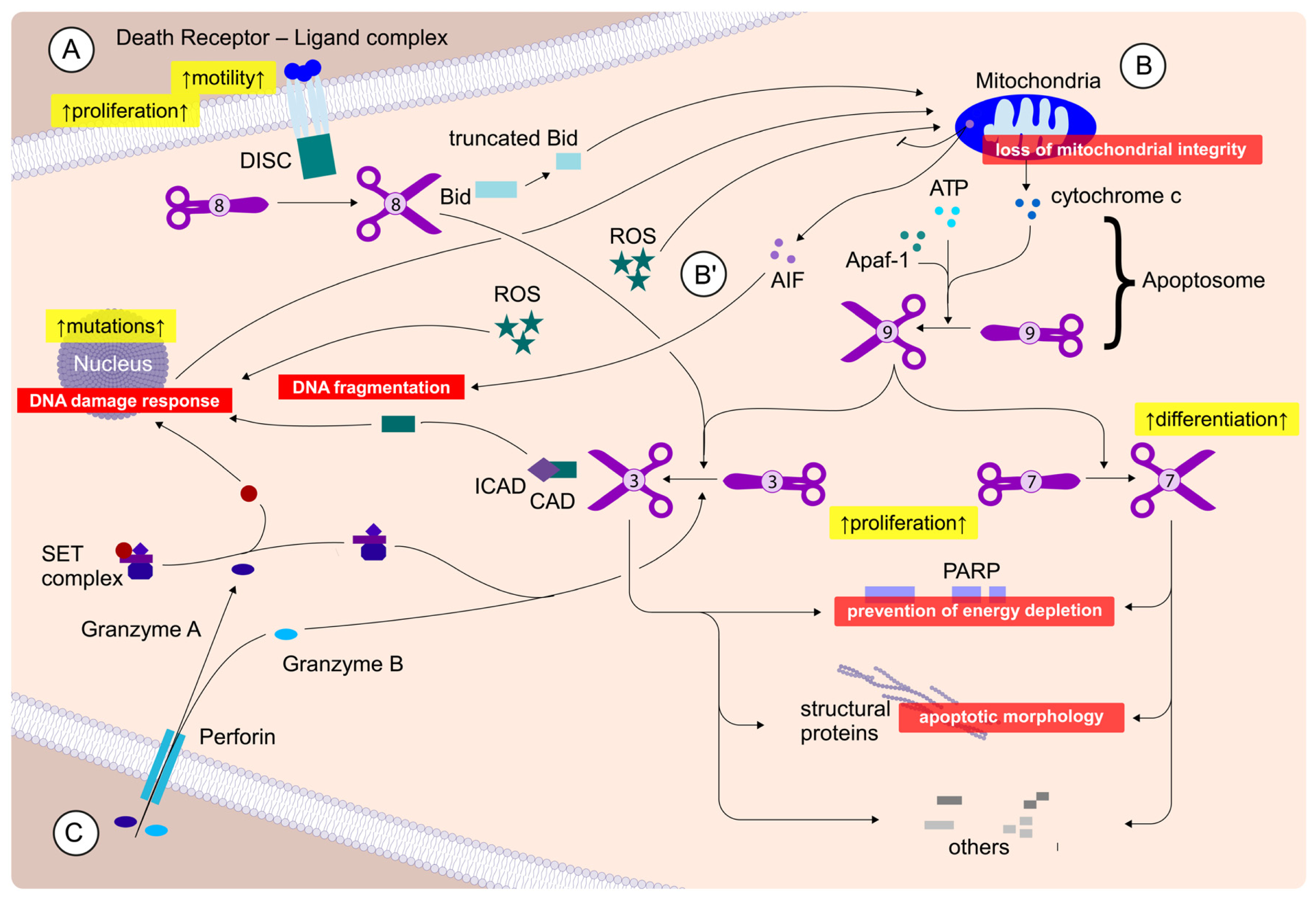
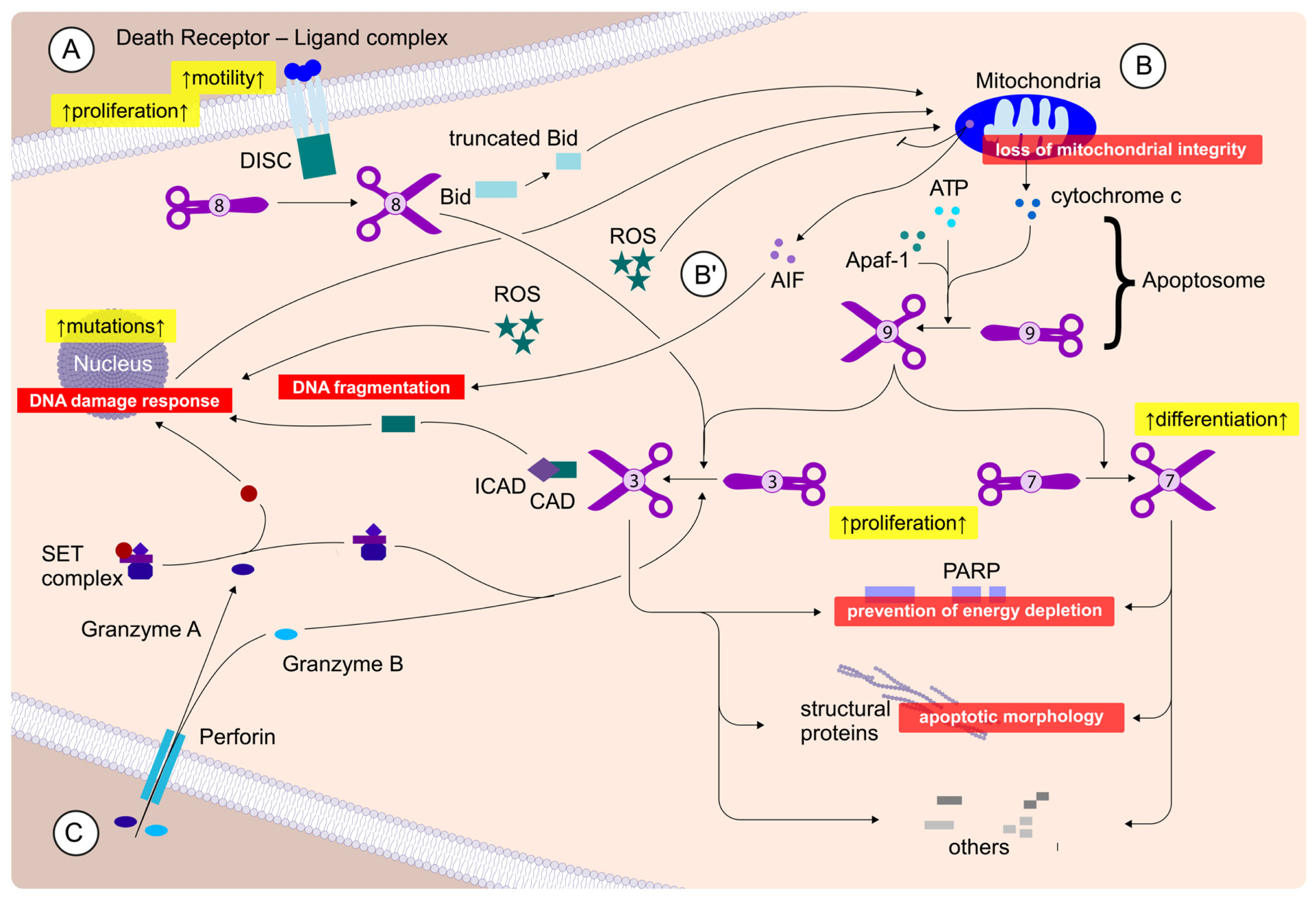
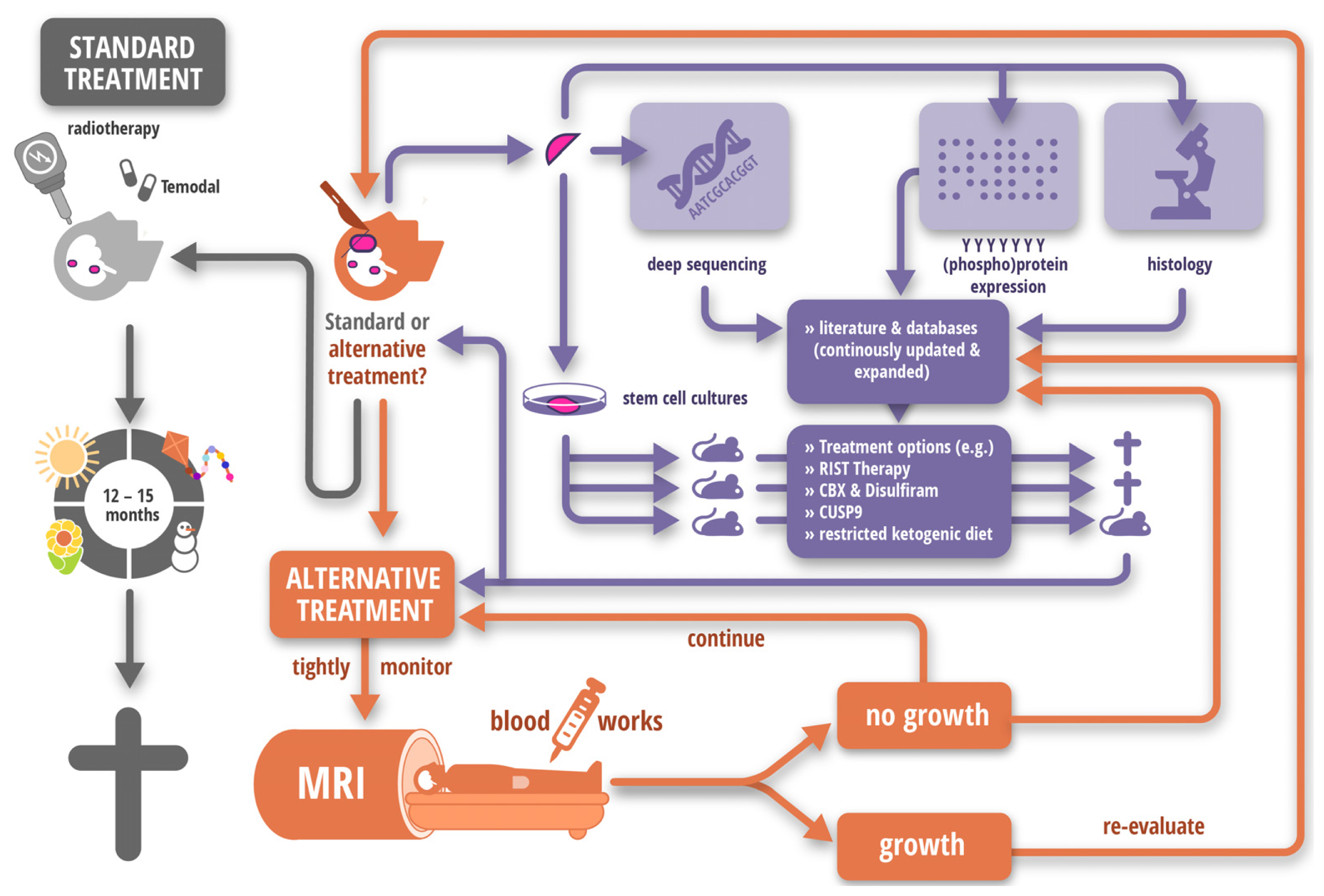
2.3. Caedite eos?
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar]
- Maghsoudi, N.; Zakeri, Z.; Lockshin, R.A. Programmed cell death and apoptosis—Where it came from and where it is going: From elie metchnikoff to the control of caspases. Exp. Oncol. 2012, 34, 146–152. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Sutherland, R.M.; Durand, R.E. Radiosensitization by nifuroxime of the hypoxic cells in an in vitro tumour model. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1972, 22, 613–618. [Google Scholar]
- Durand, R.E.; Sutherland, R.M. Radiation-resistant tumor cells may be more sensitive in vitro. Cancer Res. 1972, 32, 2587–2588. [Google Scholar]
- Sutherland, R.M.; Durand, R.E. Cell contact as a possible contribution to radiation resistance of some tumours. Br. J. Radiol. 1972, 45, 788–789. [Google Scholar]
- Chiarugi, P.; Giannoni, E. Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol. 2008, 76, 1352–1364. [Google Scholar]
- Damiano, J.S.; Hazlehurst, L.A.; Dalton, W.S. Cell adhesion-mediated drug resistance (cam-dr) protects the k562 chronic myelogenous leukemia cell line from apoptosis induced by bcr/abl inhibition cytotoxic drugs and gamma-irradiation. Leukemia 2001, 15, 1232–1239. [Google Scholar]
- Schmidmaier, R.; Baumann, P.; Simsek, M.; Dayyani, F.; Emmerich, B.; Meinhardt, G. The hmg-coa reductase inhibitor simvastatin overcomes cell adhesion-mediated drug resistance in multiple myeloma by geranylgeranylation of rho protein and activation of rho kinase. Blood 2004, 104, 1825–1832. [Google Scholar]
- Hazlehurst, L.A.; Argilagos, R.F.; Emmons, M.; Boulware, D.; Beam, C.A.; Sullivan, D.M.; Dalton, W.S. Cell adhesion to fibronectin (cam-dr) influences acquired mitoxantrone resistance in u937 cells. Cancer Res. 2006, 66, 2338–2345. [Google Scholar]
- Yanamandra, N.; Colaco, N.M.; Parquet, N.A.; Buzzeo, R.W.; Boulware, D.; Wright, G.; Perez, L.E.; Dalton, W.S.; Beaupre, D.M. Tipifarnib and bortezomib are synergistic and overcome cell adhesion-mediated drug resistance in multiple myeloma and acute myeloid leukemia. Clin. Cancer Res. 2006, 12, 591–599. [Google Scholar]
- Matsunaga, T.; Fukai, F.; Miura, S.; Nakane, Y.; Owaki, T.; Kodama, H.; Tanaka, M.; Nagaya, T.; Takimoto, R.; Takayama, T.; et al. Combination therapy of an anticancer drug with the fniii14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia 2008, 22, 353–360. [Google Scholar]
- Nimmanapalli, R.; Gerbino, E.; Dalton, W.S.; Gandhi, V.; Alsina, M. Hsp70 inhibition reverses cell adhesion mediated and acquired drug resistance in multiple myeloma. Br. J. Haematol. 2008, 142, 551–561. [Google Scholar]
- Wang, X.; Wang, C.; Qin, Y.W.; Yan, S.K.; Gao, Y.R. The association of up-regulation of x-linked inhibitor of apoptosis protein with cell adhesion-mediated drug resistance in u937 cells. Hematol. Oncol. 2008, 26, 21–26. [Google Scholar]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of vla-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar]
