Rationalization of Activity Cliffs of a Sulfonamide Inhibitor of DNA Methyltransferases with Induced-Fit Docking

Abstract
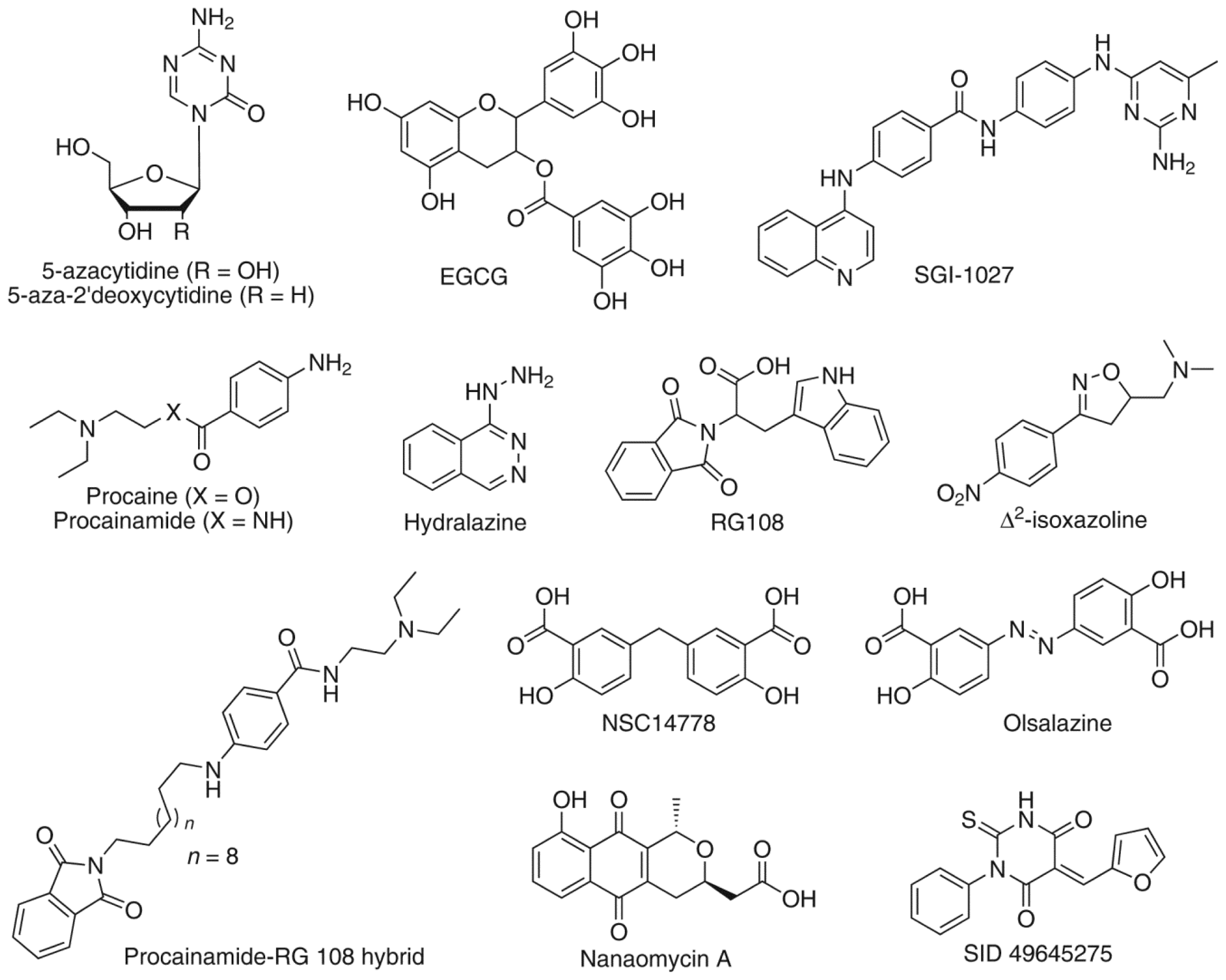
:
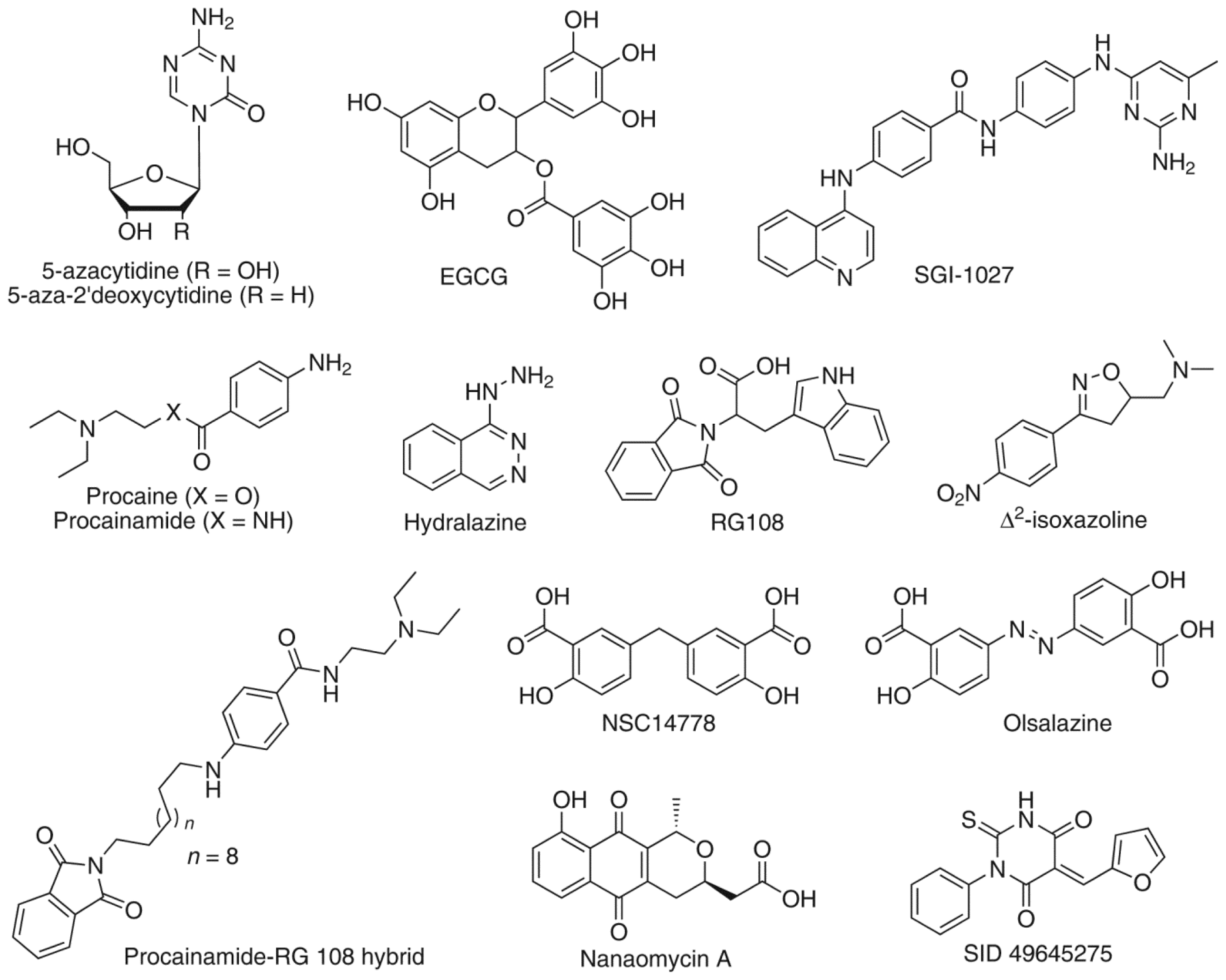
1. Introduction
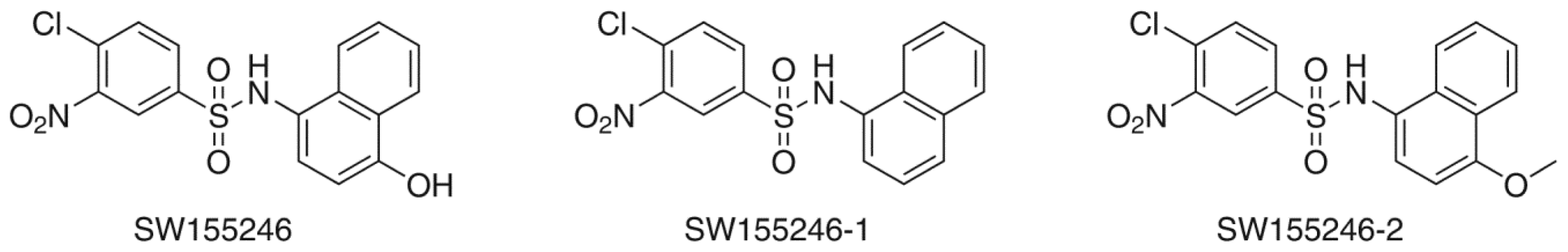
2. Results and Discussion
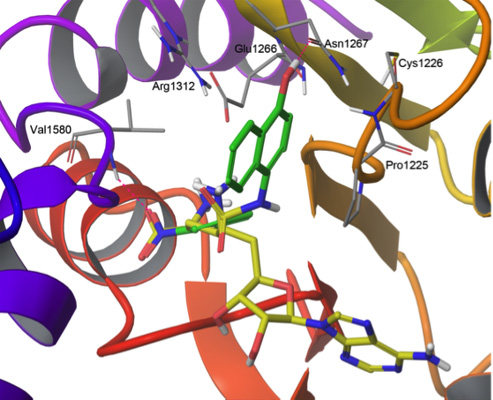
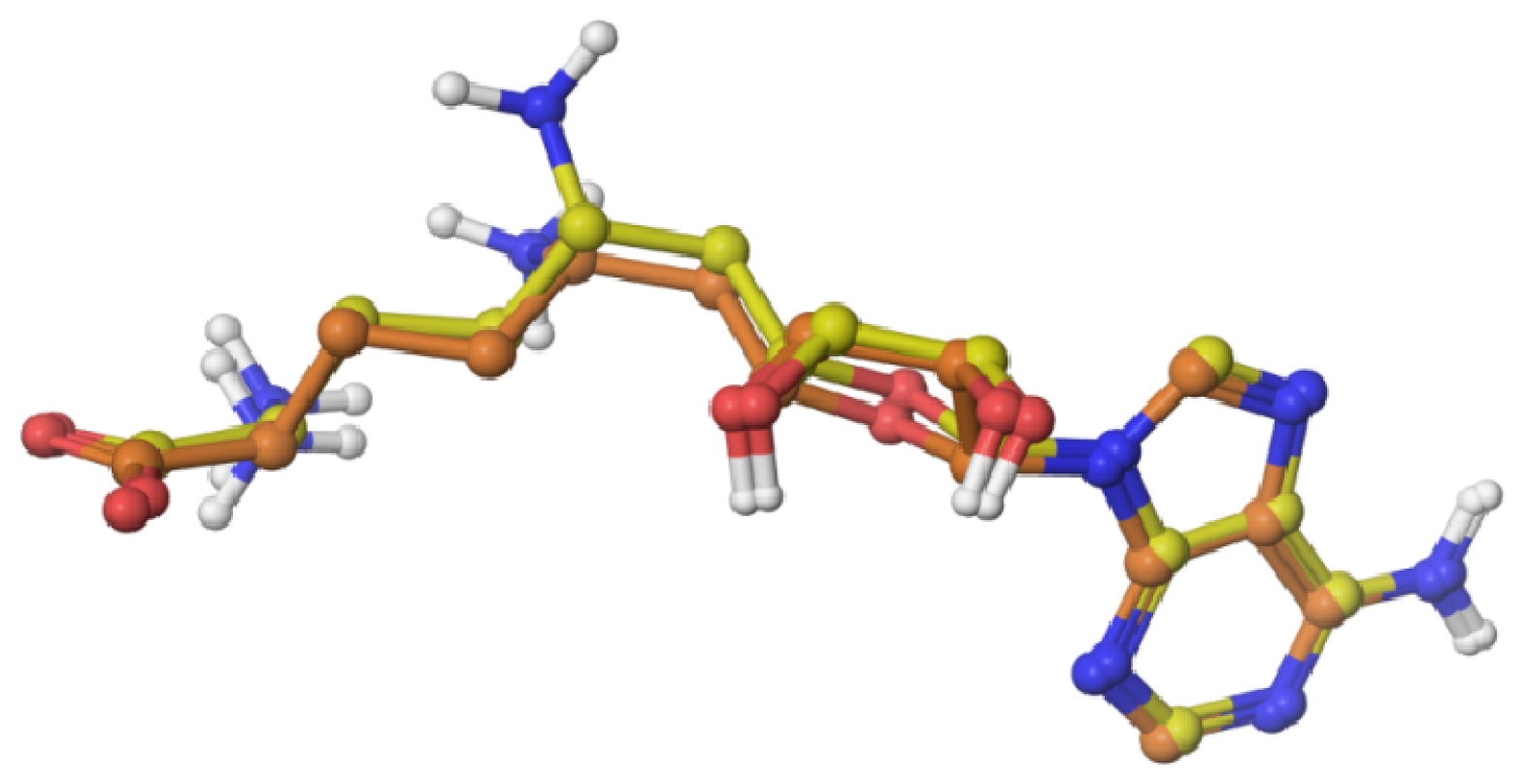
2.1. Validation of the Docking Protocol
2.2. Flexible Docking
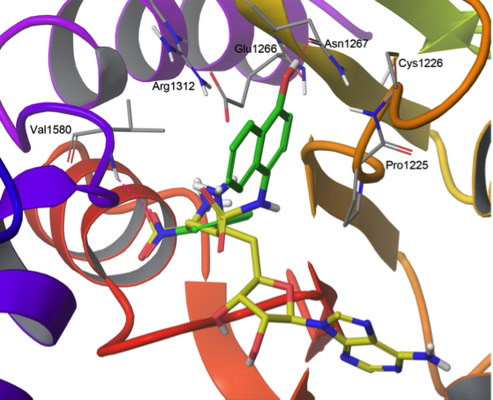
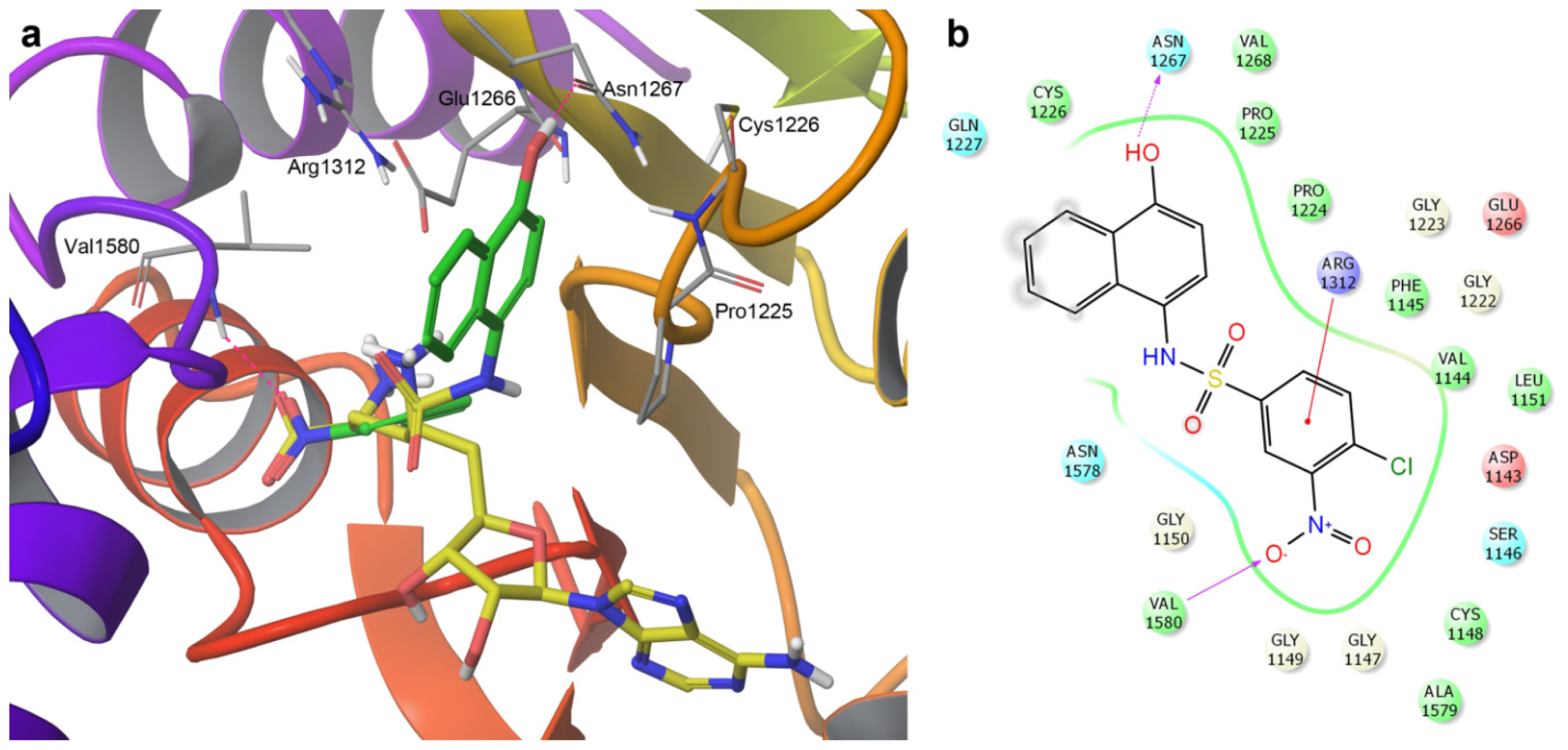
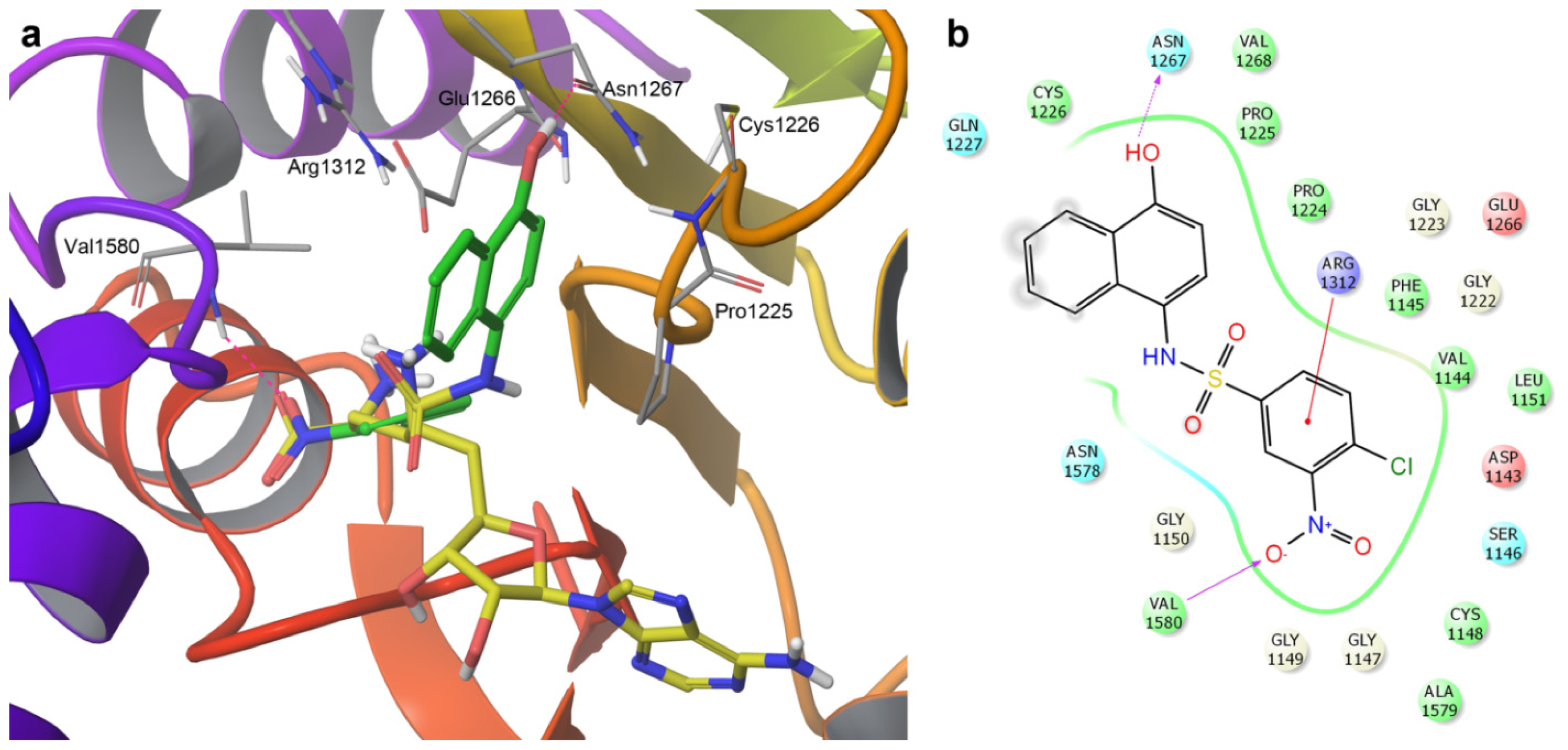
2.3. Induced-Fit Docking
3. Experimental Section
4. Conclusions
Supplementary Information
ijms-15-03253-s001.pdfAcknowledgments
