Activation of VCAM-1 and Its Associated Molecule CD44 Leads to Increased Malignant Potential of Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
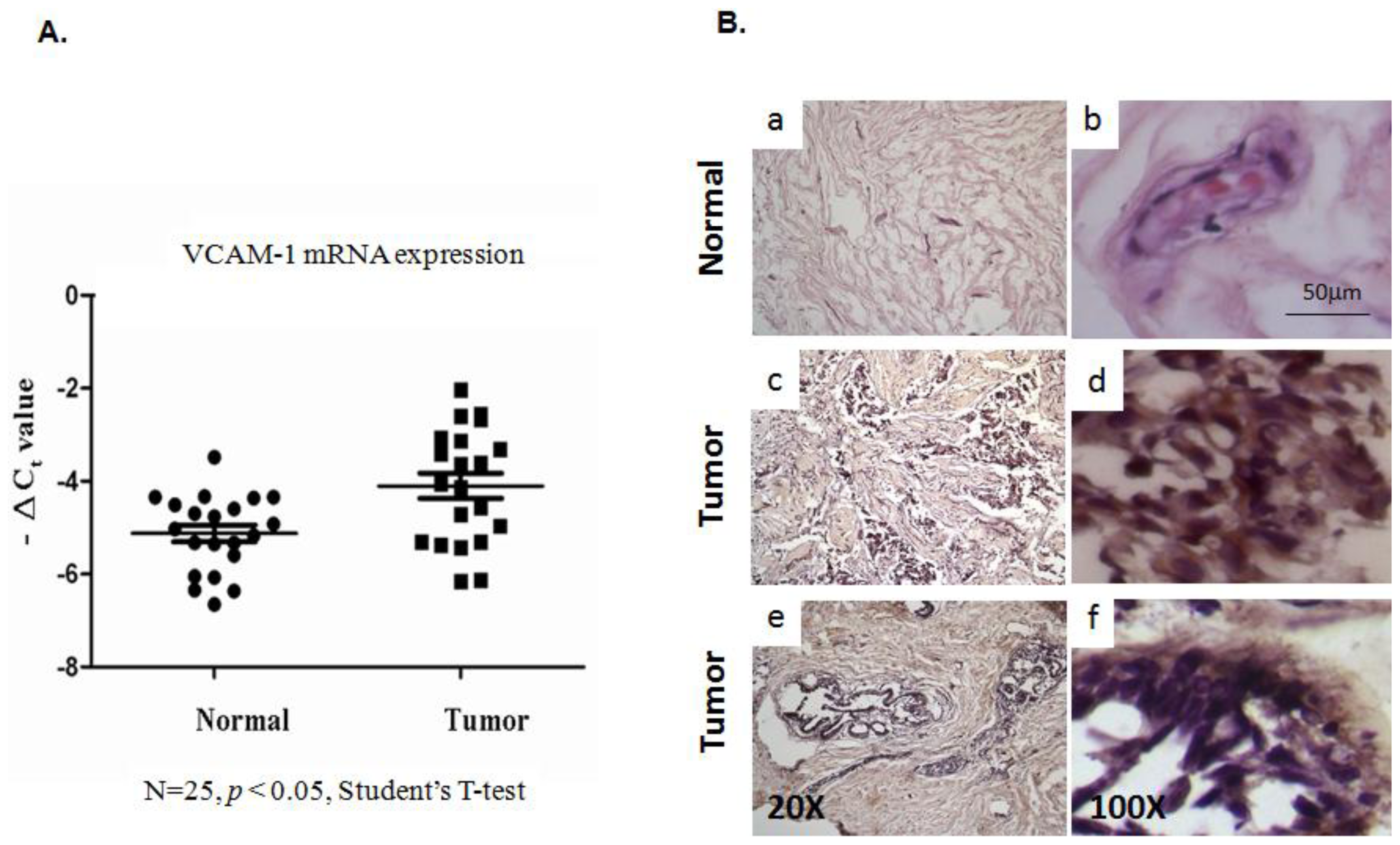
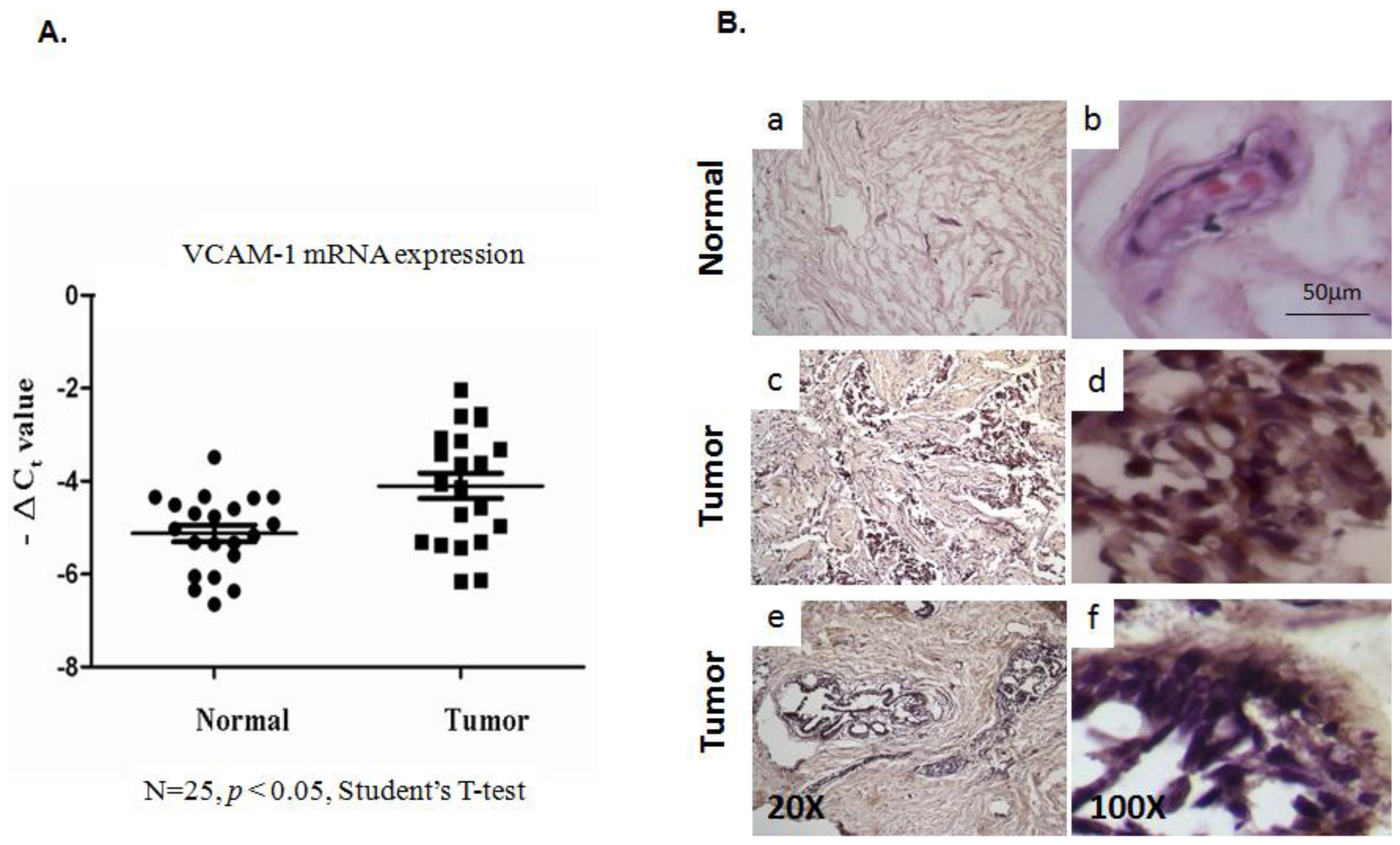
2.1.1. Evaluation of VCAM-1 Expression in Primary Human Breast Cancer
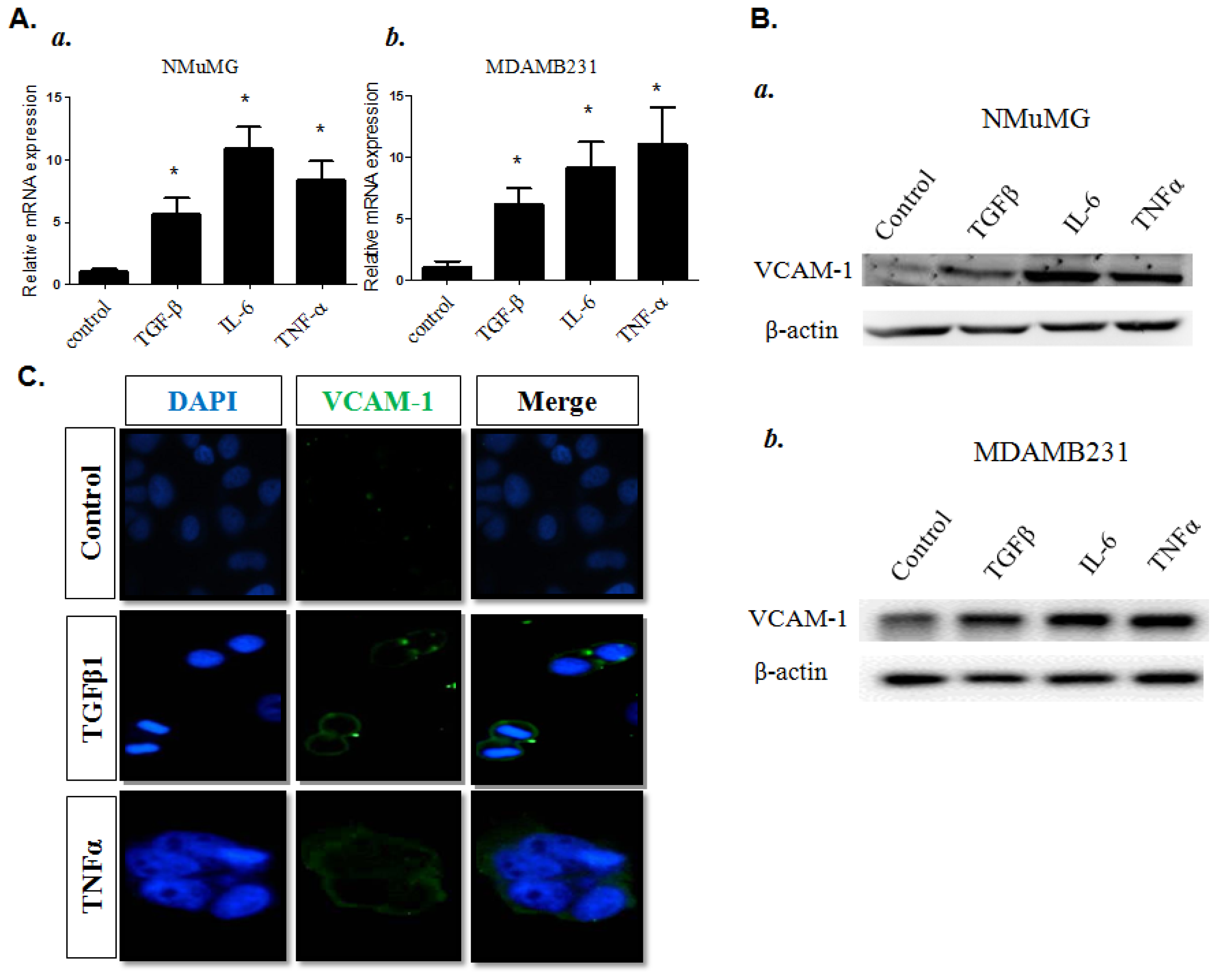