- Bourogaa, E.; Bertrand, J.; Despeaux, M.; Jarraya, R.; Fabre, N.; Payrastre, L.; Demur, C.; Fournie, J.J.; Damak, M.; Feki, A.E.; et al. Hammada scoparia flavonoids and rutin kill adherent and chemoresistant leukemic cells. Leuk. Res. 2011, 35, 1093–1101. [Google Scholar]
- Mraz, M.; Zent, C.S.; Church, A.K.; Jelinek, D.F.; Wu, X.; Pospisilova, S.; Ansell, S.M.; Novak, A.J.; Kay, N.E.; Witzig, T.E.; et al. Bone marrow stromal cells protect lymphoma b-cells from rituximab-induced apoptosis and targeting integrin α-4-β-1 (vla-4) with natalizumab can overcome this resistance. Br. J. Haematol. 2011, 155, 53–64. [Google Scholar]
- Kiziltepe, T.; Ashley, J.D.; Stefanick, J.F.; Qi, Y.M.; Alves, N.J.; Handlogten, M.W.; Suckow, M.A.; Navari, R.M.; Bilgicer, B. Rationally engineered nanoparticles target multiple myeloma cells overcome cell-adhesion-mediated drug resistance and show enhanced efficacy in vivo. Blood Cancer J. 2012, 2, e64. [Google Scholar]
- Hazlehurst, L.A.; Valkov, N.; Wisner, L.; Storey, J.A.; Boulware, D.; Sullivan, D.M.; Dalton, W.S. Reduction in drug-induced DNA double-strand breaks associated with β1 integrin-mediated adhesion correlates with drug resistance in u937 cells. Blood 2001, 98, 1897–1903. [Google Scholar]
- Zhu, B.; Zhao, L.; Zhu, L.; Wang, H.; Sha, Y.; Yao, J.; Li, Z.; You, Q.; Guo, Q. Oroxylin a reverses cam-dr of hepg2 cells by suppressing integrinβ1 and its related pathway. Toxicol. Appl. Pharmacol. 2012, 259, 387–394. [Google Scholar]
- Bjorklund, C.C.; Baladandayuthapani, V.; Lin, H.Y.; Jones, R.J.; Kuiatse, I.; Wang, H.; Yang, J.; Shah, J.J.; Thomas, S.K.; Wang, M.; et al. Evidence of a role for cd44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: Therapeutic implications. Leukemia 2014, 28, 373–383. [Google Scholar]
- Fei, M.; Hang, Q.; Hou, S.; Ruan, C. Cell adhesion to fibronectin down-regulates the expression of spy1 and contributes to drug resistance in multiple myeloma cells. Int. J. Hematol. 2013, 98, 446–455. [Google Scholar]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (cam-dr): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar]
- Westhoff, M.A.; Zhou, S.; Bachem, M.G.; Debatin, K.M.; Fulda, S. Identification of a novel switch in the dominant forms of cell adhesion-mediated drug resistance in glioblastoma cells. Oncogene 2008, 27, 5169–5181. [Google Scholar]
- Shain, K.H.; Landowski, T.H.; Dalton, W.S. Adhesion-mediated intracellular redistribution of c-fas-associated death domain-like il-1-converting enzyme-like inhibitory protein-long confers resistance to cd95-induced apoptosis in hematopoietic cancer cell lines. J. Immunol. 2002, 168, 2544–2553. [Google Scholar]
- Taylor, S.T.; Hickman, J.A.; Dive, C. Epigenetic determinants of resistance to etoposide regulation of bcl-x(l) and bax by tumor microenvironmental factors. J. Natl. Cancer Inst. 2000, 92, 18–23. [Google Scholar]
