HBx Protein Promotes Oval Cell Proliferation by Up-Regulation of Cyclin D1 via Activation of the MEK/ERK and PI3K/Akt Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
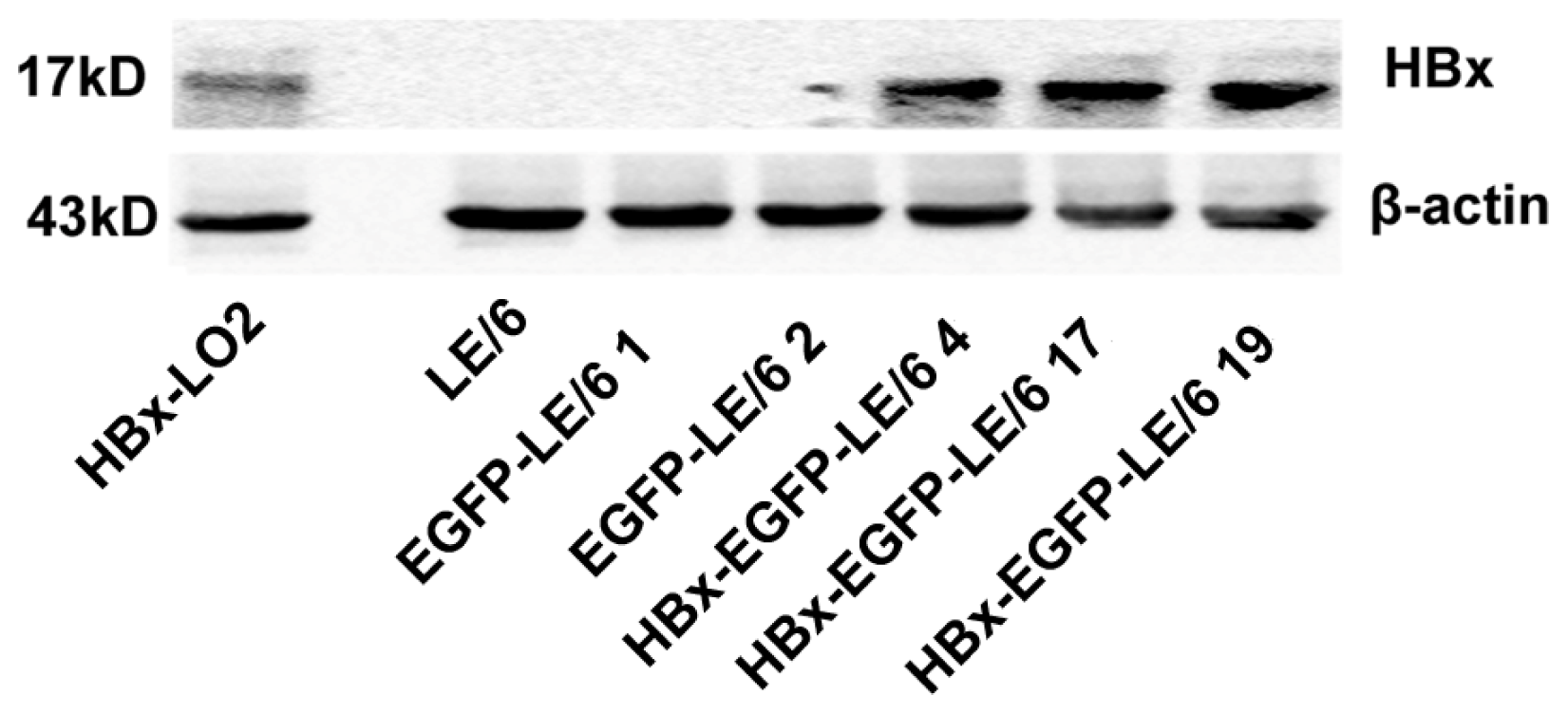
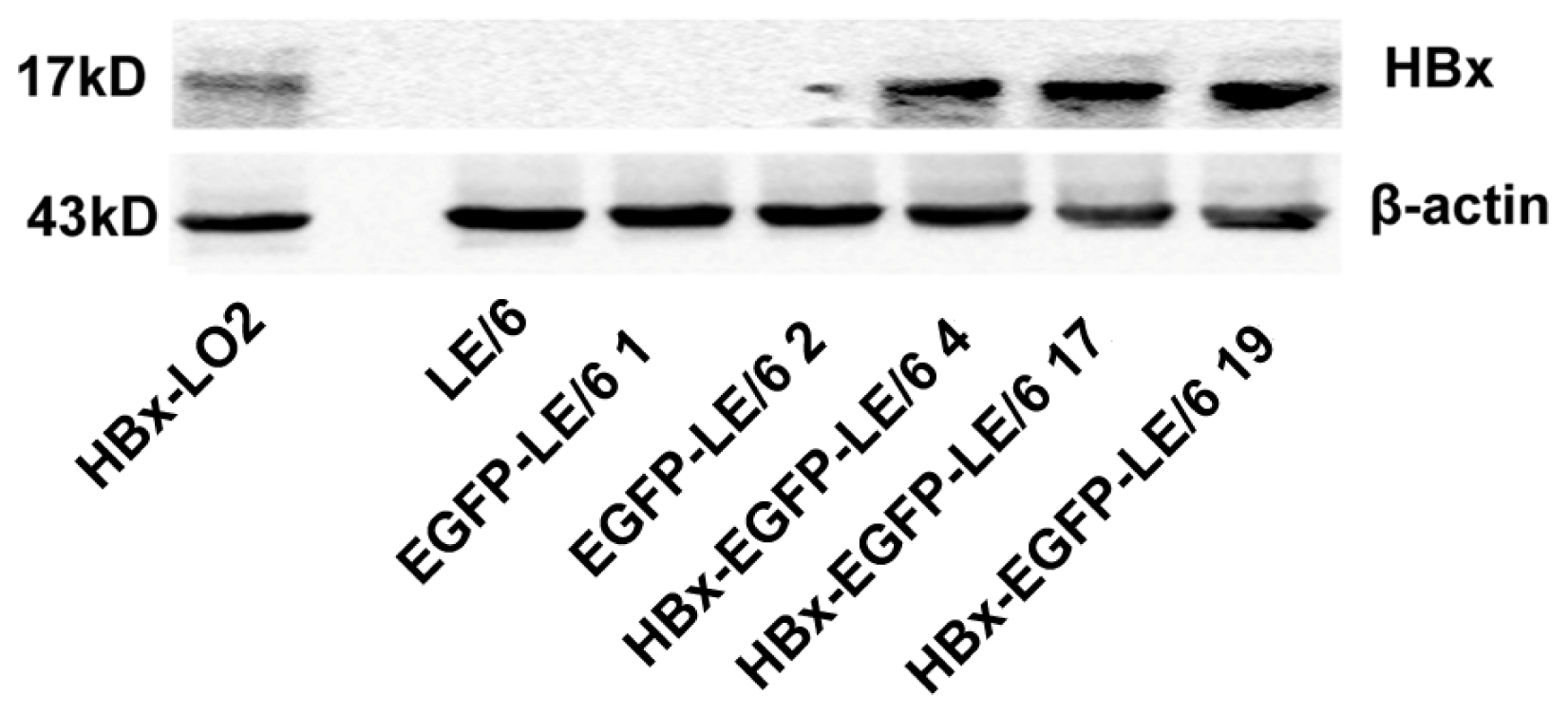
2.1. Expression of HBx in LE/6 Cells
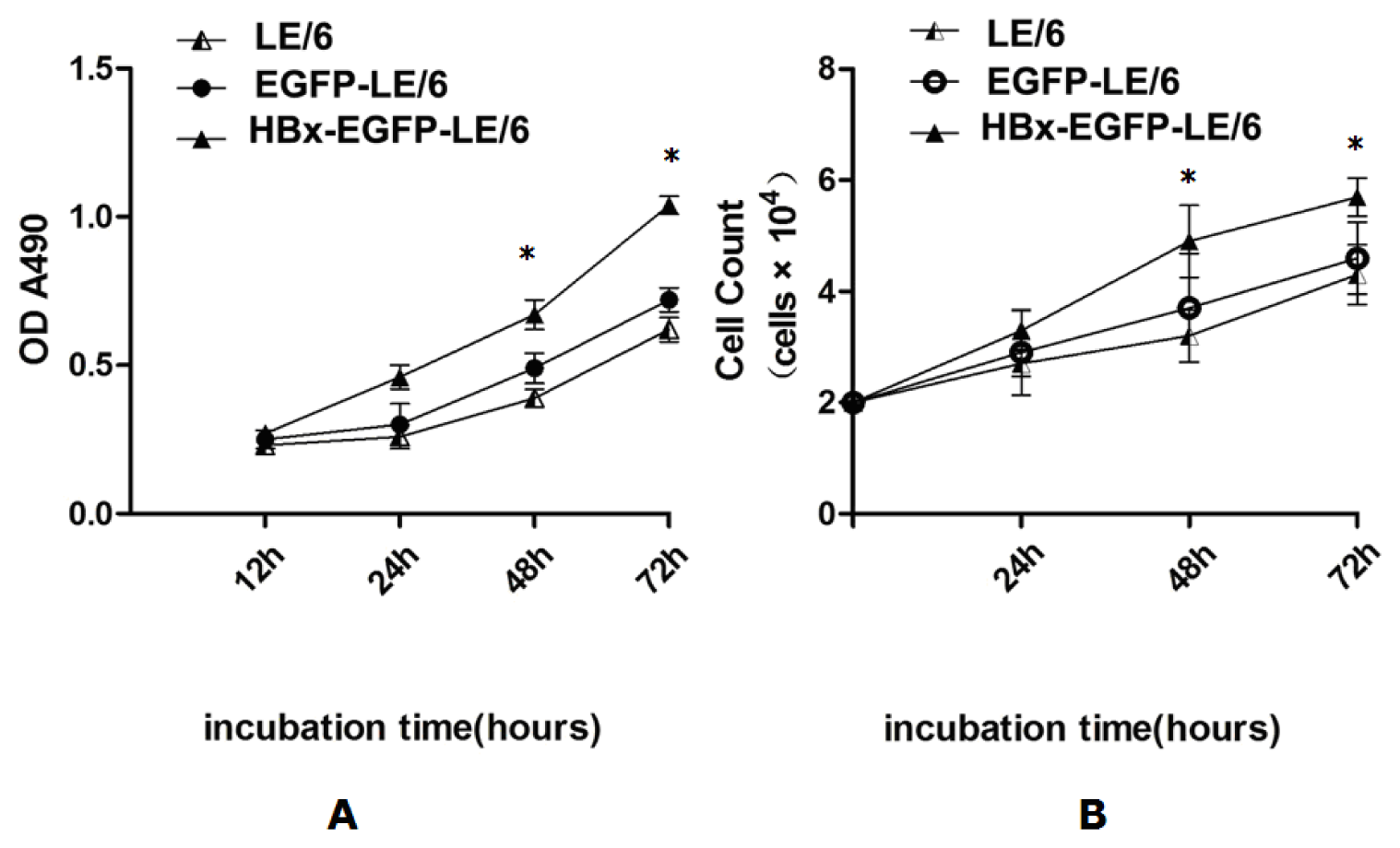
2.2. HBx Promotes Proliferation of LE/6 Cells
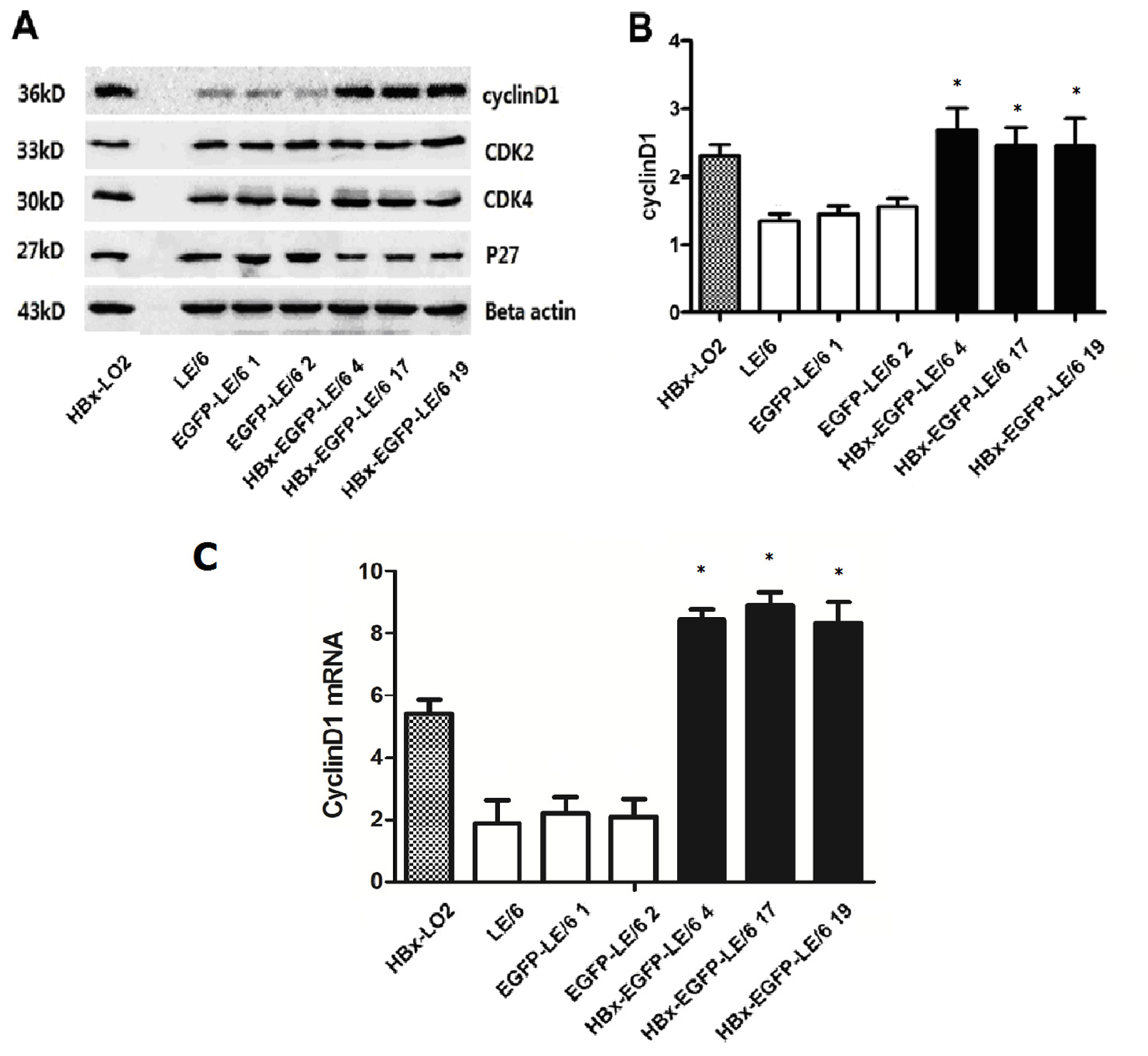
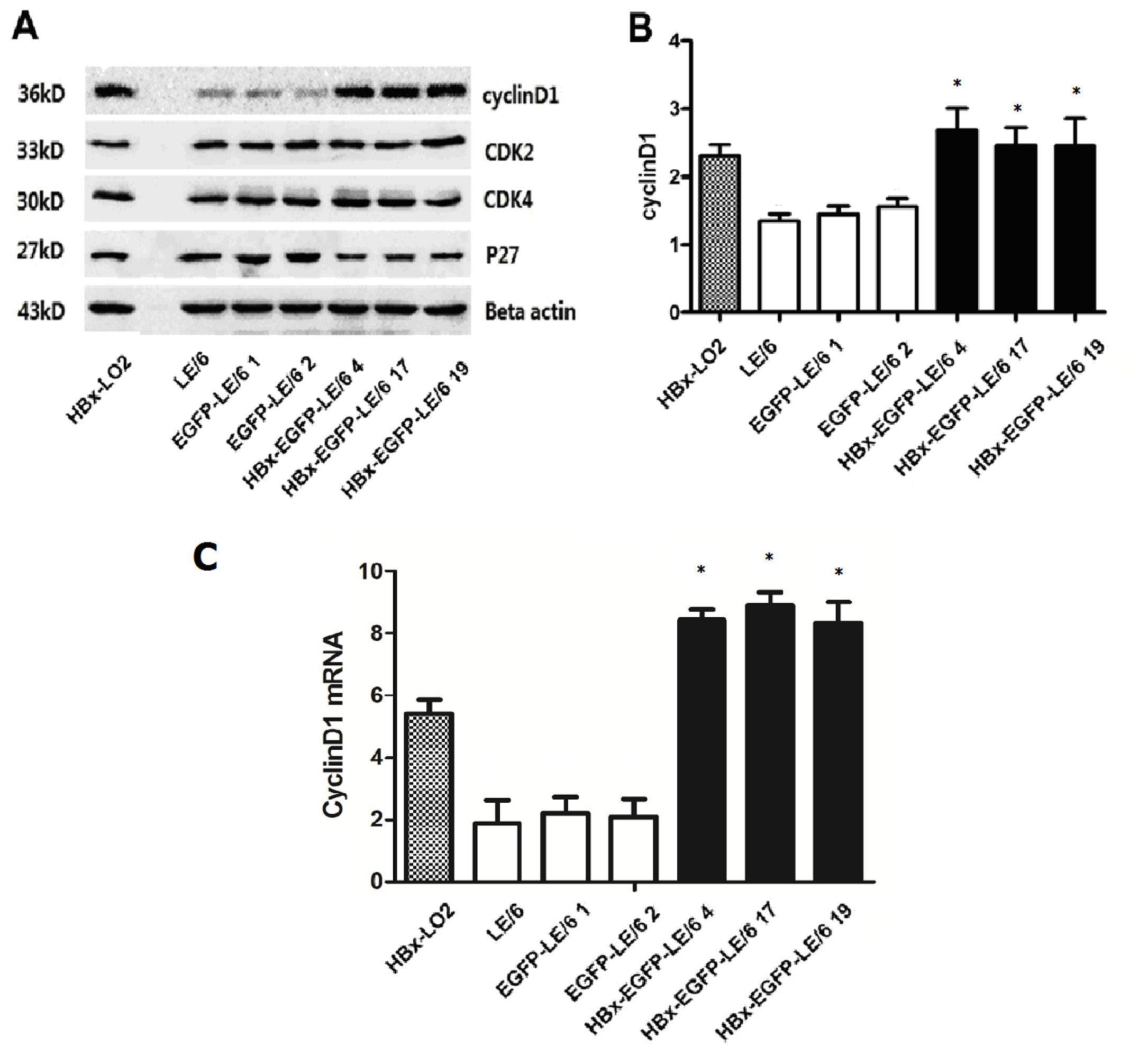
2.3. Expression of Modulators in Cell Cycle Progression in HBx Overexpressing Cells
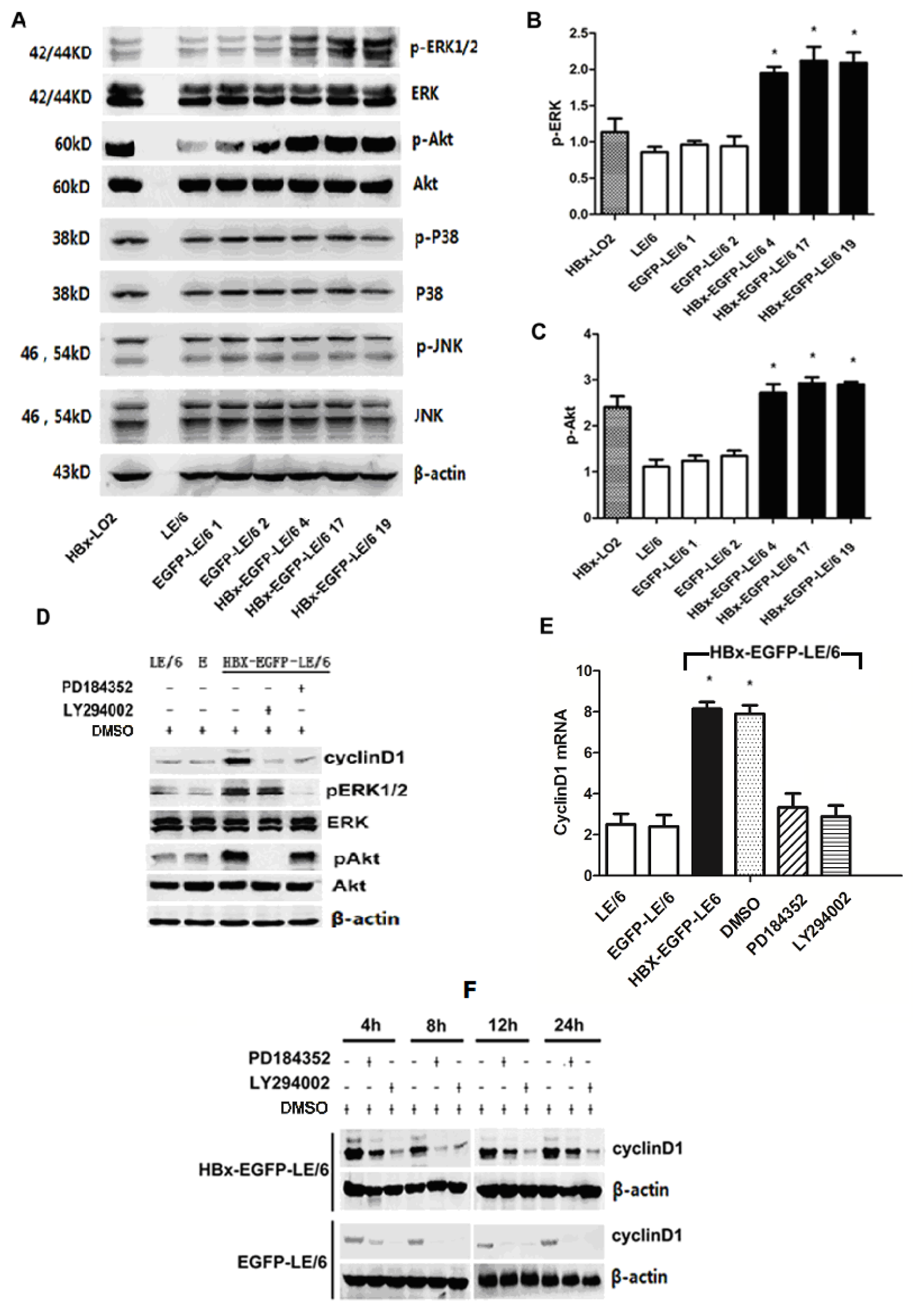
2.4. Up-Regulation of Cyclin D1 by HBx in Oval Cells Is Dependent of the Activation of MEK/ERK and PI-3K/Akt Signaling Pathways
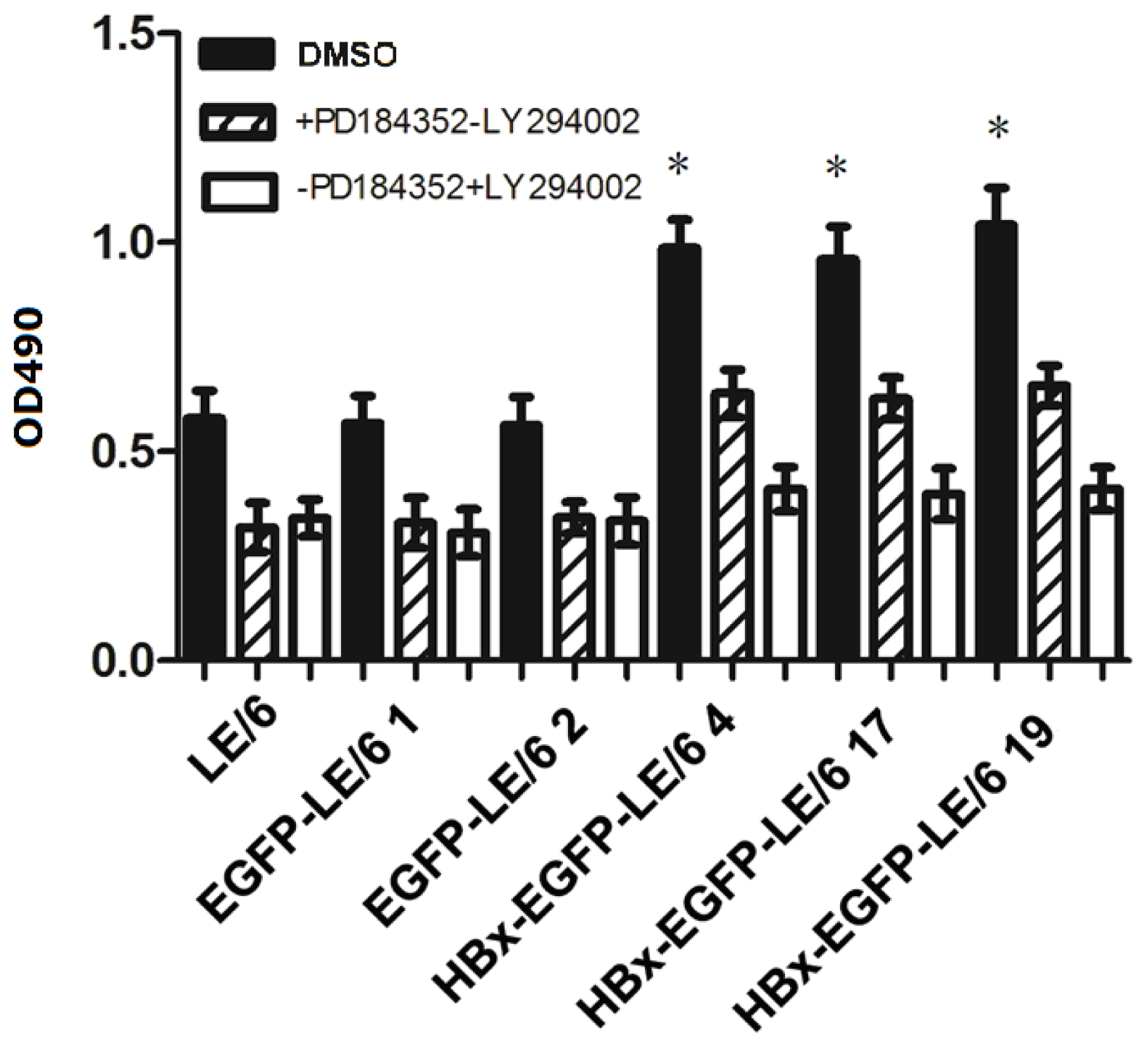
2.5. HBx Effects on Oval Cell Proliferation Are Abolished by LY294002 or PD184352
2.6. Discussion
3. Experimental Section
3.1. Cell Lines and Cell Culture
3.2. Cell Proliferation Assay
3.3. Quantitative Real-Time PCR (qPCR) Analysis
3.4. Western Blotting Analysis
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Sun, C.; Jin, X.L.; Xiao, J.C. Oval cells in hepatitis B virus-positive and hepatitis C virus-positive liver cirrhosis: Histological and ultrastructural study. Histopathology 2006, 48, 546–555. [Google Scholar]