2.1.2. Proinflammatory Cytokine-Induced VCAM-1 Over-Expression in Normal and Malignant Breast Epithelial Cells
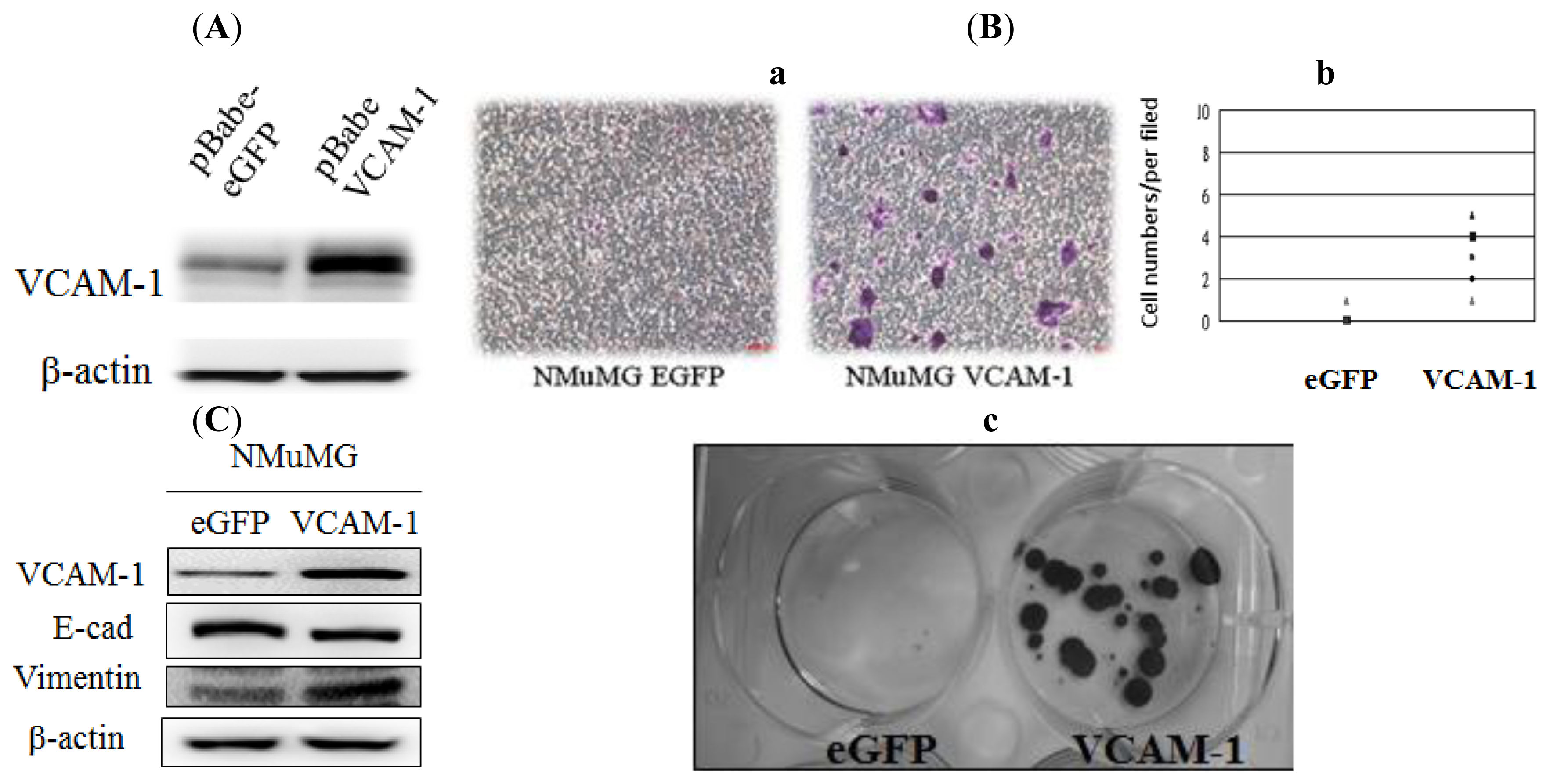
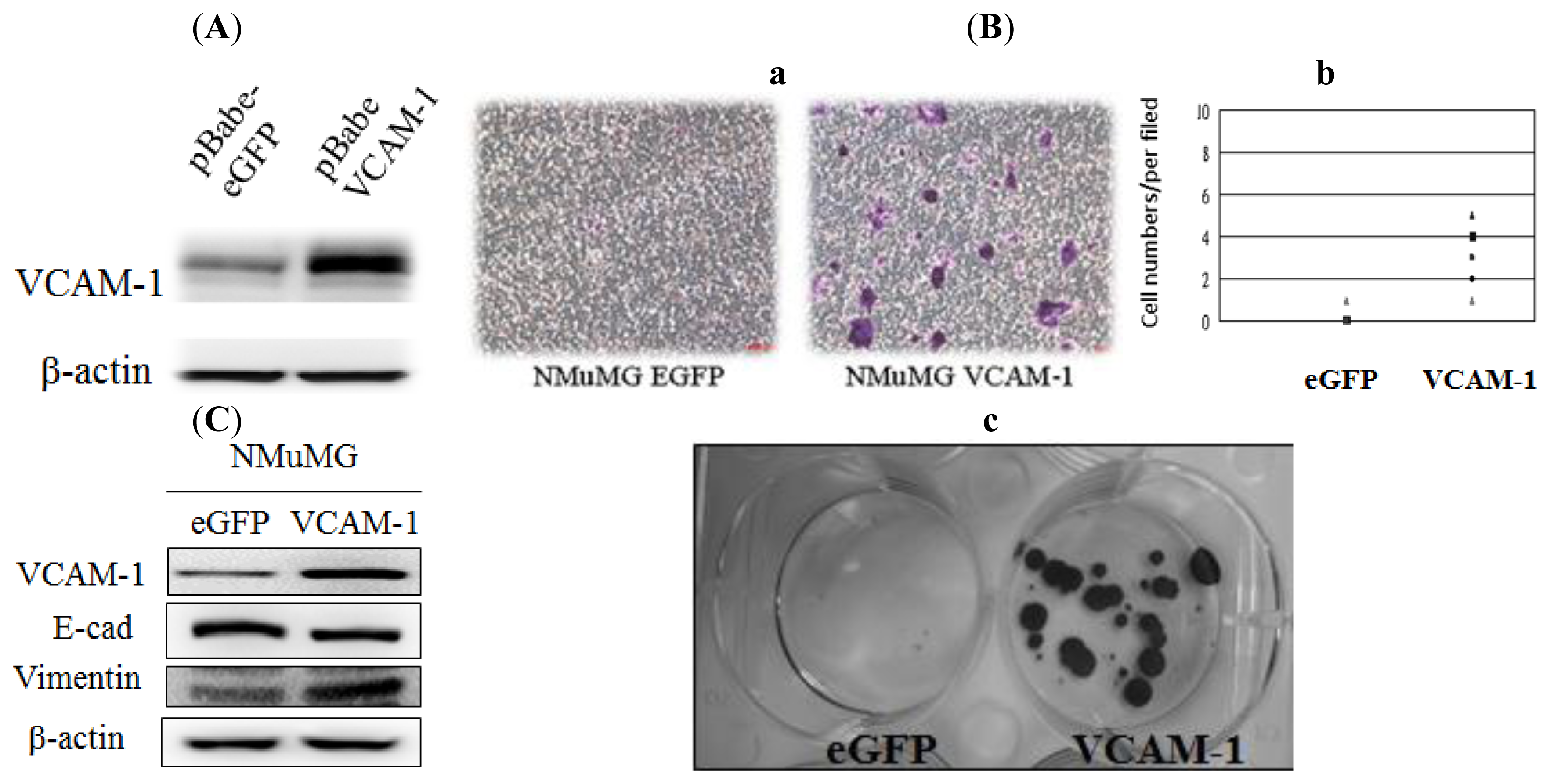
2.1.3. Effect of VCAM-1 Over-Expression on NMuMG Cell Migration
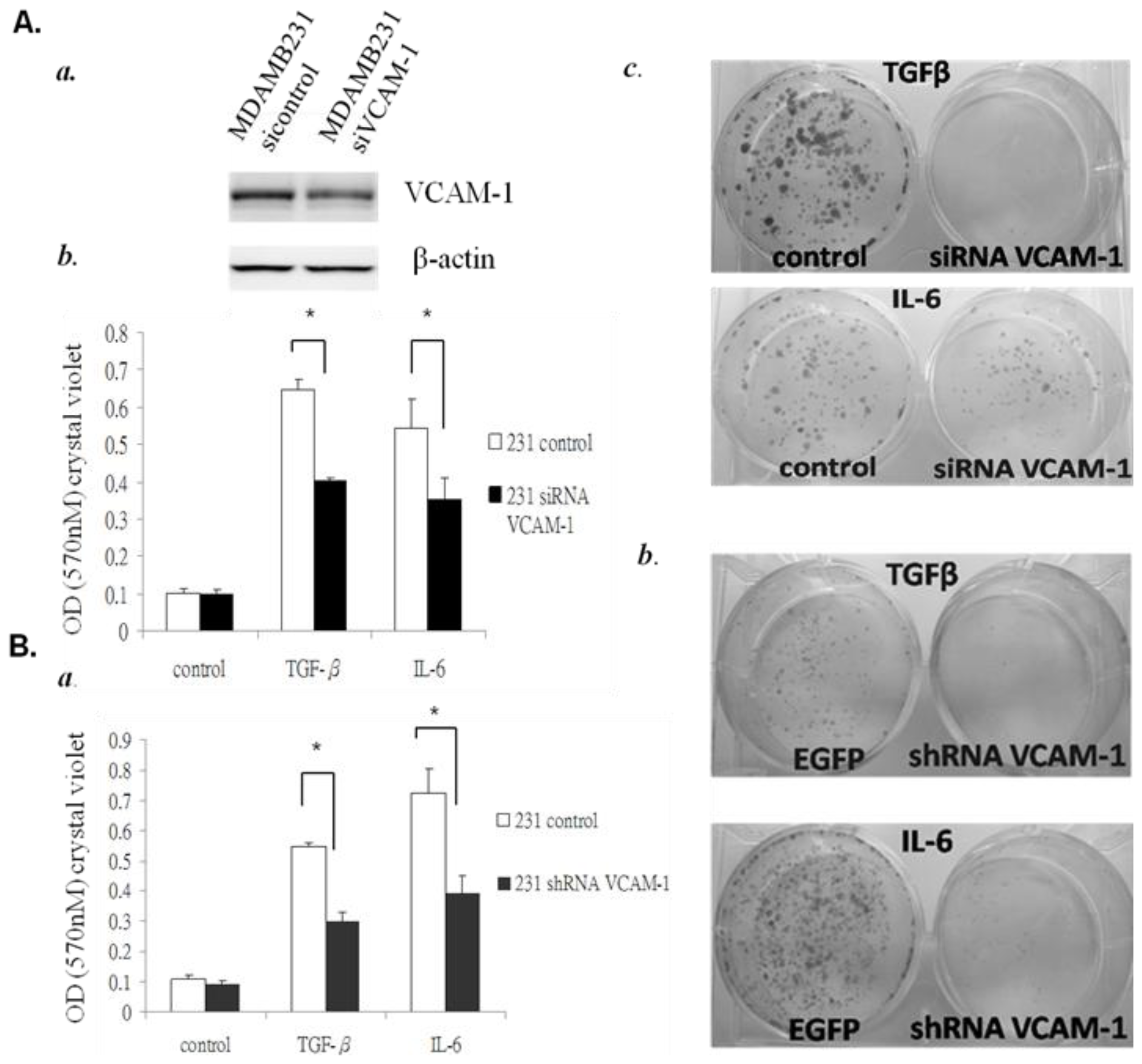
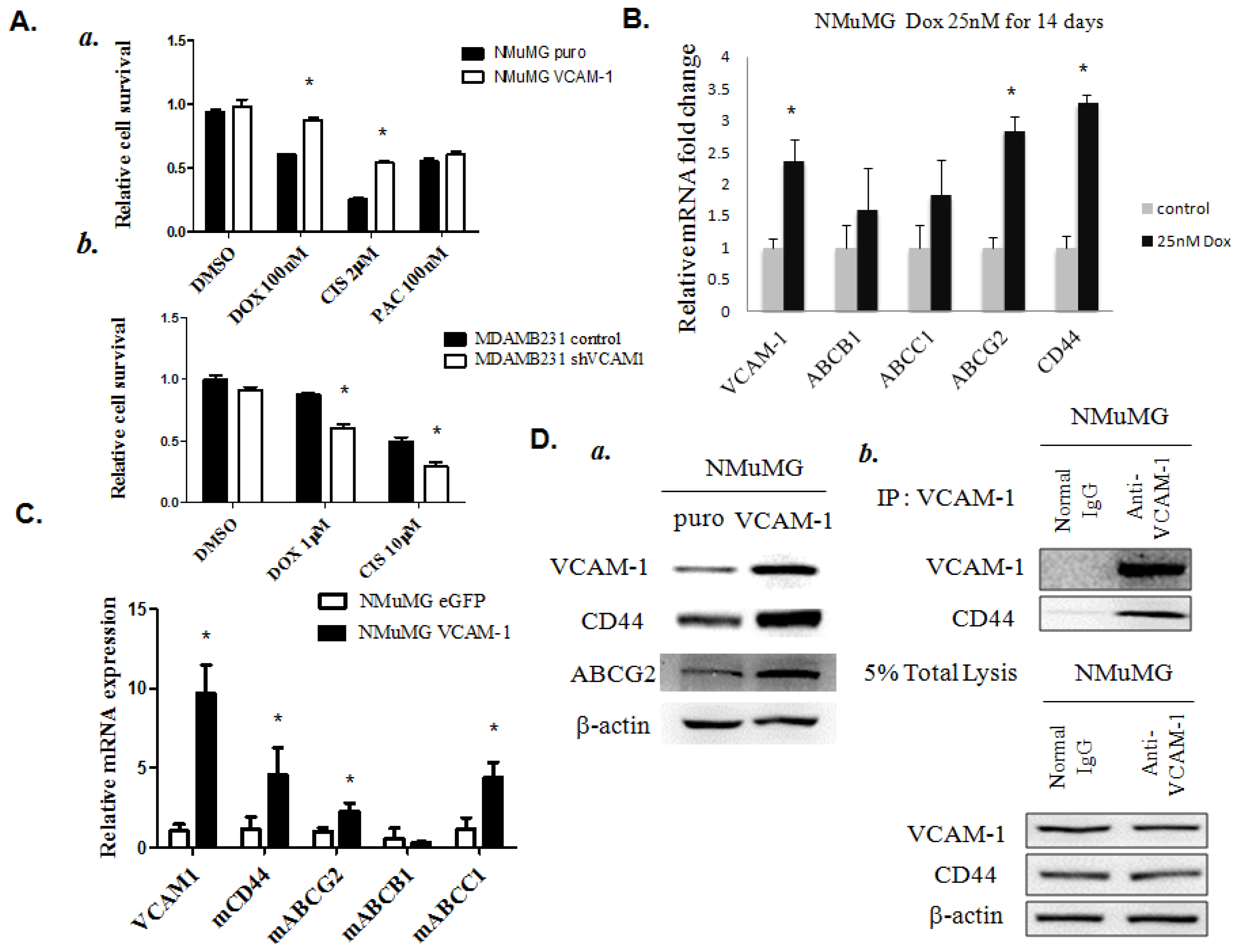