- Lwin, T.; Hazlehurst, L.A.; Li, Z.; Dessureault, S.; Sotomayor, E.; Moscinski, L.C.; Dalton, W.S.; Tao, J. Bone marrow stromal cells prevent apoptosis of lymphoma cells by upregulation of anti-apoptotic proteins associated with activation of NF-κB (relb/p52) in non-hodgkin’s lymphoma cells. Leukemia 2007, 21, 1521–1531. [Google Scholar]
- Fortney, J.E.; Zhao, W.; Wenger, S.L.; Gibson, L.F. Bone marrow stromal cells regulate caspase 3 activity in leukemic cells during chemotherapy. Leuk. Res. 2001, 25, 901–907. [Google Scholar]
- Niedermeier, M.; Hennessy, B.T.; Knight, Z.A.; Henneberg, M.; Hu, J.; Kurtova, A.V.; Wierda, W.G.; Keating, M.J.; Shokat, K.M.; Burger, J.A. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit cxcr4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: A novel therapeutic approach. Blood 2009, 113, 5549–5557. [Google Scholar]
- Aoudjit, F.; Vuori, K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene 2001, 20, 4995–5004. [Google Scholar]
- Sethi, T.; Rintoul, R.C.; Moore, S.M.; MacKinnon, A.C.; Salter, D.; Choo, C.; Chilvers, E.R.; Dransfield, I.; Donnelly, S.C.; Strieter, R.; et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 1999, 5, 662–668. [Google Scholar]
- Uhm, J.H.; Dooley, N.P.; Kyritsis, A.P.; Rao, J.S.; Gladson, C.L. Vitronectin a glioma-derived extracellular matrix protein protects tumor cells from apoptotic death. Clin. Cancer Res. 1999, 5, 1587–1594. [Google Scholar]
- Wang, L.; Li, Z.; Wang, C.; Yang, Y.; Sun, L.; Yao, W.; Cai, X.; Wu, G.; Zhou, F.; Zha, X. E-cadherin decreased human breast cancer cells sensitivity to staurosporine by up-regulating bcl-2 expression. Arch. Biochem. Biophys. 2009, 481, 116–122. [Google Scholar]
- Cordes, N.; Meineke, V. Cell adhesion-mediated radioresistance (cam-rr) Extracellular matrix-dependent improvement of cell survival in human tumor and normal cells in vitro. Strahlenther. Onkol. 2003, 179, 337–344. [Google Scholar]
- Sandfort, V.; Koch, U.; Cordes, N. Cell adhesion-mediated radioresistance revisited. Int. J. Radiat. Biol. 2007, 83, 727–732. [Google Scholar]
- Westhoff, M.A.; Fulda, S. Adhesion-mediated apoptosis resistance in cancer. Drug Resist. Updat. 2009, 12, 127–136. [Google Scholar]
- Dolberg, D.S.; Bissell, M.J. Inability of rous sarcoma virus to cause sarcomas in the avian embryo. Nature 1984, 309, 552–556. [Google Scholar]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar]
- Hoelzinger, D.B.; Demuth, T.; Berens, M.E. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J. Natl. Cancer Inst. 2007, 99, 1583–1593. [Google Scholar]
- Pullan, S.; Wilson, J.; Metcalfe, A.; Edwards, G.M.; Goberdhan, N.; Tilly, J.; Hickman, J.A.; Dive, C.; Streuli, C.H. Requirement of basement membrane for the suppression of programmed cell death in mammary epithelium. J. Cell. Sci. 1996, 109, 631–642. [Google Scholar]
