Recent Progress in Understanding Subtype Specific Regulation of NMDA Receptors by G Protein Coupled Receptors (GPCRs)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Introduction of G Protein Coupled Receptors (GPCRs)
2. Introduction to NMDARs (NMDA Receptors)
2.1. GluN1 Subunits
2.2. GluN2 Subunits
2.2.1. Intracellular Association of GluN2 Subunits
2.2.2. Distinct Functional Roles of GluN2 Subunits in Synaptic Plasticity
2.2.3. GluN2 Subunits in Metaplasticity
2.2.4. Tri-heteromeric GluN1/GluN2A/GluN2B Receptors in Synaptic Plasticity
3. The Regulation of NMDARs by G Protein Coupled Receptor (GPCR)
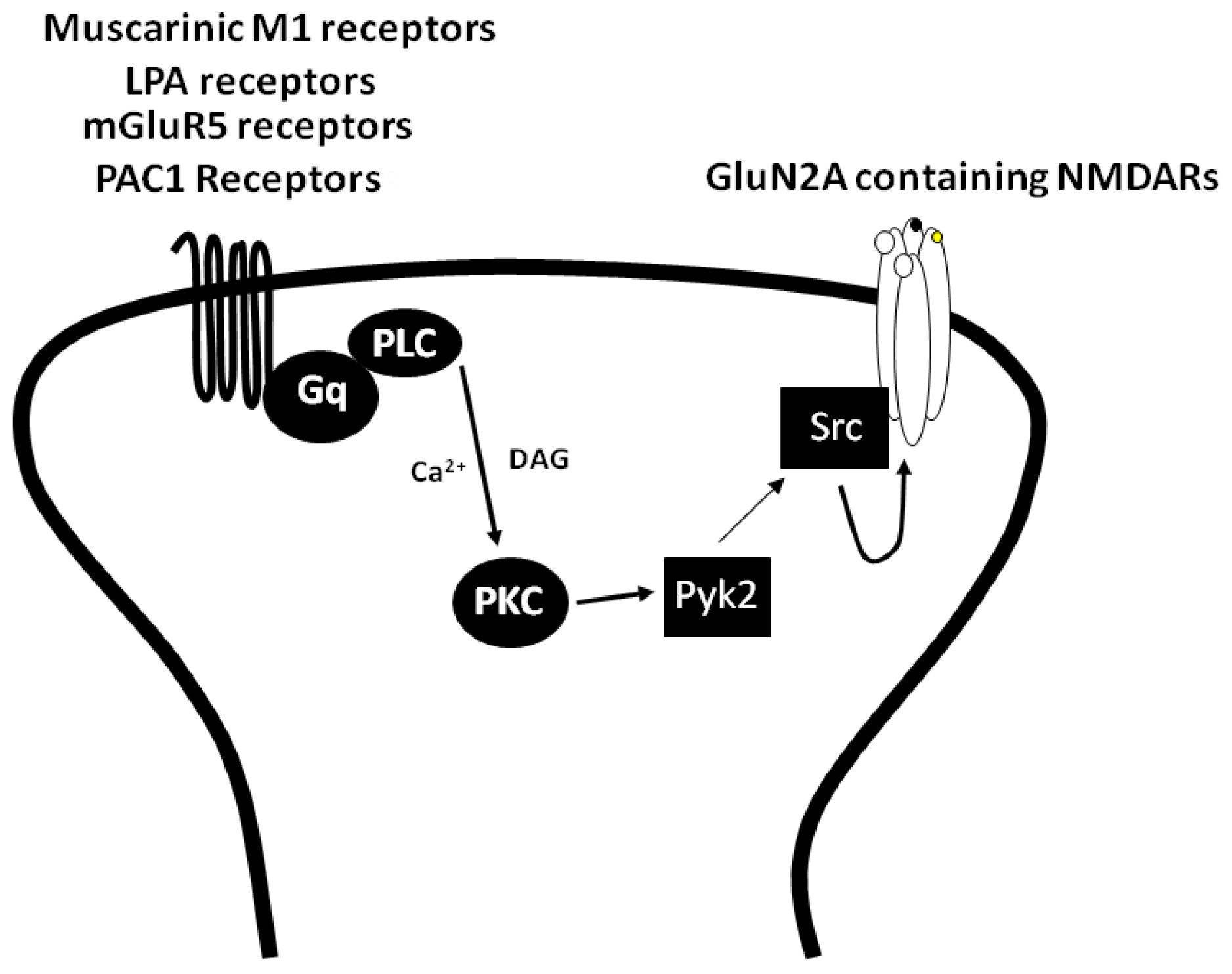
3.1. The Regulation of NMDARs by Gαq Containing GPCRs
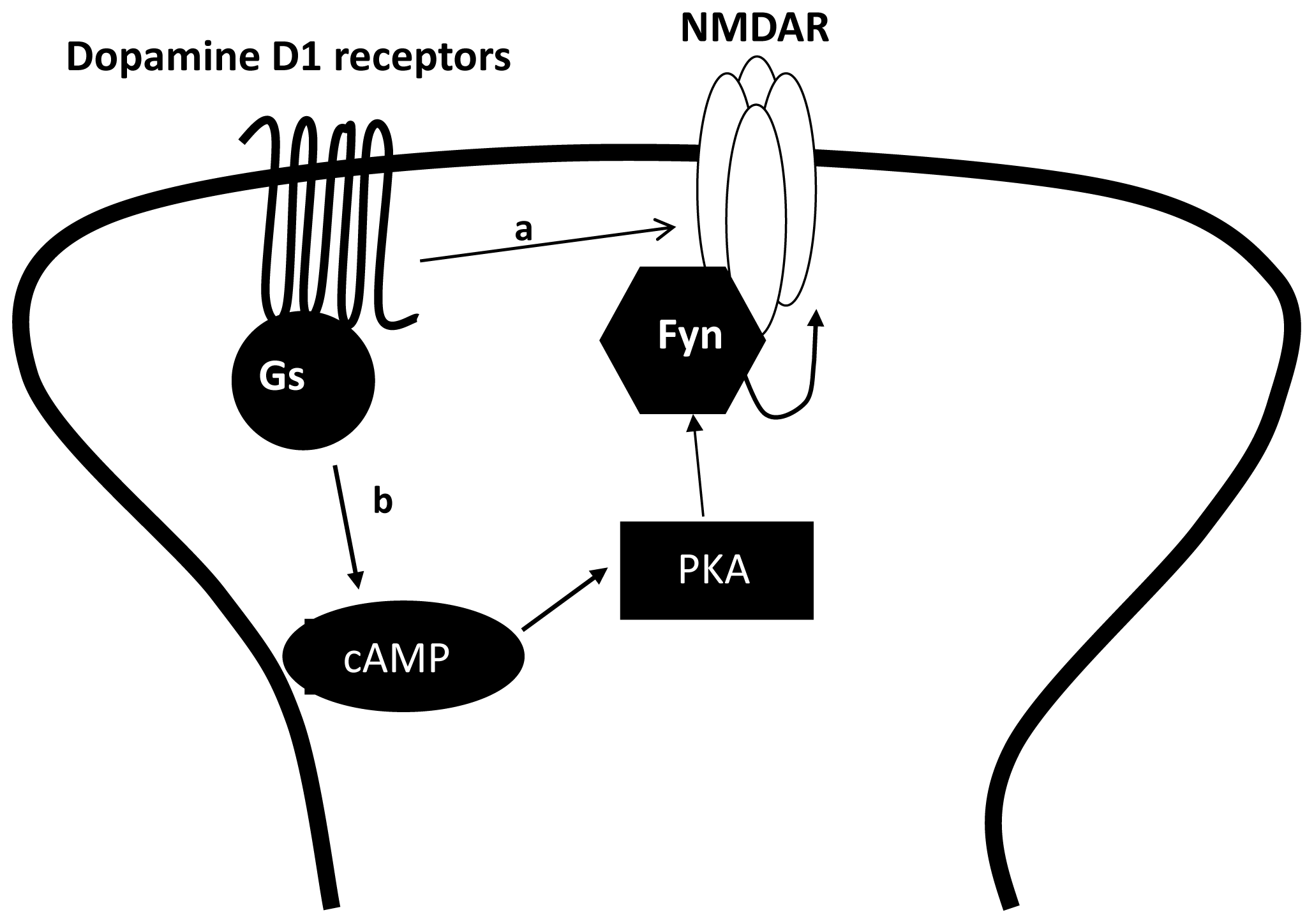
3.2. The Regulation of NMDAR by Gαs Containing GPCRs
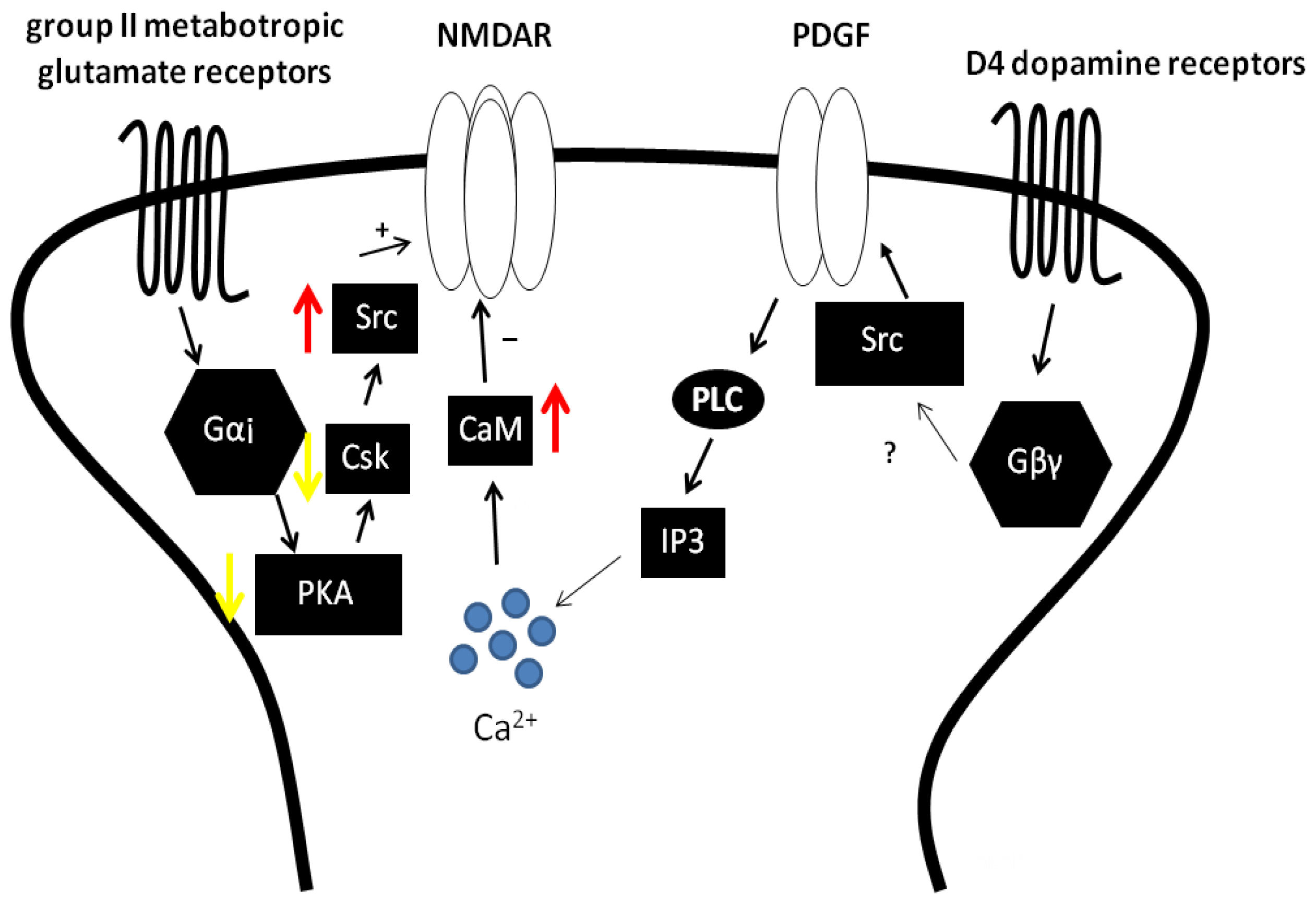
3.3. The Regulation of NMDAR by Gαi Containing GPCRs
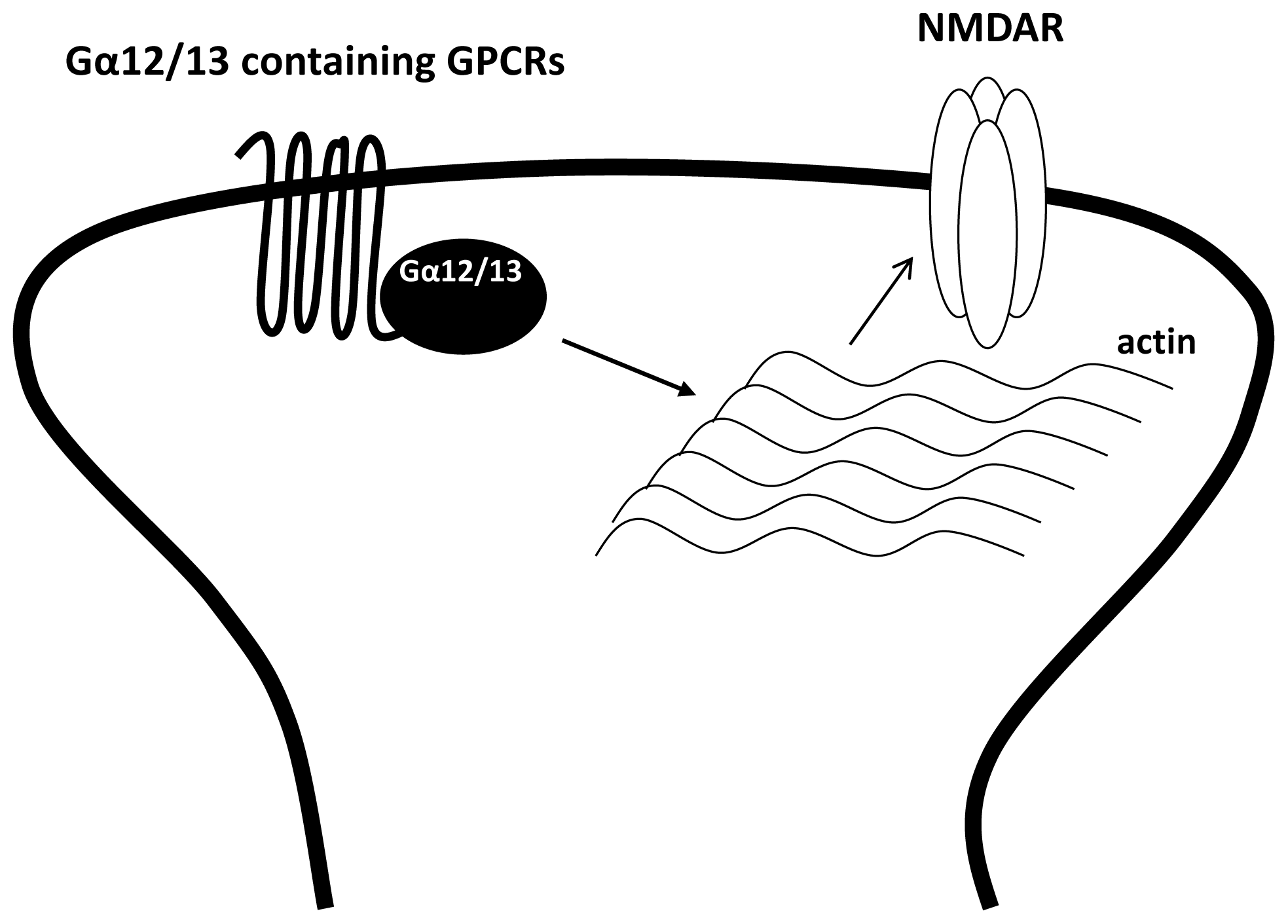
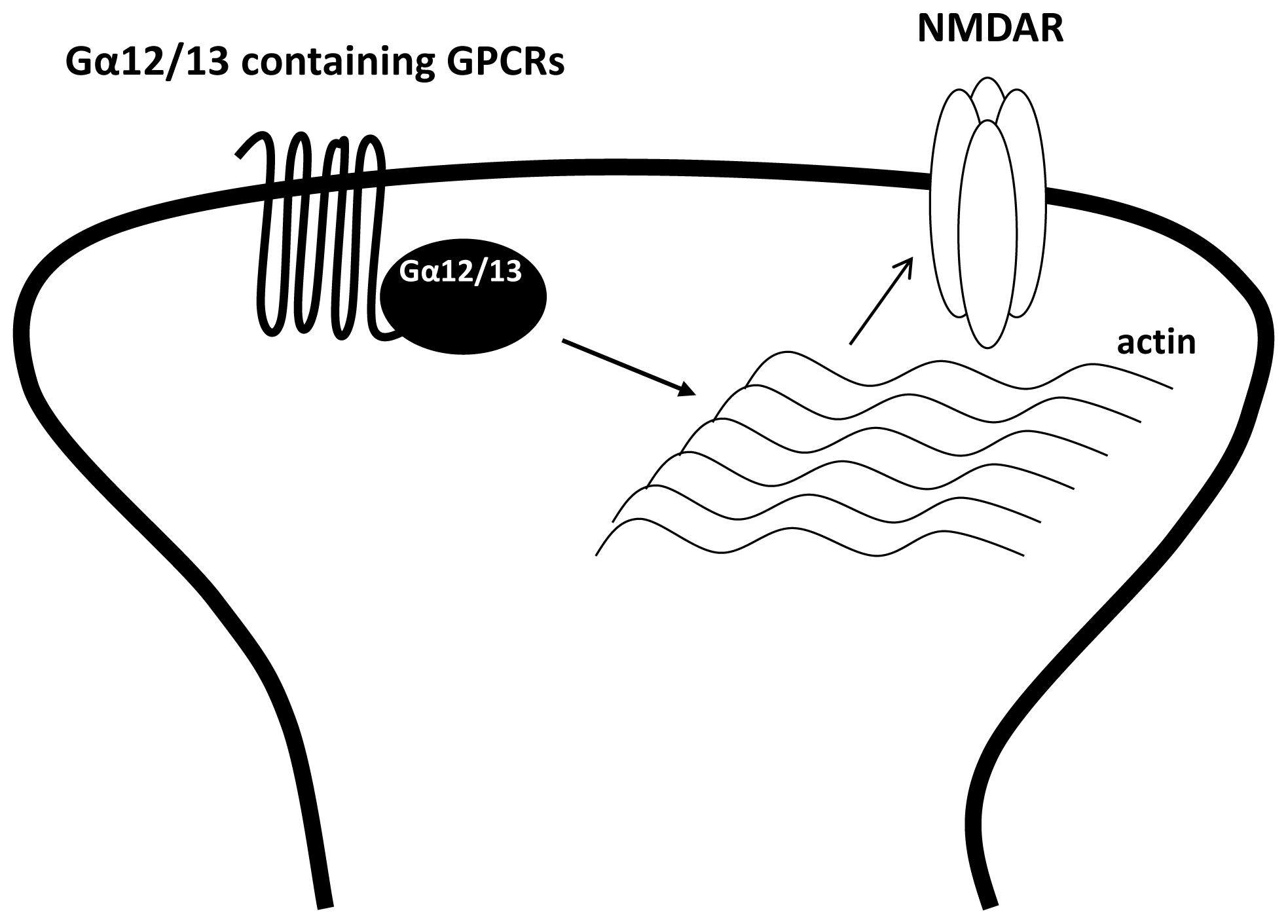
3.4. The Regulation of NMDAR by Gα12/13 Containing GPCRs
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jacoby, E.; Bouhelal, R.; Gerspacher, M.; Seuwen, K. The 7 TM G-protein-coupled receptor target family. ChemMedChem 2006, 1, 761–782. [Google Scholar]
- Gether, U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev 2000, 21, 90–113. [Google Scholar]
- Berman, D.M.; Gilman, A.G. Mammalian RGS proteins: Barbarians at the gate. J. Biol. Chem 1998, 273, 1269–1272. [Google Scholar]
- Ferguson, S.S. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol. Rev 2001, 53, 1–24. [Google Scholar]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol 2002, 3, 639–650. [Google Scholar]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar]
- Anagnostaras, S.G.; Murphy, G.G.; Hamilton, S.E.; Mitchell, S.L.; Rahnama, N.P.; Nathanson, N.M.; Silva, A.J. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat. Neurosci 2003, 6, 51–58. [Google Scholar]
- Meltzer, H.Y.; Li, Z.; Kaneda, Y.; Ichikawa, J. Serotonin receptors: Their key role in drugs to treat schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 1159–1172. [Google Scholar]
- Meltzer, H.Y. Update on typical and atypical antipsychotic drugs. Annu. Rev. Med 2013, 64, 393–406. [Google Scholar]
- Harrison, P.J.; Weinberger, D.R. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol. Psychiatry 2005, 10, 40–68. [Google Scholar]
- Cull-Candy, S.G.; Leszkiewicz, D.N. Role of distinct NMDA receptor subtypes at central synapses. Sci. Signal 2004. [Google Scholar] [CrossRef]
- McBain, C.J.; Mayer, M.L. N-methyl-d-aspartic acid receptor structure and function. Physiol. Rev 1994, 74, 723–760. [Google Scholar]
- Herin, G.A.; Aizenman, E. Amino terminal domain regulation of NMDA receptor function. Eur. J. Pharmacol 2004, 500, 101–111. [Google Scholar]