- Conigliaro, A.; Brenner, D.A.; Kisseleva, T. Hepatic progenitors for liver disease: Current position. Stem Cells Cloning 2010, 3, 39–47. [Google Scholar]
- Eleazar, J.A.; Memeo, L.; Jhang, J.S.; Mansukhani, M.M.; Chin, S.; Park, S.M.; Lefkowitch, J.H.; Bhagat, G. Progenitor cell expansion: An important source of hepatocyte regeneration in chronic hepatitis. J. Hepatol. 2004, 41, 983–991. [Google Scholar]
- Fotiadu, A.; Tzioufa, V.; Vrettou, E.; Koufogiannis, D.; Papadimitriou, C.S.; Hytiroglou, P. Progenitor cell activation in chronic viralhepatitis. Liver Int. 2004, 24, 268–274. [Google Scholar]
- Lowes, K.N.; Brennan, B.A.; Yeoh, G.C.; Olynyk, J.K. Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am. J. Pathol. 1999, 154, 537–541. [Google Scholar]
- Knight, B.; Yeoh, G.C.; Husk, K.L.; Ly, T.; Abraham, L.J.; Yu, C.; Rhim, J.A.; Fausto, N. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J. Exp. Med. 2000, 192, 1809–1818. [Google Scholar]
- Lowes, K.N.; Croager, E.J.; Olynyk, J.K.; Abraham, L.J.; Yeoh, G.C. Oval cell-mediated liver regeneration: Role of cytokines and growth factors. J. Gastroenterol. Hepatol. 2003, 18, 4–12. [Google Scholar]