- Chidgey, M.; Dawson, C. Desmosomes: A role in cancer? Br. J. Cancer 2007, 96, 1783–1787. [Google Scholar]
- Morin, P.J. Claudin proteins in human cancer: Promising new targets for diagnosis and therapy. Cancer Res. 2005, 65, 9603–9606. [Google Scholar]
- Langlois, M.J.; Bergeron, S.; Bernatchez, G.; Boudreau, F.; Saucier, C.; Perreault, N.; Carrier, J.C.; Rivard, N. The pten phosphatase controls intestinal epithelial cell polarity and barrier function: Role in colorectal cancer progression. PLoS One 2010, 5, e15742. [Google Scholar]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The ras-erk and pi3k-mtor pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar]
- Westhoff, M.A.; Zhou, S.; Nonnenmacher, L.; Karpel-Massler, G.; Jennewein, C.; Schneider, M.; Halatsch, M.E.; Carragher, N.O.; Baumann, B.; Krause, A.; et al. Inhibition of NF-κB signaling ablates the invasive phenotype of glioblastoma. Mol. Cancer Res. 2013, 11, 1611–1623. [Google Scholar]
- Paolillo, M.; Russo, M.A.; Serra, M.; Colombo, L.; Schinelli, S. Small molecule integrin antagonists in cancer therapy. Mini Rev. Med. Chem. 2009, 9, 1439–1446. [Google Scholar]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar]
- Miller, D.H.; Weber, T.; Grove, R.; Wardell, C.; Horrigan, J.; Graff, O.; Atkinson, G.; Dua, P.; Yousry, T.; Macmanus, D.; et al. Firategrast for relapsing remitting multiple sclerosis: A phase 2 randomised double-blind placebo-controlled trial. Lancet Neurol. 2012, 11, 131–139. [Google Scholar]
- Tomlinson, I.P.; Novelli, M.R.; Bodmer, W.F. The mutation rate and cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 14800–14803. [Google Scholar]
- Hu, M.; Polyak, K. Microenvironmental regulation of cancer development. Curr. Opin. Genet. Dev. 2008, 18, 27–34. [Google Scholar]
- Kaplan, R.N.; Rafii, S.; Lyden, D. Preparing the “Soil”: The premetastatic niche. Cancer Res. 2006, 66, 11089–11093. [Google Scholar]
- Sceneay, J.; Smyth, M.J.; Moller, A. The pre-metastatic niche: Finding common ground. Cancer Metastasis Rev. 2013, 32, 449–464. [Google Scholar]
- Schultz, G.S.; Davidson, J.M.; Kirsner, R.S.; Bornstein, P.; Herman, I.M. Dynamic reciprocity in the wound microenvironment. Wound Repair Regen. 2011, 19, 134–148. [Google Scholar]
- Paget, S. The distribution of secondary growths in cancer of the breast 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- An, X.; Tiwari, A.K.; Sun, Y.; Ding, P.R.; Ashby, C.R., Jr; Chen, Z.S. Bcr-abl tyrosine kinase inhibitors in the treatment of philadelphia chromosome positive chronic myeloid leukemia: A review. Leuk. Res. 2010, 34, 1255–1268. [Google Scholar]
- Gately, S.; Kerbel, R. Antiangiogenic scheduling of lower dose cancer chemotherapy. Cancer J. 2001, 7, 427–436. [Google Scholar]
- Kerbel, R.; Folkman, J. Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2002, 2, 727–739. [Google Scholar]
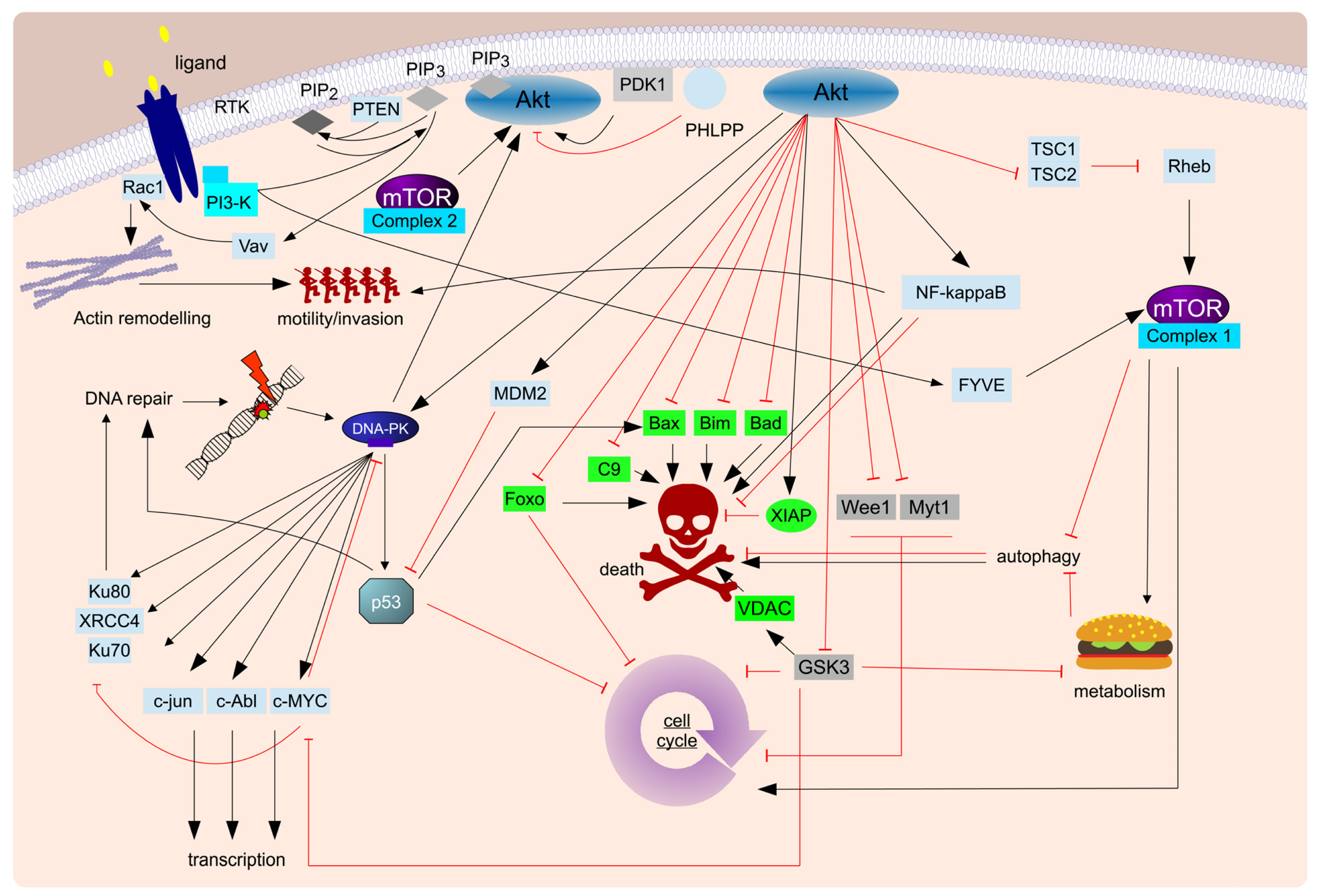
- Yuan, T.L.; Cantley, L.C. Pi3k pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar]