- Mony, L.; Kew, J.N.; Gunthorpe, M.J.; Paoletti, P. Allosteric modulators of NR2B-containing NMDA receptors: Molecular mechanisms and therapeutic potential. Br. J. Pharmacol 2009, 157, 1301–1317. [Google Scholar]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: Function and pharmacology. Curr. Opin. Pharmacol 2007, 7, 39–47. [Google Scholar]
- Furukawa, H.; Singh, S.K.; Mancusso, R.; Gouaux, E. Subunit arrangement and function in NMDA receptors. Nature 2005, 438, 185–192. [Google Scholar]
- Zukin, R.S.; Bennett, M.V. Alternatively spliced isoforms of the NMDARI receptor subunit. Trends Neurosci 1995, 18, 306–313. [Google Scholar]
- Standley, S.; Roche, K.W.; McCallum, J.; Sans, N.; Wenthold, R.J. PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 2000, 28, 887–898. [Google Scholar]
- Wenthold, R.J.; Prybylowski, K.; Standley, S.; Sans, N.; Petralia, R.S. Trafficking of NMDA receptors. Annu. Rev. Pharmacol. Toxicol 2003, 43, 335–358. [Google Scholar]
- Grosshans, D.R.; Browning, M.D. Protein kinase C activation induces tyrosine phosphorylation of the NR2A and NR2B subunits of the NMDA receptor. J. Neurochem 2001, 76, 737–744. [Google Scholar]
- Tingley, W.G.; Ehlers, M.D.; Kameyama, K.; Doherty, C.; Ptak, J.B.; Riley, C.T.; Huganir, R.L. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J. Biol. Chem 1997, 272, 5157–5166. [Google Scholar]
- Scott, D.B.; Blanpied, T.A.; Swanson, G.T.; Zhang, C.; Ehlers, M.D. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J. Neurosci 2001, 21, 3063–3072. [Google Scholar]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 2013, 14, 383–400. [Google Scholar]
- Kohr, G. NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res 2006, 326, 439–446. [Google Scholar]
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci 2004, 5, 771–781. [Google Scholar]
- Townsend, M.; Yoshii, A.; Mishina, M.; Constantine-Paton, M. Developmental loss of miniature N-methyl-d-aspartate receptor currents in NR2A knockout mice. Proc. Natl. Acad. Sci. USA 2003, A100, 1340–1345. [Google Scholar]
- Al-Hallaq, R.A.; Conrads, T.P.; Veenstra, T.D.; Wenthold, R.J. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J. Neurosci 2007, 27, 8334–8343. [Google Scholar]
- Chung, H.J.; Huang, Y.H.; Lau, L.F.; Huganir, R.L. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J. Neurosci 2004, 24, 10248–10259. [Google Scholar]
- Sanz-Clemente, A.; Matta, J.A.; Isaac, J.T.; Roche, K.W. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 2010, 67, 984–996. [Google Scholar]
- Gardoni, F.; Polli, F.; Cattabeni, F.; Di, L.M. Calcium-calmodulin-dependent protein kinase II phosphorylation modulates PSD-95 binding to NMDA receptors. Eur. J. Neurosci 2006, 24, 2694–2704. [Google Scholar]
- Prybylowski, K.; Chang, K.; Sans, N.; Kan, L.; Vicini, S.; Wenthold, R.J. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 2005, 47, 845–857. [Google Scholar]