2.1.4. Down-Regulation of VCAM-1 Expression by siRNA or shRNA Inhibits Proliferation and Migration of MDAMB231 Cells
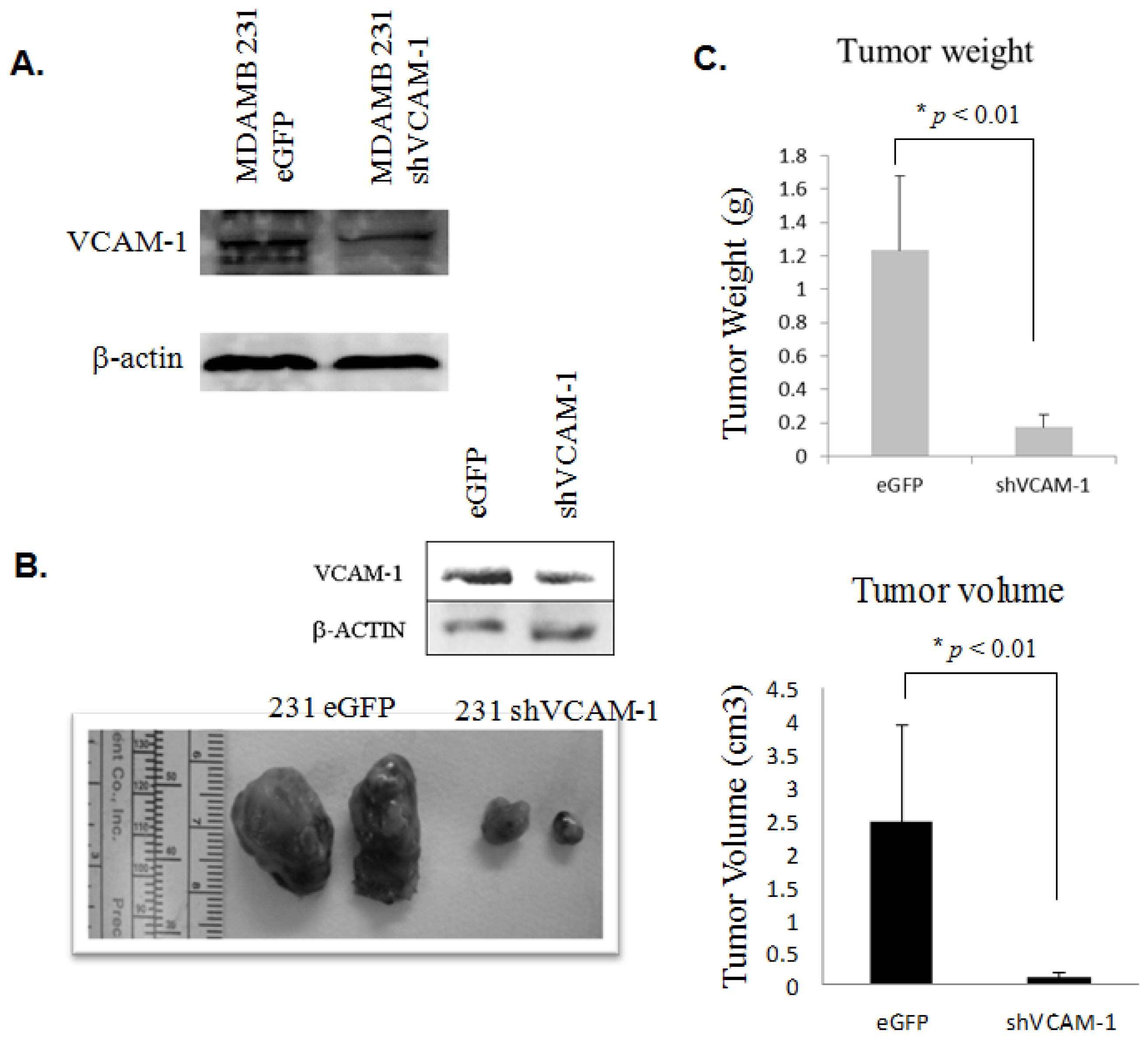
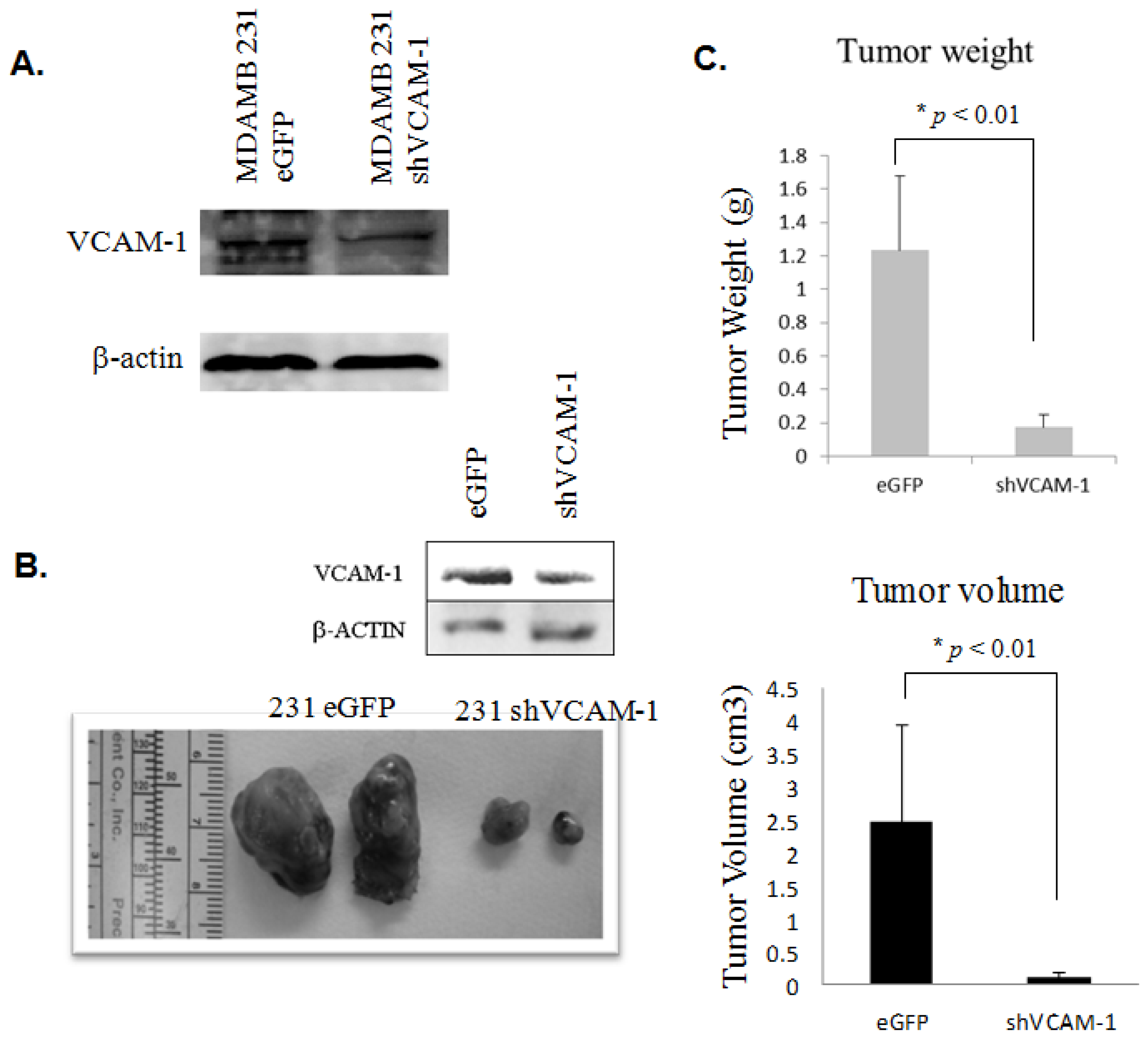
2.1.5. Knockdown of VCAM-1 Inhibits the Growth of Human MDAMB231 Breast Cancer Xenografts in SCID Mice
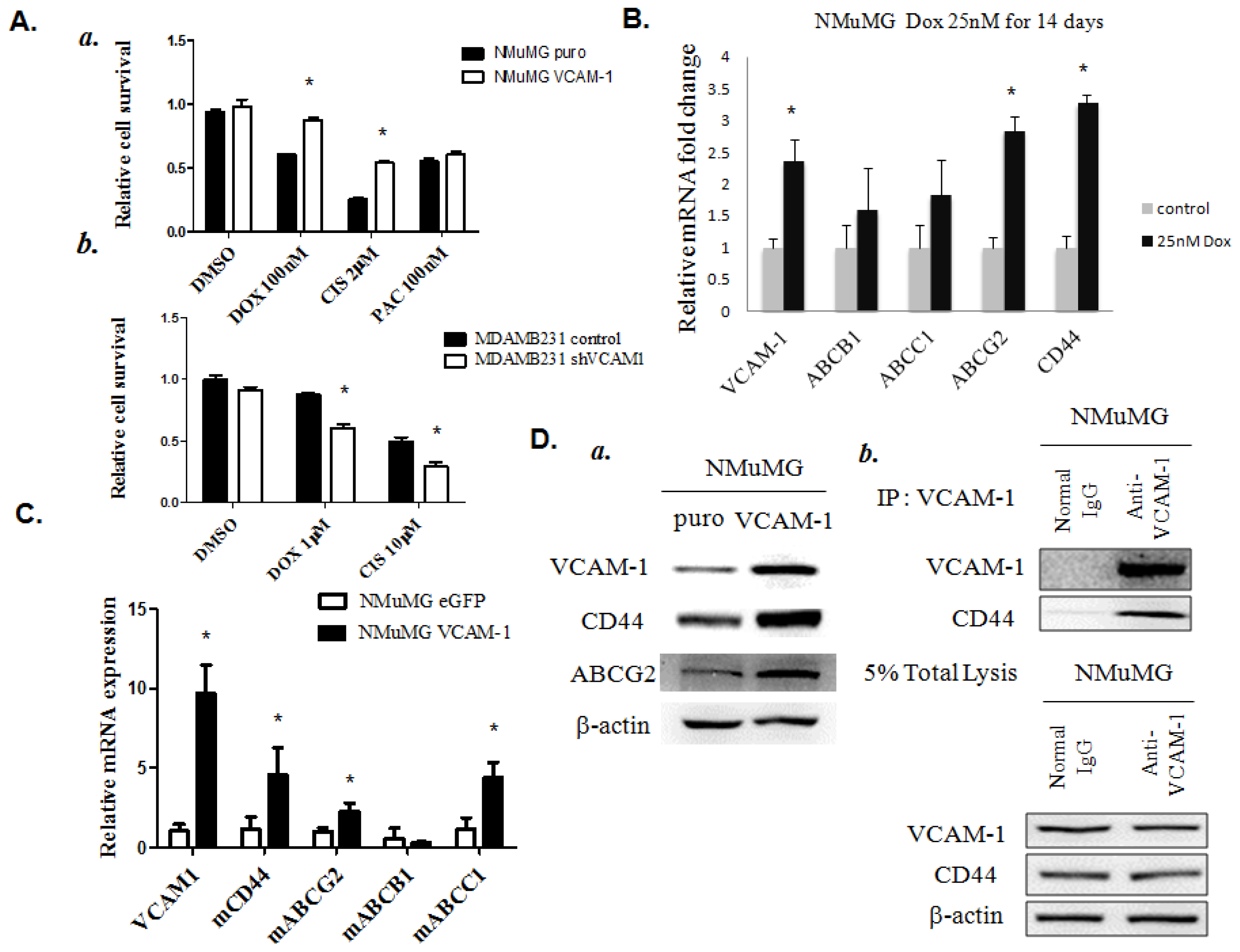