- Knight, B.; Matthews, V.B.; Akhurst, B.; Croager, E.J.; Klinken, E.; Abraham, L.J.; Olynyk, J.K.; Yeoh, G. Liver inflammation and cytokine production but not acute phase protein synthesis accompany the adult liver progenitor (oval) cell response to chronic liver injury. Immunol. Cell Biol. 2005, 83, 364–374. [Google Scholar]
- Brooling, J.T.; Campbell, J.S.; Mitchell, C.; Yeoh, G.C.; Fausto, N. Differential regulation of rodent hepatocyte and oval cell proliferation by interferon gamma. Hepatology 2005, 41, 906–915. [Google Scholar]
- Nguyen, L.N.; Furuya, M.H.; Wolfraim, L.A.; Nguyen, A.P.; Holdren, M.S.; Campbell, J.S.; Knight, B.; Yeoh, G.C.; Fausto, N.; Parks, W.T. Transforming growth factor-beta differentially regulates oval cell and hepatocyte proliferation. Hepatology 2007, 45, 31–41. [Google Scholar]
- Hwang, G.Y.; Lin, C.Y.; Huang, L.M.; Wang, Y.H.; Wang, J.C.; Hsu, C.T.; Yang, S.S.; Wu, C.C. Detection of the hepatitis B virus X protein (HBx) antigen and anti-HBx antibodies in cases of human hepatocellular carcinoma. J. Clin. Microbiol. 2003, 41, 5598–5603. [Google Scholar]