- Rong, Y.; Lu, X.; Bernard, A.; Khrestchatisky, M.; Baudry, M. Tyrosine phosphorylation of ionotropic glutamate receptors by Fyn or Src differentially modulates their susceptibility to calpain and enhances their binding to spectrin and PSD-95. J. Neurochem 2001, 79, 382–390. [Google Scholar]
- Bi, R.; Rong, Y.; Bernard, A.; Khrestchatisky, M.; Baudry, M. Src-mediated tyrosine phosphorylation of NR2 subunits of N-methyl-d-aspartate receptors protects from calpain-mediated truncation of their C-terminal domains. J. Biol. Chem 2000, 275, 26477–26483. [Google Scholar]
- Strack, S.; Colbran, R.J. Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem 1998, 273, 20689–20692. [Google Scholar]
- Bayer, K.U.; De, K.P.; Leonard, A.S.; Hell, J.W.; Schulman, H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 2001, 411, 801–805. [Google Scholar]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Ivanov, A.; Tyzio, R.; Pellegrino, C.; Ben-Ari, Y.; Clapham, D.E.; Medina, I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 2003, 40, 775–784. [Google Scholar]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 2004, 304, 1021–1024. [Google Scholar]
- Massey, P.V.; Johnson, B.E.; Moult, P.R.; Auberson, Y.P.; Brown, M.W.; Molnar, E.; Collingridge, G.L.; Bashir, Z.I. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J. Neurosci 2004, 24, 7821–7828. [Google Scholar]
- Morishita, W.; Lu, W.; Smith, G.B.; Nicoll, R.A.; Bear, M.F.; Malenka, R.C. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology 2007, 52, 71–76. [Google Scholar]
- Hendricson, A.W.; Miao, C.L.; Lippmann, M.J.; Morrisett, R.A. Ifenprodil and ethanol enhance NMDA receptor-dependent long-term depression. J. Pharmacol. Exp. Ther 2002, 301, 938–944. [Google Scholar]
- Shipton, O.A.; Paulsen, O. GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos. Trans. R. Soc. B 2014. [Google Scholar] [CrossRef]
- Barria, A.; Malinow, R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 2005, 48, 289–301. [Google Scholar]
- Tang, Y.P.; Shimizu, E.; Dube, G.R.; Rampon, C.; Kerchner, G.A.; Zhuo, M.; Liu, G.; Tsien, J.Z. Genetic enhancement of learning and memory in mice. Nature 1999, 401, 63–69. [Google Scholar]
- Weitlauf, C.; Honse, Y.; Auberson, Y.P.; Mishina, M.; Lovinger, D.M.; Winder, D.G. Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J. Neurosci 2005, 25, 8386–8390. [Google Scholar]
- Abraham, W.C. Metaplasticity: Tuning synapses and networks for plasticity. Nat. Rev. Neurosci 2008. [Google Scholar] [CrossRef]
- Philpot, B.D.; Sekhar, A.K.; Shouval, H.Z.; Bear, M.F. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 2001, 29, 157–169. [Google Scholar]
- Philpot, B.D.; Espinosa, J.S.; Bear, M.F. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J. Neurosci 2003, 23, 5583–5588. [Google Scholar]
- Philpot, B.D.; Cho, K.K.; Bear, M.F. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron 2007, 53, 495–502. [Google Scholar]