Conflicts of Interest
References
- Rius, M.; Lyko, F. Epigenetic cancer therapy: Rationales, targets and drugs. Oncogene 2012, 31, 4257–4265. [Google Scholar]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar]
- Jones, P.A.; Liang, G.N. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet 2009, 10, 805–811. [Google Scholar]
- Karpf, A.R. Epigenomic reactivation screening to identify genes silenced by DNA hypermethylation in human cancer. Curr. Opin. Mol. Ther 2007, 9, 231–241. [Google Scholar]
- Foulks, J.M.; Parnell, K.M.; Nix, R.N.; Chau, S.; Swierczek, K.; Saunders, M.; Wright, K.; Hendrickson, T.F.; Ho, K.-K.; McCullar, M.V.; et al. Epigenetic drug discovery. J. Biomol. Screening 2012, 17, 2–17. [Google Scholar]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar]
- Kuck, D.; Singh, N.; Lyko, F.; Medina-Franco, J.L. Novel and selective DNA methyltransferase inhibitors: Docking-based virtual screening and experimental evaluation. Bioorg. Med. Chem 2010, 18, 822–829. [Google Scholar]
- Méndez-Lucio, O.; Tran, J.; Medina-Franco, J.L.; Meurice, N.; Muller, M. Towards drug repurposing in epigenetics: Olsalazine as a novel hypomethylating compound active in a cellular context. ChemMedChem 2014. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Caulfield, T. Advances in the computational development of DNA methyltransferase inhibitors. Drug Discovery Today 2011, 16, 418–425. [Google Scholar]