- Cheng, A.S.; Chan, H.L.; Leung, W.K.; To, K.F.; Go, M.Y.; Chan, J.Y.; Liew, C.T.; Sung, J.J. Expression of HBx and COX-2 in chronic hepatitis B cirrhosis and hepatocellular carcinoma: Implication of HBx in upregulation of COX-2. Modern Pathol. 2004, 17, 1169–1179. [Google Scholar]
- Zhou, Y.M.; Cao, L.; Li, B.; Zhang, X.Z.; Yin, Z.F. Expression of HBx protein in hepatitis B virus-infected intrahepatic cholangiocarcinoma. Hepatobiliary Pancreat. Dis. Int. 2012, 11, 532–535. [Google Scholar]
- Hsia, C.C.; Thorgeirsson, S.S.; Tabor, E. Expression of hepatitis B surface and core antigens and transforming growth factor-alpha in “oval cells” of the liver in patients with hepatocellular carcinoma. J. Med. Virol. 1994, 43, 216–221. [Google Scholar]
- Watanabe, M.; Umenai, T.; Ohori, H.; Ishida, N. Evidence for the multiplication of hepatitis B virus in “oval cell” culture originated from human embryonic liver. Br. J. Exp. Pathol. 1976, 57, 211–216. [Google Scholar]
- Chung, T.W.; Lee, Y.C.; Kim, C.H. Hepatitis B viral HBx induces matrix metalloproteinase-9 gene expression through activation of ERK and PI-3K/AKT pathways: Involvement of invasive potential. FASEB J. 2004, 18, 1123–1125. [Google Scholar]