- Kirkwood, A.; Rioult, M.C.; Bear, M.F. Experience-dependent modification of synaptic plasticity in visual cortex. Nature 1996, 381, 526–528. [Google Scholar]
- Xu, Z.; Chen, R.Q.; Gu, Q.H.; Yan, J.Z.; Wang, S.H.; Liu, S.Y.; Lu, W. Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J. Neurosci 2009, 29, 8764–8773. [Google Scholar]
- Yang, K.; Trepanier, C.; Sidhu, B.; Xie, Y.F.; Li, H.; Lei, G.; Salter, M.W.; Orser, B.A.; Nakazawa, T.; Yamamoto, T.; et al. Metaplasticity gated through differential regulation of GluN2A versus GluN2B receptors by Src family kinases. EMBO J 2012, 31, 805–816. [Google Scholar]
- Chen, W.S.; Bear, M.F. Activity-dependent regulation of NR2B translation contributes to metaplasticity in mouse visual cortex. Neuropharmacology 2007, 52, 200–214. [Google Scholar]
- Longordo, F.; Kopp, C.; Mishina, M.; Lujan, R.; Luthi, A. NR2A at CA1 synapses is obligatory for the susceptibility of hippocampal plasticity to sleep loss. J. Neurosci 2009, 29, 9026–9041. [Google Scholar]
- Kotecha, S.A.; Jackson, M.F.; Al-Mahrouki, A.; Roder, J.C.; Orser, B.A.; Macdonald, J.F. Co-stimulation of mGluR5 and N-methyl-d-aspartate receptors is required for potentiation of excitatory synaptic transmission in hippocampal neurons. J. Biol. Chem 2003, 278, 27742–27749. [Google Scholar]
- Lu, W.Y.; Xiong, Z.G.; Lei, S.; Orser, B.A.; Dudek, E.; Browning, M.D.; Macdonald, J.F. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat. Neurosci 1999, 2, 331–338. [Google Scholar]
- Rauner, C.; Kohr, G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-d-aspartate receptor population in adult hippocampal synapses. J. Biol. Chem 2011, 286, 7558–7566. [Google Scholar]
- Hatton, C.J.; Paoletti, P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron 2005, 46, 261–274. [Google Scholar]
- Neyton, J.; Paoletti, P. Relating NMDA receptor function to receptor subunit composition: Limitations of the pharmacological approach. J. Neurosci 2006, 26, 1331–1333. [Google Scholar]
- Volianskis, A.; Bannister, N.; Collett, V.J.; Irvine, M.W.; Monaghan, D.T.; Fitzjohn, S.M.; Jensen, M.S.; Jane, D.E.; Collingridge, G.L. Different NMDA receptor subtypes mediate induction of long-term potentiation and two forms of short-term potentiation at CA1 synapses in rat hippocampus in vitro. J. Physiol. 2013, 591, 955–972. [Google Scholar]
- Chen, B.S.; Roche, K.W. Regulation of NMDA receptors by phosphorylation. Neuropharmacology 2007, 53, 362–368. [Google Scholar]
- Lee, H.K. Synaptic plasticity and phosphorylation. Pharmacol. Ther 2006, 112, 810–832. [Google Scholar]
- Salter, M.W.; Kalia, L.V. Src kinases: A hub for NMDA receptor regulation. Nat. Rev. Neurosci 2004, 5, 317–328. [Google Scholar]
- Rojas, A.; Dingledine, R. Ionotropic glutamate receptors: Regulation by G-protein-coupled receptors. Mol. Pharmacol 2013, 83, 746–752. [Google Scholar]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol Rev 2008, 88, 1341–1378. [Google Scholar]