2.1.6. VCAM-1 Expression Enhances the Chemoresistant Phenotype in Breast Cancer
2.2. Discussion
3. Experimental Section
3.1. Cell Culture, Tumor Tissues, Chemo Drugs Treatment, RNA Isolation and cDNA Synthesis
3.2. Plasmid Construction
3.4. Oligonucleotide Transfection
3.5. Lentivirus Production and Shrna for Gene Knockdown
3.6. Western Blot and Immunofluorescence
3.7. Quantitative RT-PCR Analysis
3.8. Transient Transfections and Luciferase Reporter Assays
3.9. Cell Proliferation Assay
3.10. Colony Formation Assay
3.11. In Vitro Cell Migration/Invasion Assay
3.12. Immunoprecipitation (IP)
3.13. Mice and Injection
3.14. Mice Surgery, Necropsy, Histopathology and Immunohistochemistry
3.15. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-15-03560-s001.pdfAcknowledgments
- Authors ContributionsConception and Experimental design: M.F.H. and K.H.C. Acquisition of data: P.C.W, C.C.W., Y.S.H., S.F.J. and K.T.F. Analysis and interpretation of data: P.C.W., C.C.W. Writing the manuscript: M.F.H, K.T.F. and K.H.C. Material support: M.F.H. and K.T.F. Study supervision: M.F.H. and K.H.C.