- Tang, R.X.; Kong, F.Y.; Fan, B.F.; Liu, X.M.; You, H.J.; Zhang, P.; Zheng, K.Y. HBx activates FasL and mediates HepG2 cell apoptosis through MLK3-MKK7-JNKs signal module. World J. Gastroenterol. 2012, 18, 1485–1495. [Google Scholar]
- Park, Y.H.; Shin, H.J.; Kim, S.U.; Kim, J.M.; Kim, J.H.; Bang, D.H.; Chang, K.T.; Kim, B.Y.; Yu, D.Y. iNOS promotes HBx-induced hepatocellular carcinoma via upregulation of JNK activation. Biochem. Biophys. Res. Commun. 2013, 435, 244–249. [Google Scholar]
- Sze, K.M.; Chu, G.K.; Lee, J.M.; Ng, I.O. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar]
- Neuzillet, C.; Hammel, P.; Tijeras-Raballand, A.; Couvelard, A.; Raymond, E. Targeting the Ras-ERK pathway in pancreatic adenocarcinoma. Cancer Metastasis Rev. 2013, 32, 147–162. [Google Scholar]
- Dent, P. Crosstalk between ERK AKT and cell survival. Cancer Biol. Ther. 2014, 15. [Google Scholar] [CrossRef]
- Pal, I.; Mandal, M. PI3K and Akt as molecular targets for cancer therapy: Current clinical outcomes. Acta Pharmacol. Sin. 2012, 33, 1441–1458. [Google Scholar]
- Li, C.H.; Wang, Y.J.; Dong, W.; Xiang, S.; Liang, H.F.; Wang, H.Y.; Dong, H.H.; Chen, L.; Chen, X.P. Hepatic oval cell lines generate hepatocellular carcinoma following transfection with HBx gene and treatment with aflatoxin B1 in vivo. Cancer Lett. 2011, 311, 1–10. [Google Scholar]