- Chen, B.S.; Braud, S.; Badger, J.D.; Isaac, J.T.; Roche, K.W. Regulation of NR1/NR2C N-methyl-d-aspartate (NMDA) receptors by phosphorylation. J. Biol. Chem 2006, 281, 16583–16590. [Google Scholar]
- Jones, M.L.; Leonard, J.P. PKC site mutations reveal differential modulation by insulin of NMDA receptors containing NR2A or NR2B subunits. J. Neurochem 2005, 92, 1431–1438. [Google Scholar]
- Liao, G.Y.; Wagner, D.A.; Hsu, M.H.; Leonard, J.P. Evidence for direct protein kinase-C mediated modulation of N-methyl-d-aspartate receptor current. Mol. Pharmacol 2001, 59, 960–964. [Google Scholar]
- Zheng, X.; Zhang, L.; Wang, A.P.; Bennett, M.V.; Zukin, R.S. Protein kinase C potentiation of N-methyl-d-aspartate receptor activity is not mediated by phosphorylation of N-methyl-d-aspartate receptor subunits. Proc. Natl. Acad. Sci. USA 1999, 96, 15262–15267. [Google Scholar]
- Huang, Y.; Lu, W.; Ali, D.W.; Pelkey, K.A.; Pitcher, G.M.; Lu, Y.M.; Aoto, H.; Roder, J.C.; Sasaki, T.; Salter, M.W.; et al. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 2001, 29, 485–496. [Google Scholar]
- Carroll, R.C.; Zukin, R.S. NMDA-receptor trafficking and targeting: Implications for synaptic transmission and plasticity. Trends Neurosci 2002, 25, 571–577. [Google Scholar]
- Lan, J.Y.; Skeberdis, V.A.; Jover, T.; Grooms, S.Y.; Lin, Y.; Araneda, R.C.; Zheng, X.; Bennett, M.V.; Zukin, R.S. Protein kinase C modulates NMDA receptor trafficking and gating. Nat. Neurosci 2001, 4, 382–390. [Google Scholar]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci 2007, 8, 413–426. [Google Scholar]
- Lin, Y.; Jover-Mengual, T.; Wong, J.; Bennett, M.V.; Zukin, R.S. PSD-95 and PKC converge in regulating NMDA receptor trafficking and gating. Proc. Natl. Acad. Sci. USA 2006, 103, 19902–19907. [Google Scholar]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev 2009, 61, 283–357. [Google Scholar]
- Macdonald, D.S.; Weerapura, M.; Beazely, M.A.; Martin, L.; Czerwinski, W.; Roder, J.C.; Orser, B.A.; Macdonald, J.F. Modulation of NMDA receptors by pituitary adenylate cyclase activating peptide in CA1 neurons requires G alpha q, protein kinase C, and activation of Src. J. Neurosci 2005, 25, 11374–11384. [Google Scholar]
- Benquet, P.; Gee, C.E.; Gerber, U. Two distinct signaling pathways upregulate NMDA receptor responses via two distinct metabotropic glutamate receptor subtypes. J. Neurosci 2002, 22, 9679–9686. [Google Scholar]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 2008, 57, 121–134. [Google Scholar]
- De Fernandez, S.D.; Buno, W. The muscarinic long-term enhancement of NMDA and AMPA receptor-mediated transmission at Schaffer collateral synapses develop through different intracellular mechanisms. J. Neurosci 2010, 30, 11032–11042. [Google Scholar]