Conflicts of Interest
References
- Hartge, P. Abortion breast cancer and epidemiology. N. Engl. J. Med. 1997, 336, 127–128. [Google Scholar]
- Mettlin, C.J.; Menck, H.R.; Winchester, D.P.; Murphy, G.P. A comparison of breast colorectal lung and prostate cancers reported to the National Cancer Data Base and the Surveillance Epidemiology and End Results Program. Cancer 1997, 79, 2052–2061. [Google Scholar]
- Bostner, J.; Ahnstrom-Waltersson, M.; Fornander, T.; Skoog, L.; Nordenskjold, B.; Stal, O. Amplification of CCND1 and PAK1 as predictors of recurrence and tamoxifen resistance in postmenopausal breast cancer. Oncogene 2007, 26, 6997–7005. [Google Scholar]
- Hui, R.; Campbell, D.H.; Lee, C.S.; McCaul, K.; Horsfall, D.J.; Musgrove, E.A.; Daly, R.J.; Seshadri, R.; Sutherland, R.L. EMS1 amplification can occur independently of CCND1 or INT-2 amplification at 11q13 and may identify different phenotypes in primary breast cancer. Oncogene 1997, 15, 1617–1623. [Google Scholar]
- Mukherjee, S.; Conrad, S.E. c-Myc suppresses p21WAF1/CIP1 expression during estrogen signaling and antiestrogen resistance in human breast cancer cells. J. Biol. Chem. 2005, 280, 17617–17625. [Google Scholar]
- Fujita, T.; Liu, W.; Doihara, H.; Wan, Y. An in vivo study of Cdh1/APC in breast cancer formation. Int. J. Cancer 2009, 125, 826–836. [Google Scholar]
- Lei, H.; Sjoberg-Margolin, S.; Salahshor, S.; Werelius, B.; Jandakova, E.; Hemminki, K.; Lindblom, A.; Vorechovsky, I. CDH1 mutations are present in both ductal and lobular breast cancer but promoter allelic variants show no detectable breast cancer risk. Int. J. Cancer 2002, 98, 199–204. [Google Scholar]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar]
- Inokuchi, S.; Aoyama, T.; Miura, K.; Osterreicher, C.H.; Kodama, Y.; Miyai, K.; Akira, S.; Brenner, D.A.; Seki, E. Disruption of TAK1 in hepatocytes causes hepatic injury inflammation fibrosis and carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 107, 844–849. [Google Scholar]
- Mariani, F.; Sena, P.; Marzona, L.; Riccio, M.; Fano, R.; Manni, P.; Gregorio, C.D.; Pezzi, A.; Leon, M.P.; Monni, S.; et al. Cyclooxygenase-2 and hypoxia-inducible factor-1α protein expression is related to inflammation and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett. 2009, 279, 221–229. [Google Scholar]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar]
- Trapani, J.A. The dual adverse effects of TGF-β secretion on tumor progression. Cancer Cell 2005, 8, 349–350. [Google Scholar]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar]
- Bogiatzi, S.I.; Fernandez, I.; Bichet, J.C.; Marloie-Provost, M.A.; Volpe, E.; Sastre, X.; Soumelis, V. Cutting Edge: Proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. J. Immunol. 2007, 178, 3373–3377. [Google Scholar]
- Romieu-Mourez, R.; Francois, M.; Boivin, M.N.; Bouchentouf, M.; Spaner, D.E.; Galipeau, J. Cytokine modulation of TLR expression and activation in mesenchymal stromal cells leads to a proinflammatory phenotype. J. Immunol. 2009, 182, 7963–7973. [Google Scholar]
- Yamaji-Kegan, K.; Su, Q.; Angelini, D.J.; Champion, H.C.; Johns, R.A. Hypoxia-induced mitogenic factor has proangiogenic and proinflammatory effects in the lung via VEGF and VEGF receptor-2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1159–L1168. [Google Scholar]
- Lee, C.G.; Link, H.; Baluk, P.; Homer, R.J.; Chapoval, S.; Bhandari, V.; Kang, M.J.; Cohn, L.; Kim, Y.K.; McDonald, D.M.; et al. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat. Med. 2004, 10, 1095–1103. [Google Scholar]
- Hagos, G.K.; Carroll, R.E.; Kouznetsova, T.; Li, Q.; Toader, V.; Fernandez, P.A.; Swanson, S.M.; Thatcher, G.R. Colon cancer chemoprevention by a novel NO chimera that shows anti-inflammatory and antiproliferative activity in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 2230–2239. [Google Scholar]
- Surh, Y.J.; Na, H.K. NF-kappaB and Nrf2 as prime molecular targets for chemoprevention and cytoprotection with anti-inflammatory and antioxidant phytochemicals. Genes Nutr. 2008, 2, 313–317. [Google Scholar]
- Polte, T.; Newman, W.; Raghunathan, G.; Gopal, T.V. Structural and functional studies of full-length vascular cell adhesion molecule-1: Internal duplication and homology to several adhesion proteins. DNA Cell Biol. 1991, 10, 349–357. [Google Scholar]
- Williams, A.J.; Atkins, R.C.; Fries, J.W.; Gimbrone, M.A., Jr; Cybulsky, M.I.; Collins, T. Nucleotide sequence of rat vascular cell adhesion molecule-1 cDNA. Biochim. Biophys. Acta 1992, 1131, 214–216. [Google Scholar]
- Woodside, D.G.; Kram, R.M.; Mitchell, J.S.; Belsom, T.; Billard, M.J.; McIntyre, B.W.; Vanderslice, P. Contrasting roles for domain 4 of VCAM-1 in the regulation of cell adhesion and soluble VCAM-1 binding to integrin α4β1. J. Immunol. 2006, 176, 5041–5049. [Google Scholar]
- Feuerbach, D.; Feyen, J.H. Expression of the cell-adhesion molecule VCAM-1 by stromal cells is necessary for osteoclastogenesis. FEBS Lett. 1997, 402, 21–24. [Google Scholar]
- Stanley, A.C.; Dalton, J.E.; Rossotti, S.H.; MacDonald, K.P.; Zhou, Y.; Rivera, F.; Schroder, W.A.; Maroof, A.; Hill, G.R.; Kaye, P.M.; et al. VCAM-1 and VLA-4 modulate dendritic cell IL-12p40 production in experimental visceral leishmaniasis. PLoS Pathog. 2008, 4, e1000158. [Google Scholar]
- Yakubenko, V.P.; Lobb, R.R.; Plow, E.F.; Ugarova, T.P. Differential induction of gelatinase B (MMP-9) and gelatinase A (MMP-2) in T lymphocytes upon α4β1-mediated adhesion to VCAM-1 and the CS-1 peptide of fibronectin. Exp. Cell Res. 2000, 260, 73–84. [Google Scholar]
- Van Dinther-Janssen, A.C.; Horst, E.; Koopman, G.; Newmann, W.; Scheper, R.J.; Meijer, C.J.; Pals, S.T. The VLA-4/VCAM-1 pathway is involved in lymphocyte adhesion to endothelium in rheumatoid synovium. J. Immunol. 1991, 147, 4207–4210. [Google Scholar]
- Devine, L.; Lightman, S.L.; Greenwood, J. Role of LFA-1 ICAM-1 VLA-4 and VCAM-1 in lymphocyte migration across retinal pigment epithelial monolayers in vitro. Immunology 1996, 88, 456–462. [Google Scholar]