- Shan, C.; Xu, F.; Zhang, S.; You, J.; You, X.; Qiu, L.; Zheng, J.; Ye, L.; Zhang, X. Hepatitis B virus X protein promotes liver cell proliferation via a positive cascade loop involving arachidonic acid metabolism and p-ERK1/2. Cell Res. 2010, 20, 563–575. [Google Scholar]
- Qiao, L.; Leach, K.; McKinstry, R.; Gilfor, D.; Yacoub, A.; Park, J.S.; Grant, S.; Hylemon, P.B.; Fisher, P.B.; Dent, P. Hepatitis B virus X protein increases expression of p21(Cip-1/WAF1/MDA6) and p27(Kip-1) in primary mouse hepatocytes leading to reduced cell cycle progression. Hepatology 2001, 34, 906–917. [Google Scholar]
- Walker, J.L.; Assoian, R.K. Integrin-dependent signal transduction regulating cyclin D1 expression and G1 phase cell cycle progression. Cancer Metastasis Rev. 2005, 24, 383–393. [Google Scholar]
- Tashiro, E.; Tsuchiya, A.; Imoto, M. Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci. 2007, 98, 629–635. [Google Scholar]
- Jun, E.K.; Zhang, Q.; Yoon, B.S.; Moon, J.H.; Lee, G.; Park, G.; Kang, P.J.; Lee, J.H.; Kim, A.; You, S. Hypoxic conditioned medium from human amniotic fluid-derived mesenchymal stem cells accelerates skin wound healing through TGF-beta/SMAD2 and PI3K/Akt pathways. Int. J. Mol. Sci. 2013, 15, 605–628. [Google Scholar]