- Medina-Franco, J.L.; Yoo, J. Molecular modeling and virtual screening of DNA methyltransferase inhibitors. Curr. Pharm. Des 2013, 19, 2138–2147. [Google Scholar]
- Medina-Franco, J.L.; Yoo, J. Docking of a novel DNA methyltransferase inhibitor identified from high-throughput screening: Insights to unveil inhibitors in chemical databases. Mol. Div 2013, 17, 337–344. [Google Scholar]
- Kilgore, J.A.; Du, X.L.; Melito, L.; Wei, S.G.; Wang, C.G.; Chin, H.G.; Posner, B.; Pradhan, S.; Ready, J.M.; Williams, N.S. Identification of DNMT1 selective antagonists using a novel scintillation proximity assay. J. Biol. Chem 2013, 288, 19673–19684. [Google Scholar]
- Maggiora, G.M. On outliers and activity cliffs-why QSAR often disappoints. J. Chem. Inf. Model 2006, 46, 1535. [Google Scholar]
- Medina-Franco, J.L. Scanning structure–activity relationships with structure–activity similarity and related maps: From consensus activity cliffs to selectivity switches. J. Chem. Inf. Model 2012, 52, 2485–2493. [Google Scholar]
- Yoo, J.; Choi, S.; Medina-Franco, J.L. Molecular modeling studies of the novel inhibitors of DNA methyltransferases SGI-1027 and CBC12: Implications for the mechanism of inhibition of DNMTs. PLoS One 2013, 8, e62152. [Google Scholar]