- Cassinelli, G.; Zuco, V.; Gatti, L.; Lanzi, C.; Zaffaroni, N.; Colombo, D.; Perego, P. Targeting the akt kinase to modulate survival invasiveness and drug resistance of cancer cells. Curr. Med. Chem. 2013, 20, 1923–1945. [Google Scholar]
- Franke, T.F. Pi3k/akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar]
- Hemmings, B.A.; Restuccia, D.F. Pi3k-pkb/akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar]
- Chalhoub, N.; Baker, S.J. Pten and the pi3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar]
- Brana, I.; Siu, L.L. Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med. 2012, 10, 161. [Google Scholar]
- Yin, Y.; Shen, W.H. Pten: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar]
- Burris, H.A., 3rd. Overcoming acquired resistance to anticancer therapy: Focus on the pi3k/akt/mtor pathway. Cancer Chemother. Pharmacol. 2013, 71, 829–842. [Google Scholar]
- Figlin, R.A.; Kaufmann, I.; Brechbiel, J. Targeting pi3k and mtorc2 in metastatic renal cell carcinoma: New strategies for overcoming resistance to vegfr and mtorc1 inhibitors. Int. J. Cancer 2013, 133, 788–796. [Google Scholar]
- Fulda, S. The pi3k/akt/mtor pathway as therapeutic target in neuroblastoma. Curr. Cancer Drug Targets 2009, 9, 729–737. [Google Scholar]
- Wolin, E.M. Pi3k/akt/mtor pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013, 335, 1–8. [Google Scholar]
- Okumura, N.; Yoshida, H.; Kitagishi, Y.; Murakami, M.; Nishimura, Y.; Matsuda, S. Pi3k/akt/pten signaling as a molecular target in leukemia angiogenesis. Adv. Hematol. 2012, 2012, 843085. [Google Scholar]
- Wen, P.Y.; Lee, E.Q.; Reardon, D.A.; Ligon, K.L.; Alfred Yung, W.K. Current clinical development of pi3k pathway inhibitors in glioblastoma. Neuro Oncol. 2012, 14, 819–829. [Google Scholar]
- Lee, M.J.; Ye, A.S.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; MacBeath, G.; Yaffe, M.B. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012, 149, 780–794. [Google Scholar]
- Westhoff, M.A.; Faham, N.; Marx, D.; Nonnenmacher, L.; Jennewein, C.; Enzenmüller, S.; Gonzalez, P.; Fulda, S.; Debatin, K.M. Sequential dosing in chemosensitization: Targeting the PI3K/Akt/mTOR pathway in neuroblastoma. PLoS One 2013, 8, e83128. [Google Scholar]
- Hoelzinger, D.B.; Mariani, L.; Weis, J.; Woyke, T.; Berens, T.J.; McDonough, W.S.; Sloan, A.; Coons, S.W.; Berens, M.E. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia 2005, 7, 7–16. [Google Scholar]
- Mullighan, C.G.; Phillips, L.A.; Su, X.; Ma, J.; Miller, C.B.; Shurtleff, S.A.; Downing, J.R. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322, 1377–1380. [Google Scholar]
- Denison, T.A.; Bae, Y.H. Tumor heterogeneity and its implication for drug delivery. J. Control. Release 2012, 164, 187–191. [Google Scholar]
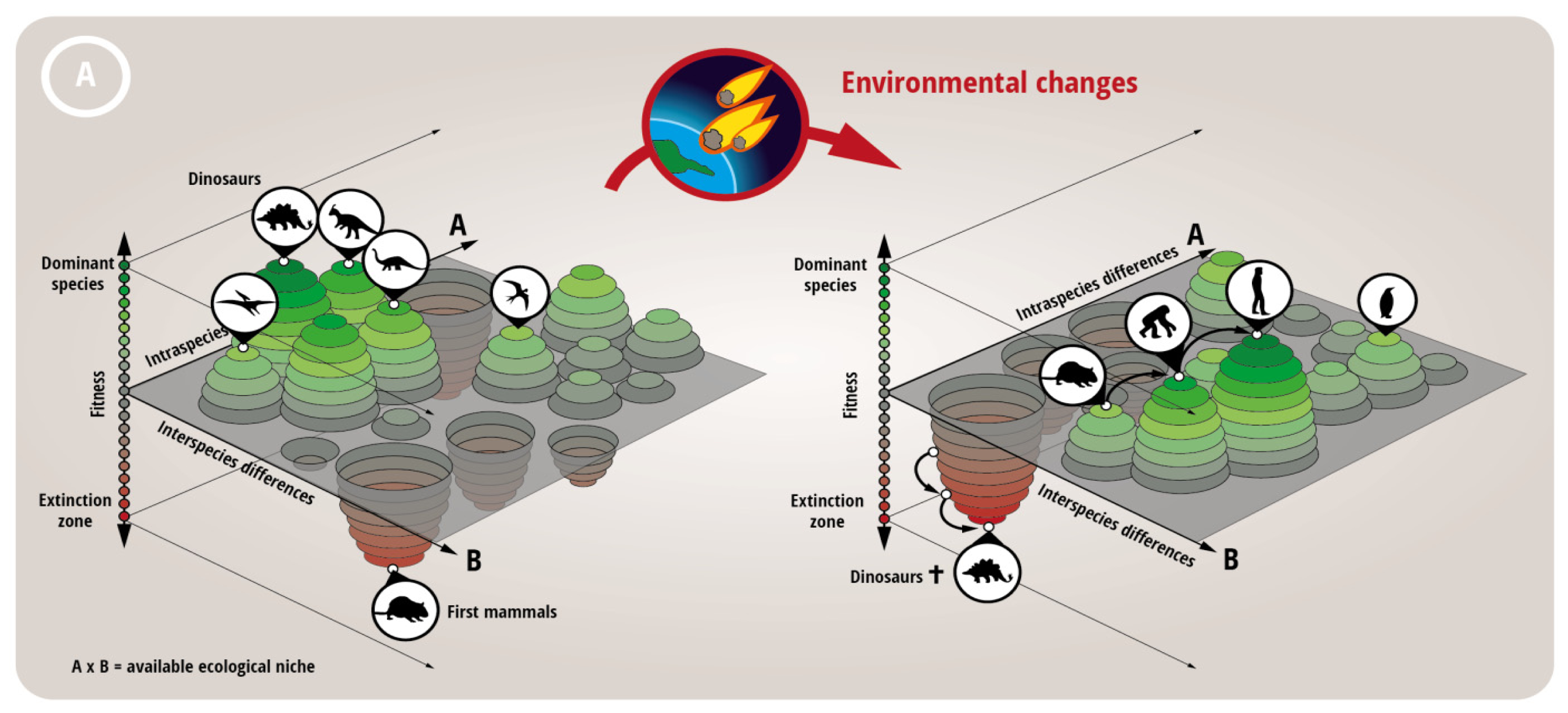
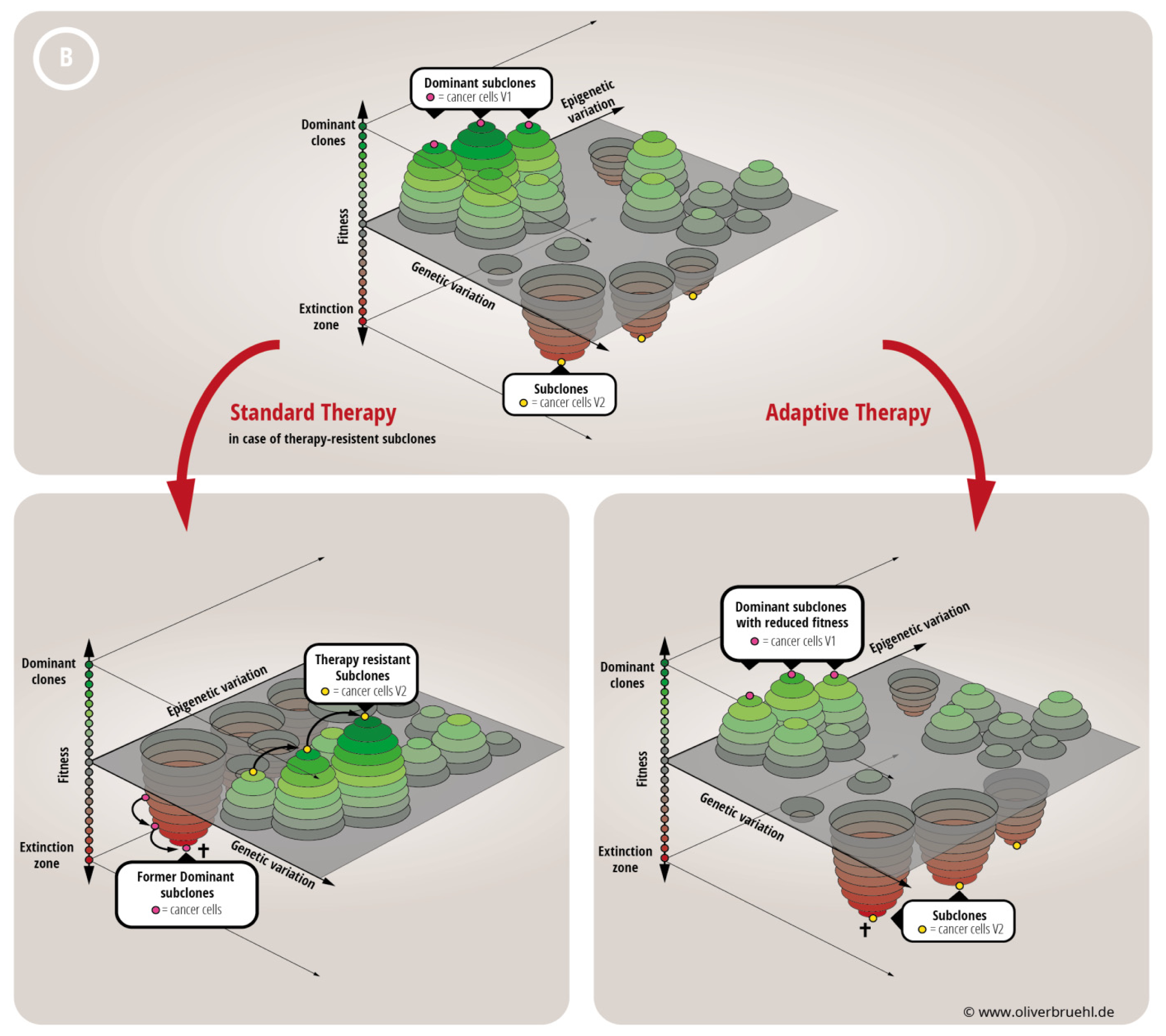
- Huang, S. Genetic and non-genetic instability in tumor progression: Link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev. 2013, 32, 423–448. [Google Scholar]