- Yokota, J.; Chosa, N.; Sawada, S.; Okubo, N.; Takahashi, N.; Hasegawa, T.; Kondo, H.; Ishisaki, A. PDGF-induced PI3K-mediated signaling enhances the TGFbetainduced osteogenic differentiation of human mesenchymal stem cells in a TGF-beta-activated MEK-dependent manner. Int. J. Mol. Med. 2014, 33, 534–542. [Google Scholar]
- Jo, C.; Koh, Y.H. Cadmium induces N-cadherin cleavage via ERK-mediated gamma-secretase activation in C6 astroglia cells. Toxicol. Lett. 2013, 222, 117–121. [Google Scholar]
- Sakamoto, T.; Ozaki, K.; Fujio, K.; Kajikawa, S.H.; Uesato, S.; Watanabe, K.; Tanimura, S.; Koji, T.; Kohno, M. Blockade of the ERK pathway enhances the therapeutic efficacy of the histone deacetylase inhibitor MS-275 in human tumor xenograft models. Biochem. Biophys. Res. Commun. 2013, 433, 456–462. [Google Scholar]
- Park, S.G.; Chung, C.; Kang, H.; Kim, J.Y.; Jung, G. Up-regulation of cyclin D1 by HBx is mediated by NF-kappaB2/BCL3 complex through kappaB site of cyclin D1 promoter. J. Biol. Chem. 2006, 281, 31770–31777. [Google Scholar]