- Shah, S.S.; Rivera, G.; Ashfaq, M. Recent advances in medicinal chemistry of sulfonamides. Rational design as anti-tumoral, anti-bacterial and anti-inflammatory agents. Mini Rev. Med. Chem 2013, 13, 70–86. [Google Scholar]
- Bello, M.; Martinez-Archundia, M.; Correa-Basurto, J. Automated docking for novel drug discovery. Expert Opin. Drug Discovery 2013, 8, 821–834. [Google Scholar]
- Jeltsch, A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. ChemBioChem 2002, 3, 274–293. [Google Scholar]
- Yoo, J.; Kim, J.H.; Robertson, K.D.; Medina-Franco, J.L. Molecular modeling of inhibitors of human DNA methyltransferase with a crystal structure: Discovery of a novel DNMT1 inhibitor. Adv.Protein Chem.Struct.Biol 2012, 87, 219–247. [Google Scholar]
- Hu, Y.; Furtmann, N.; Gütschow, M.; Bajorath, J. Systematic identification and classification of three-dimensional activity cliffs. J. Chem. Inf. Model 2012, 52, 1490–1498. [Google Scholar]
- Mendez-Lucio, O.; Perez-Villanueva, J.; Castillo, R.; Medina-Franco, J.L. Identifying activity cliff generators of PPAR ligands using SAS maps. Mol. Inf 2012, 31, 837–846. [Google Scholar]
- Schrödinger Suite 2012 Protein Preparation Wizard; Epik Version 2.3; Schrödinger, LLC: New York, NY, USA, 2012.Impact, version 5.8; Schrödinger, LLC: New York, NY, USA, 2005.Prime, Version 3.1; Schrödinger, LLC: New York, NY, USA, 2012.