- Jackson, M.F.; Konarski, J.Z.; Weerapura, M.; Czerwinski, W.; Macdonald, J.F. Protein kinase C enhances glycine-insensitive desensitization of NMDA receptors independently of previously identified protein kinase C sites. J. Neurochem 2006, 96, 1509–1518. [Google Scholar]
- Lu, W.Y.; Jackson, M.F.; Bai, D.; Orser, B.A.; Macdonald, J.F. In CA1 pyramidal neuronsof the hippocampus protein kinase C regulates calcium-dependent inactivation of NMDA receptors. J. Neurosci 2000, 20, 4452–4462. [Google Scholar]
- Macdonald, J.F.; Kotecha, S.A.; Lu, W.Y.; Jackson, M.F. Convergence of PKC-dependent kinase signal cascades on NMDA receptors. Curr. Drug Targets 2001, 2, 299–312. [Google Scholar]
- Paoletti, P.; Ascher, P.; Neyton, J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci 1997, 17, 5711–5725. [Google Scholar]
- Borgland, S.L.; Taha, S.A.; Sarti, F.; Fields, H.L.; Bonci, A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron 2006, 49, 589–601. [Google Scholar]
- Leonard, A.S.; Hell, J.W. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J. Biol. Chem 1997, 272, 12107–12115. [Google Scholar]
- Skeberdis, V.A.; Chevaleyre, V.; Lau, C.G.; Goldberg, J.H.; Pettit, D.L.; Suadicani, S.O.; Lin, Y.; Bennett, M.V.; Yuste, R.; Castillo, P.E.; et al. Protein kinase A regulates calcium permeability of NMDA receptors. Nat. Neurosci 2006, 9, 501–510. [Google Scholar]
- Svenningsson, P.; Nishi, A.; Fisone, G.; Girault, J.A.; Nairn, A.C.; Greengard, P. DARPP-32: An integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol 2004, 44, 269–296. [Google Scholar]
- Scott, D.B.; Blanpied, T.A.; Ehlers, M.D. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 2003, 45, 755–767. [Google Scholar]
- Dunah, A.W.; Sirianni, A.C.; Fienberg, A.A.; Bastia, E.; Schwarzschild, M.A.; Standaert, D.G. Dopamine D1-dependent trafficking of striatal N-methyl-d-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol. Pharmacol 2004, 65, 121–129. [Google Scholar]
- Hallett, P.J.; Spoelgen, R.; Hyman, B.T.; Standaert, D.G.; Dunah, A.W. Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J. Neurosci 2006, 26, 4690–4700. [Google Scholar]
- Snyder, G.L.; Fienberg, A.A.; Huganir, R.L.; Greengard, P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J. Neurosci 1998, 18, 10297–10303. [Google Scholar]
- Cepeda, C.; Levine, M.S. Where do you think you are going? The NMDA-D1 receptor trap. Sci. STKE 2006. [Google Scholar] [CrossRef]
- Hu, J.L.; Liu, G.; Li, Y.C.; Gao, W.J.; Huang, Y.Q. Dopamine D1 receptor-mediated NMDA receptor insertion depends on Fyn but not Src kinase pathway in prefrontal cortical neurons. Mol. Brain 2010. [Google Scholar] [CrossRef]
- Schilstrom, B.; Yaka, R.; Argilli, E.; Suvarna, N.; Schumann, J.; Chen, B.T.; Carman, M.; Singh, V.; Mailliard, W.S.; Ron, D.; et al. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J. Neurosci 2006, 26, 8549–8558. [Google Scholar]