- Lazaro, C.A.; Rhim, J.A.; Yamada, Y.; Fausto, N. Generation of hepatocytes from oval cell precursors in culture. Cancer Res. 1998, 58, 5514–5522. [Google Scholar]
- Yang, S.; Pan, X.; Xiong, Z.; Wei, B.; Yao, H. The influence of hepatitis B virus X protein on the clock genes in liver cells and its significance. Chin.-Ger. J. Clin. Oncol. 2011, 10, 468–471. [Google Scholar]



© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, H.-Y.; Yang, S.-L.; Liang, H.-F.; Li, C.-H. HBx Protein Promotes Oval Cell Proliferation by Up-Regulation of Cyclin D1 via Activation of the MEK/ERK and PI3K/Akt Pathways. Int. J. Mol. Sci. 2014, 15, 3507-3518. https://doi.org/10.3390/ijms15033507
Wang H-Y, Yang S-L, Liang H-F, Li C-H. HBx Protein Promotes Oval Cell Proliferation by Up-Regulation of Cyclin D1 via Activation of the MEK/ERK and PI3K/Akt Pathways. International Journal of Molecular Sciences. 2014; 15(3):3507-3518. https://doi.org/10.3390/ijms15033507
Chicago/Turabian StyleWang, Heng-Yi, Sheng-Li Yang, Hui-Fang Liang, and Chang-Hai Li. 2014. "HBx Protein Promotes Oval Cell Proliferation by Up-Regulation of Cyclin D1 via Activation of the MEK/ERK and PI3K/Akt Pathways" International Journal of Molecular Sciences 15, no. 3: 3507-3518. https://doi.org/10.3390/ijms15033507