- Maestro, version 9.3; Schrödinger, LLC: New York, NY, USA, 2012.
- Molecular Operating Environment (MOE), version 2013.08; Chemical Computing Group Inc: Montreal, QC, Canada, 2013.
- Glide, Version 5.8; Schrödinger, LLC: New York, NY, USA, 2012.
- Schrödinger Suite 2012 Induced Fit Docking Protocol; Glide Version 5.8; Schrödinger, LLC: New York, NY, USA, 2012.Prime, Version 3.1; Schrödinger, LLC: New York, NY, USA, 2012.
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem 2006, 49, 534–553. [Google Scholar]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des 2006, 67, 83–84. [Google Scholar]
- Medina-Franco, J.L. Activity cliffs: Facts or artifacts? Chem. Biol. Drug Des 2013, 81, 553–556. [Google Scholar]
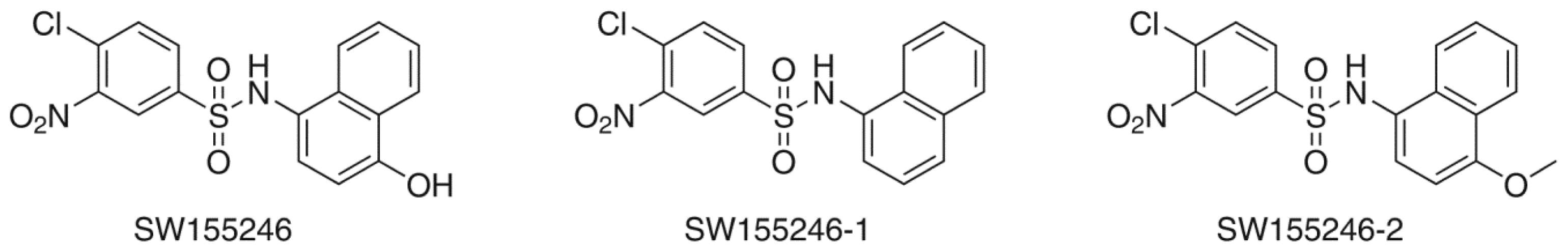


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Flexible docking | IFD | |
|---|---|---|---|
| Glide XP | Glide XP | IFDScore | |
| SW155246 | −4.817 | −5.858 | −1009.528 |
| SW155246-1 | −4.312 | −5.244 | −1008.860 |
| SW155246-2 | −4.370 | −5.819 | −1009.967 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Medina-Franco, J.L.; Méndez-Lucio, O.; Yoo, J. Rationalization of Activity Cliffs of a Sulfonamide Inhibitor of DNA Methyltransferases with Induced-Fit Docking. Int. J. Mol. Sci. 2014, 15, 3253-3261. https://doi.org/10.3390/ijms15023253
Medina-Franco JL, Méndez-Lucio O, Yoo J. Rationalization of Activity Cliffs of a Sulfonamide Inhibitor of DNA Methyltransferases with Induced-Fit Docking. International Journal of Molecular Sciences. 2014; 15(2):3253-3261. https://doi.org/10.3390/ijms15023253
Chicago/Turabian StyleMedina-Franco, José L., Oscar Méndez-Lucio, and Jakyung Yoo. 2014. "Rationalization of Activity Cliffs of a Sulfonamide Inhibitor of DNA Methyltransferases with Induced-Fit Docking" International Journal of Molecular Sciences 15, no. 2: 3253-3261. https://doi.org/10.3390/ijms15023253
APA StyleMedina-Franco, J. L., Méndez-Lucio, O., & Yoo, J. (2014). Rationalization of Activity Cliffs of a Sulfonamide Inhibitor of DNA Methyltransferases with Induced-Fit Docking. International Journal of Molecular Sciences, 15(2), 3253-3261. https://doi.org/10.3390/ijms15023253