- Stramiello, M.; Wagner, J.J. D1/5 receptor-mediated enhancement of LTP requires PKA, Src family kinases, and NR2B-containing NMDARs. Neuropharmacology 2008, 55, 871–877. [Google Scholar]
- Trepanier, C.; Lei, G.; Xie, Y.F.; Macdonald, J.F. Group II metabotropic glutamate receptors modify N-methyl-d-aspartate receptors via Src kinase. Sci. Rep 2013. [Google Scholar] [CrossRef]
- Kotecha, S.A.; Oak, J.N.; Jackson, M.F.; Perez, Y.; Orser, B.A.; van Tol, H.H.; Macdonald, J.F. A D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission. Neuron 2002, 35, 1111–1122. [Google Scholar]
- Cheng, J.; Liu, W.; Duffney, L.J.; Yan, Z. SNARE proteins are essential in the potentiation of NMDA receptors by group II metabotropic glutamate receptors. J. Physiol 2013, 591, 3935–3947. [Google Scholar]
- Krupp, J.J.; Vissel, B.; Thomas, C.G.; Heinemann, S.F.; Westbrook, G.L. Interactions of calmodulin and α-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J. Neurosci 1999, 19, 1165–1178. [Google Scholar]
- Wechsler, A.; Teichberg, V.I. Brain spectrin binding to the NMDA receptor is regulated by phosphorylation, calcium and calmodulin. EMBO J 1998, 17, 3931–3939. [Google Scholar]
- Lei, S.; Lu, W.Y.; Xiong, Z.G.; Orser, B.A.; Valenzuela, C.F.; Macdonald, J.F. Platelet-derived growth factor receptor-induced feed-forward inhibition of excitatory transmission between hippocampal pyramidal neurons. J. Biol. Chem 1999, 274, 30617–30623. [Google Scholar]
- Beazely, M.A.; Tong, A.; Wei, W.L.; Van, T.H.; Sidhu, B.; Macdonald, J.F. D2-class dopamine receptor inhibition of NMDA currents in prefrontal cortical neurons is platelet-derived growth factor receptor-dependent. J. Neurochem 2006, 98, 1657–1663. [Google Scholar]
- Wang, X.; Zhong, P.; Gu, Z.; Yan, Z. Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J. Neurosci 2003, 23, 9852–9861. [Google Scholar]
- Liu, X.Y.; Chu, X.P.; Mao, L.M.; Wang, M.; Lan, H.X.; Li, M.H.; Zhang, G.C.; Parelkar, N.K.; Fibuch, E.E.; Haines, M.; et al. Modulation of D2R-NR2B interactions in response to cocaine. Neuron 2006, 52, 897–909. [Google Scholar]
- Tyszkiewicz, J.P.; Gu, Z.; Wang, X.; Cai, X.; Yan, Z. Group II metabotropic glutamate receptors enhance NMDA receptor currents via a protein kinase C-dependent mechanism in pyramidal neurones of rat prefrontal cortex. J. Physiol 2004, 554, 765–777. [Google Scholar]
- Suzuki, N.; Hajicek, N.; Kozasa, T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 2009, 17, 55–70. [Google Scholar]
- Wyszynski, M.; Lin, J.; Rao, A.; Nigh, E.; Beggs, A.H.; Craig, A.M.; Sheng, M. Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature 1997, 385, 439–442. [Google Scholar]
- Rosenmund, C.; Westbrook, G.L. Rundown of N-methyl-d-aspartate channels during whole-cell recording in rat hippocampal neurons: Role of Ca2+ and ATP. J. Physiol 1993, 470, 705–729. [Google Scholar]