- Panettieri, R.A., Jr; Lazaar, A.L.; Pure, E.; Albelda, S.M. Activation of cAMP-dependent pathways in human airway smooth muscle cells inhibits TNF-α-induced ICAM-1 and VCAM-1 expression and T lymphocyte adhesion. J. Immunol. 1995, 154, 2358–2365. [Google Scholar]
- Gamble, J.R.; Bradley, S.; Noack, L.; Vadas, M.A. TGF-β and endothelial cells inhibit VCAM-1 expression on human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 949–955. [Google Scholar]
- Fukushi, J.; Ono, M.; Morikawa, W.; Iwamoto, Y.; Kuwano, M. The activity of soluble VCAM-1 in angiogenesis stimulated by IL-4 and IL-13. J. Immunol. 2000, 165, 2818–2823. [Google Scholar]
- Griffioen, A.W.; Damen, C.A.; Blijham, G.H.; Groenewegen, G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood 1996, 88, 667–673. [Google Scholar]
- Lee, Y.W.; Kuhn, H.; Hennig, B.; Neish, A.S.; Toborek, M. IL-4-induced oxidative stress upregulates VCAM-1 gene expression in human endothelial cells. J. Mol. Cell. Cardiol. 2001, 33, 83–94. [Google Scholar]
- Jeong, H.J.; Lee, H.H.; Kim, Y.S.; Kim, S.I.; Moon, J.I.; Park, K. Expression of ICAM-1 and VCAM-1 in renal allograft rejection. Transplant. Proc. 1998, 30, 2953–2954. [Google Scholar]
- Hemmerlein, B.; Scherbening, J.; Kugler, A.; Radzun, H.J. Expression of VCAM-1 ICAM-1 E- and P-selectin and tumour-associated macrophages in renal cell carcinoma. Histopathology 2000, 37, 78–83. [Google Scholar]
- Vasselli, J.R.; Shih, J.H.; Iyengar, S.R.; Maranchie, J.; Riss, J.; Worrell, R.; Torres-Cabala, C.; Tabios, R.; Mariotti, A.; Stearman, R.; et al. Predicting survival in patients with metastatic kidney cancer by gene-expression profiling in the primary tumor. Proc. Natl. Acad. Sci. USA 2003, 100, 6958–6963. [Google Scholar]
- Regidor, P.A.; Callies, R.; Regidor, M.; Schindler, A.E. Expression of the cell adhesion molecules ICAM-1 and VCAM-1 in the cytosol of breast cancer tissue benign breast tissue and corresponding sera. Eur. J. Gynaecol. Oncol. 1998, 19, 377–383. [Google Scholar]
- Sansone, P.; Bromberg, J. Environment inflammation and cancer. Curr. Opin. Genet. Dev. 2011, 21, 80–85. [Google Scholar]
- Ben-Baruch, A. Host microenvironment in breast cancer development: Inflammatory cells cytokines and chemokines in breast cancer progression: Reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003, 5, 31–36. [Google Scholar]
- Raman, D.; Baugher, P.J.; Thu, Y.M.; Richmond, A. Role of chemokines in tumor growth. Cancer Lett. 2007, 256, 137–165. [Google Scholar]
- Yurkovetsky, Z.; Skates, S.; Lomakin, A.; Nolen, B.; Pulsipher, T.; Modugno, F.; Marks, J.; Godwin, A.; Gorelik, E.; Jacobs, I.; et al. Development of a multimarker assay for early detection of ovarian cancer. J. Clin. Oncol. 2011, 28, 2159–2166. [Google Scholar]
- Slack-Davis, J.K.; Atkins, K.A.; Harrer, C.; Hershey, E.D.; Conaway, M. Vascular cell adhesion molecule-1 is a regulator of ovarian cancer peritoneal metastasis. Cancer Res. 2009, 69, 1469–1476. [Google Scholar]
- Pitteri, S.J.; JeBailey, L.; Faca, V.M.; Thorpe, J.D.; Silva, M.A.; Ireton, R.C.; Horton, M.B.; Wang, H.; Pruitt, L.C.; Zhang, Q.; et al. Integrated proteomic analysis of human cancer cells and plasma from tumor bearing mice for ovarian cancer biomarker discovery. PLoS One 2009, 4, e7916. [Google Scholar]
- Cheng, K.H.; Dinulescu, D.M. Molecular analysis of genes associated with the phenotype of stem cell-like side population in ovarian cancer cells. PLoS One 2014. to be submitted for publication. [Google Scholar]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. Oncomine: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar]
- O’Hanlon, D.M.; Fitzsimons, H.; Lynch, J.; Tormey, S.; Malone, C.; Given, H.F. Soluble adhesion molecules (E-selectin ICAM-1 and VCAM-1) in breast carcinoma. Eur. J. Cancer 2002, 38, 2252–2257. [Google Scholar]
- Smith, L.; Watson, M.B.; O’Kane, S.L.; Drew, P.J.; Lind, M.J.; Cawkwell, L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol. Cancer Ther. 2006, 5, 2115–2120. [Google Scholar]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell. Biol. 2003, 4, 33–45. [Google Scholar]
- Abraham, B.K.; Fritz, P.; McClellan, M.; Hauptvogel, P.; Athelogou, M.; Brauch, H. Prevalence of CD44+/CD24−/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin. Cancer Res. 2005, 11, 1154–1159. [Google Scholar]
- Cheng, K.H.; Ponte, J.F.; Thiagalingam, S. Elucidation of epigenetic inactivation of SMAD8 in cancer using targeted expressed gene display. Cancer Res. 2004, 64, 1639–1646. [Google Scholar]
- Papageorgis, P.; Cheng, K.; Ozturk, S.; Gong, Y.; Lambert, A.W.; Abdolmaleky, H.M.; Zhou, J.R.; Thiagalingam, S. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011, 71, 998–1008. [Google Scholar]
- Chiu, C.Y.; Kuo, K.K.; Kuo, T.L.; Lee, K.T.; Cheng, K.H. The activation of MEK/ERK signaling pathway by bone morphogenetic protein 4 to increase hepatocellular carcinoma cell proliferation and migration. Mol. Cancer Res. 2012, 10, 415–427. [Google Scholar]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar]
- Su, H.T.; Weng, C.C.; Hsiao, P.J.; Chen, L.H.; Kuo, T.L.; Chen, Y.W.; Kuo, K.K.; Cheng, K.H. Stem cell marker nestin is critical for TGF-β1-mediated tumor progression in pancreatic cancer. Mol. Cancer Res. 2013, 11, 768–779. [Google Scholar]


© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, P.-C.; Weng, C.-C.; Hou, Y.-S.; Jian, S.-F.; Fang, K.-T.; Hou, M.-F.; Cheng, K.-H. Activation of VCAM-1 and Its Associated Molecule CD44 Leads to Increased Malignant Potential of Breast Cancer Cells. Int. J. Mol. Sci. 2014, 15, 3560-3579. https://doi.org/10.3390/ijms15033560
Wang P-C, Weng C-C, Hou Y-S, Jian S-F, Fang K-T, Hou M-F, Cheng K-H. Activation of VCAM-1 and Its Associated Molecule CD44 Leads to Increased Malignant Potential of Breast Cancer Cells. International Journal of Molecular Sciences. 2014; 15(3):3560-3579. https://doi.org/10.3390/ijms15033560
Chicago/Turabian StyleWang, Pei-Chen, Ching-Chieh Weng, You-Syuan Hou, Shu-Fang Jian, Kuan-Te Fang, Ming-Feng Hou, and Kuang-Hung Cheng. 2014. "Activation of VCAM-1 and Its Associated Molecule CD44 Leads to Increased Malignant Potential of Breast Cancer Cells" International Journal of Molecular Sciences 15, no. 3: 3560-3579. https://doi.org/10.3390/ijms15033560