- Brock, A.; Chang, H.; Huang, S. Non-genetic heterogeneity—A mutation-independent driving force for the somatic evolution of tumours. Nat. Rev. Genet. 2009, 10, 336–342. [Google Scholar]
- Foo, J.; Leder, K.; Mumenthaler, S.M. Cancer as a moving target: Understanding the composition and rebound growth kinetics of recurrent tumors. Evol. Appl. 2013, 6, 54–69. [Google Scholar]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar]
- Gatenby, R.A.; Brown, J.; Vincent, T. Lessons from applied ecology: Cancer control using an evolutionary double bind. Cancer Res. 2009, 69, 7499–7502. [Google Scholar]
- Durrett, R. Population genetics of neutral mutations in exponentially growing cancer cell populations. Ann. Appl. Probab. 2013, 23, 230–250. [Google Scholar]
- Nonnenmacher, L.; Westhoff, M.A.; Fulda, S.; Karpel-Massler, G.; Halatsch, M.; Engelke, J.; Simmet, T.; Gorbacioglu, S.; Debatin, K.M. RIST: A potent new combination therapy for glioblastoma. Clin. Cancer Res. 2014. to be submitted. [Google Scholar]
- Zhou, J.; Bi, C.; Janakakumara, J.V.; Liu, S.C.; Chng, W.J.; Tay, K.G.; Poon, L.F.; Xie, Z.; Palaniyandi, S.; Yu, H.; et al. Enhanced activation of stat pathways and overexpression of survivin confer resistance to flt3 inhibitors and could be therapeutic targets in aml. Blood 2009, 113, 4052–4062. [Google Scholar]
- Stolzel, F.; Steudel, C.; Oelschlagel, U.; Mohr, B.; Koch, S.; Ehninger, G.; Thiede, C. Mechanisms of resistance against pkc412 in resistant flt3-itd positive human acute myeloid leukemia cells. Ann. Hematol. 2010, 89, 653–662. [Google Scholar]
- Herrmann, M.D.; Lennerz, J.K.; Bullinger, L.; Bartholomae, S.; Holzmann, K.; Westhoff, M.A.; Corbacioglu, S.; Debatin, K.M. Transitory dasatinib-resistant states in kitmut t(8;21) acute myeloid leukemia cells correlate with altered kit expression. Exp. Hematol. 2014, 42, 90–100. [Google Scholar]
- Saito, N.; Ishihara, S.; Kaneko, K. Baldwin effect under multipeaked fitness landscapes: Phenotypic fluctuation accelerates evolutionary rate. Phys. Rev. E 2013, 87, 052701. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the united states before and during the temozolomide era. J. Neurooncol. 2011, 107, 359–364. [Google Scholar]
- Kim, M.; Lee, K.H.; Yoon, S.W.; Kim, B.S.; Chun, J.; Yi, H. Analytical tools and databases for metagenomics in the next-generation sequencing era. Genomics Inform. 2013, 11, 102–113. [Google Scholar]
- Lee, S.H.; Sim, S.H.; Kim, J.Y.; Cha, S.; Song, A. Application of cancer genomics to solve unmet clinical needs. Genomics Inform. 2013, 11, 174–179. [Google Scholar]
- Van Oostrum, J.; Calonder, C.; Rechsteiner, D.; Ehrat, M.; Mestan, J.; Fabbro, D.; Voshol, H. Tracing pathway activities with kinase inhibitors and reverse phase protein arrays. Proteomics Clin. Appl. 2009, 3, 412–422. [Google Scholar]