- Lei, S.; Czerwinska, E.; Czerwinski, W.; Walsh, M.P.; Macdonald, J.F. Regulation of NMDA receptor activity by F-actin and myosin light chain kinase. J. Neurosci 2001, 21, 8464–8472. [Google Scholar]
- Siehler, S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol 2009, 158, 41–49. [Google Scholar]
- Yuen, E.Y.; Jiang, Q.; Chen, P.; Gu, Z.; Feng, J.; Yan, Z. Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. J. Neurosci 2005, 25, 5488–5501. [Google Scholar]
- Kim, D.H.; Maneen, M.J.; Stahl, S.M. Building a better antipsychotic: receptor targets for the treatment of multiple symptom dimensions of schizophrenia. Neurotherapeutics 2009, 6, 78–85. [Google Scholar]
- Ginovart, N.; Kapur, S. Role of dopamine D(2) receptors for antipsychotic activity. Handb. Exp. Pharmacol 2012, 212, 27–52. [Google Scholar]
- Kuepper, R.; Skinbjerg, M.; bi-Dargham, A. The dopamine dysfunction in schizophrenia revisited: New insights into topography and course. Handb. Exp. Pharmacol 2012, 212, 1–26. [Google Scholar]
- Conn, P.J.; Lindsley, C.W.; Jones, C.K. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol. Sci 2009, 30, 25–31. [Google Scholar]
- Field, J.R.; Walker, A.G.; Conn, P.J. Targeting glutamate synapses in schizophrenia. Trends Mol. Med 2011, 17, 689–698. [Google Scholar]
- Harvey, R.J.; Yee, B.K. Glycine transporters as novel therapeutic targets in schizophrenia, alcohol dependence and pain. Nat. Rev. Drug Discov 2013, 12, 866–885. [Google Scholar]
- Hashimoto, K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res. Rev 2009, 61, 105–123. [Google Scholar]
- Barkus, C.; McHugh, S.B.; Sprengel, R.; Seeburg, P.H.; Rawlins, J.N.; Bannerman, D.M. Hippocampal NMDA receptors and anxiety: At the interface between cognition and emotion. Eur. J. Pharmacol 2010, 626, 49–56. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, K.; Jackson, M.F.; MacDonald, J.F. Recent Progress in Understanding Subtype Specific Regulation of NMDA Receptors by G Protein Coupled Receptors (GPCRs). Int. J. Mol. Sci. 2014, 15, 3003-3024. https://doi.org/10.3390/ijms15023003
Yang K, Jackson MF, MacDonald JF. Recent Progress in Understanding Subtype Specific Regulation of NMDA Receptors by G Protein Coupled Receptors (GPCRs). International Journal of Molecular Sciences. 2014; 15(2):3003-3024. https://doi.org/10.3390/ijms15023003
Chicago/Turabian StyleYang, Kai, Michael F. Jackson, and John F. MacDonald. 2014. "Recent Progress in Understanding Subtype Specific Regulation of NMDA Receptors by G Protein Coupled Receptors (GPCRs)" International Journal of Molecular Sciences 15, no. 2: 3003-3024. https://doi.org/10.3390/ijms15023003
APA StyleYang, K., Jackson, M. F., & MacDonald, J. F. (2014). Recent Progress in Understanding Subtype Specific Regulation of NMDA Receptors by G Protein Coupled Receptors (GPCRs). International Journal of Molecular Sciences, 15(2), 3003-3024. https://doi.org/10.3390/ijms15023003