- Carragher, N.O.; Unciti-Broceta, A.; Cameron, D.A. Advancing cancer drug discovery towards more agile development of targeted combination therapies. Future Med. Chem. 2012, 4, 87–105. [Google Scholar]
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 319–331. [Google Scholar]
- De Witt Hamer, P.C.; van Tilborg, A.A.; Eijk, P.P.; Sminia, P.; Troost, D.; van Noorden, C.J.; Ylstra, B.; Leenstra, S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar]
- Seyfried, T.N.; Marsh, J.; Shelton, L.M.; Huysentruyt, L.C.; Mukherjee, P. Is the restricted ketogenic diet a viable alternative to the standard of care for managing malignant brain cancer? Epilepsy Res. 2011, 100, 310–326. [Google Scholar]
- Kast, R.E.; Boockvar, J.A.; Bruning, A.; Cappello, F.; Chang, W.W.; Cvek, B.; Dou, Q.P.; Duenas-Gonzalez, A.; Efferth, T.; Focosi, D.; et al. A conceptually new treatment approach for relapsed glioblastoma: Coordinated undermining of survival paths with nine repurposed drugs (cusp9) by the international initiative for accelerated improvement of glioblastoma care. Oncotarget 2013, 4, 502–530. [Google Scholar]
- Amaravadi, R.K.; Thompson, C.B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar]
- Barbero, S.; Mielgo, A.; Torres, V.; Teitz, T.; Shields, D.J.; Mikolon, D.; Bogyo, M.; Barila, D.; Lahti, J.M.; Schlaepfer, D.; et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009, 69, 3755–3763. [Google Scholar]
- Gdynia, G.; Grund, K.; Eckert, A.; Bock, B.C.; Funke, B.; Macher-Goeppinger, S.; Sieber, S.; Herold-Mende, C.; Wiestler, B.; Wiestler, O.D.; et al. Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol. Cancer Res. 2007, 5, 1232–1240. [Google Scholar]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through wnt16b. Nat. Med. 2012, 18, 1359–1368. [Google Scholar]
- Basu, S.; Rajakaruna, S.; Menko, A.S. Insulin-like growth factor receptor-1 and nuclear factor kappab are crucial survival signals that regulate caspase-3-mediated lens epithelial cell differentiation initiation. J. Biol. Chem. 2012, 287, 8384–8397. [Google Scholar]
- Fernando, P.; Brunette, S.; Megeney, L.A. Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J. 2005, 19, 1671–1673. [Google Scholar]
- Esserman, L.J.; Thompson, I.M., Jr; Reid, B. Overdiagnosis and overtreatment in cancer: An opportunity for improvement. JAMA 2013, 310, 797–798. [Google Scholar]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Adhesion type | Apoptosis inducing agents investigated | Molecular basis for AMAR | Therapeutic suggestion | Reference |
|---|---|---|---|---|---|
| Multiple myeloma | He, ECM | Melphalan | enhanced HSP70 expression | no concrete suggestion | [17] |
| Multiple myeloma | ECM | Doxorubicin, etoposide | not determined | no concrete suggestion | [23] |
| Multiple myeloma | ECM | CD95 | increased soluble c-Flip | no concrete suggestion | [29] |
| Multiple myeloma | ECM | Doxorubicin, melphalan | not determined | no concrete suggestion | [27] |
| Multiple myeloma | ECM | Bortezomib | not determined | no concrete suggestion | [19] |
| Multiple myeloma | He | Doxorubicin | not determined | Integrin antagonists | [22] |
| Multiple myeloma | He, ECM | Lenalidomide | not determined | All-trans retinoic acid | [25] |
| Multiple myeloma | ECM | Doxorubicin, mitoxantrone | Up-regulation of p27Kip1 | no concrete suggestion | [26] |
| Multiple myeloma | He | Melphalan, treosulfan, doxorubicin, dexamethasone, bortezomib | Rho/Rho-Kinase signaling | Statins | [13] |
| Multiple myeloma, acute myeloid leukemia | ECM | Tipifarnib | not determined | combine with bortezomib | [15] |
| Burkett’s lymphoma | ECM | Etoposide | Bcl-2 family | no concrete suggestion | [30] |
| B-cell lymphoma | He | Rituximab | not determined | Integrin antagonists | [21] |
| Histiocytic lymphoma | ECM | Mitroxantrone | not determined | no concrete suggestion | [14] |
| Lymphoma | He | Mitroxantrone | IAPs via NF-kappaB | no concrete suggestion | [31] |
| Lymphoblastic leukemia | He | Cytarabine, etoposide | not determined | no concrete suggestion | [32] |
| Chronic lymphocytic leukemia | He | Fludarabine | PI3K/Akt mediated signaling | PI3K inhibitor | [33] |
| Acute myeloid leukemia | ECM | Cytarabine | Bcl-2 via PI3K/Akt | Integrin antagonists | [16] |
| Acute myeloid leukemia | ECM | Daunorubicin | XIAP via PI3K/Akt | no concrete suggestion | [18] |
| Acute myeloid leukemia | ECM, He | Rutin, etoposide | GSK3beta | Rutin | [20] |
| Chronic myeloid leukemia | ECM | Melphalan, cytarabin, radiation | not determined | Integrin antagonists | [12] |
| Breast cancer | ECM | Paclitaxel, vincristine | PI3K/Akt mediated signaling | no concrete suggestion | [34] |
| Breast cancer | Ho | Staurosporine | Bcl-2 family | no concrete suggestion | [37] |
| Hepatocellular carcinoma | ECM | Paclitaxel | not determined | Integrin antagonists | [24] |
| Small cell lung cancer | ECM | Doxorubicin, etoposide | Tyrosine phosphorylation | Integrin antagonists | [35] |
| Glioblastoma | ECM | Etoposide | Bcl-2 family | no concrete suggestion | [36] |
| Glioblastoma | ECM | Radiation | not determined | no concrete suggestion | [38] |
| Glioblastoma | Ho, ECM | TRAIL | not determined | Carbenoxolone | [28] |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Westhoff, M.-A.; Brühl, O.; Nonnenmacher, L.; Karpel-Massler, G.; Debatin, K.-M. Killing Me Softly—Future Challenges in Apoptosis Research. Int. J. Mol. Sci. 2014, 15, 3746-3767. https://doi.org/10.3390/ijms15033746
Westhoff M-A, Brühl O, Nonnenmacher L, Karpel-Massler G, Debatin K-M. Killing Me Softly—Future Challenges in Apoptosis Research. International Journal of Molecular Sciences. 2014; 15(3):3746-3767. https://doi.org/10.3390/ijms15033746
Chicago/Turabian StyleWesthoff, Mike-Andrew, Oliver Brühl, Lisa Nonnenmacher, Georg Karpel-Massler, and Klaus-Michael Debatin. 2014. "Killing Me Softly—Future Challenges in Apoptosis Research" International Journal of Molecular Sciences 15, no. 3: 3746-3767. https://doi.org/10.3390/ijms15033746