Paclitaxel Induces Apoptosis in Breast Cancer Cells through Different Calcium—Regulating Mechanisms Depending on External Calcium Conditions


Abstract
:1. Introduction
2. Results and Discussion
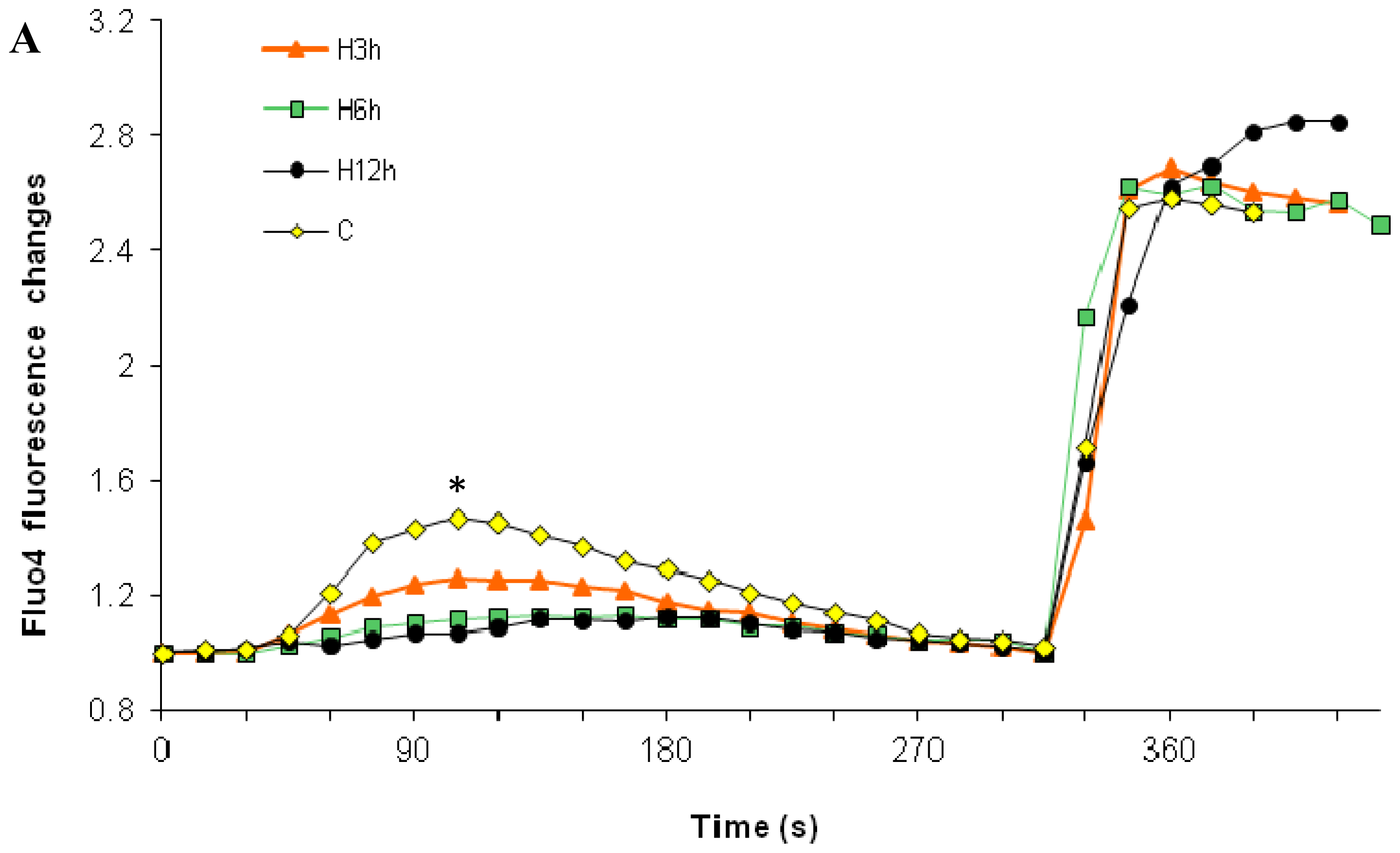
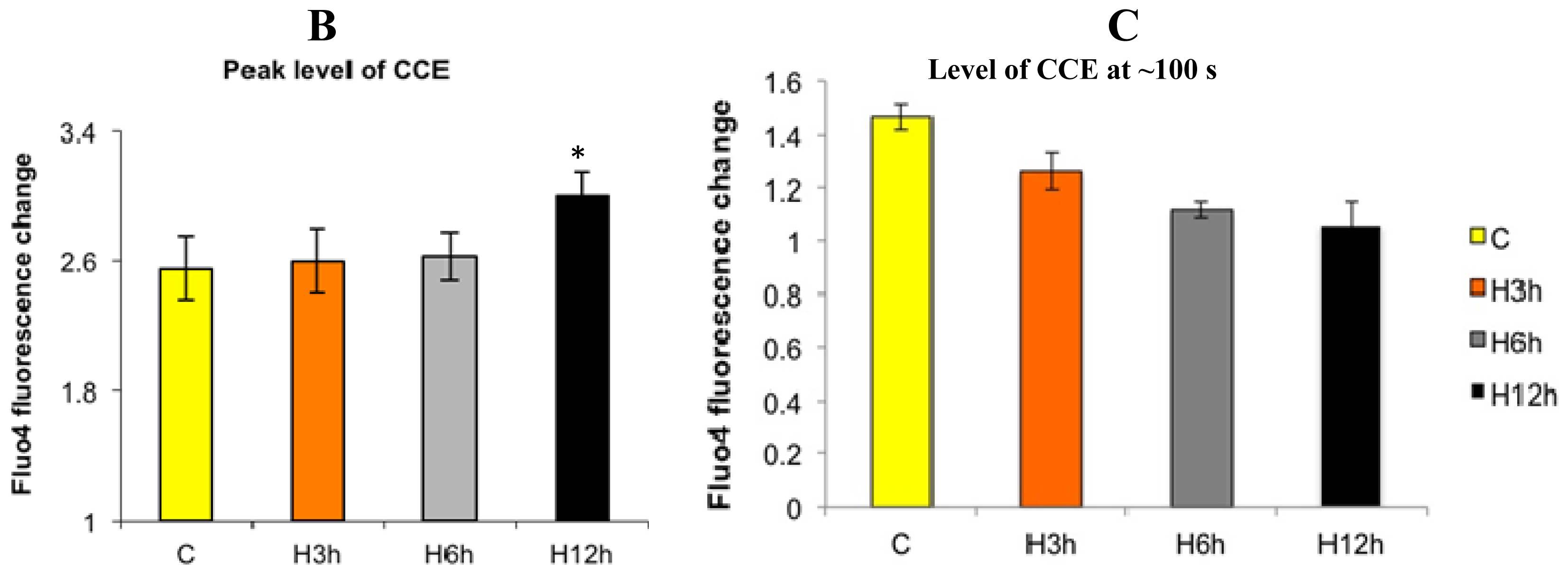
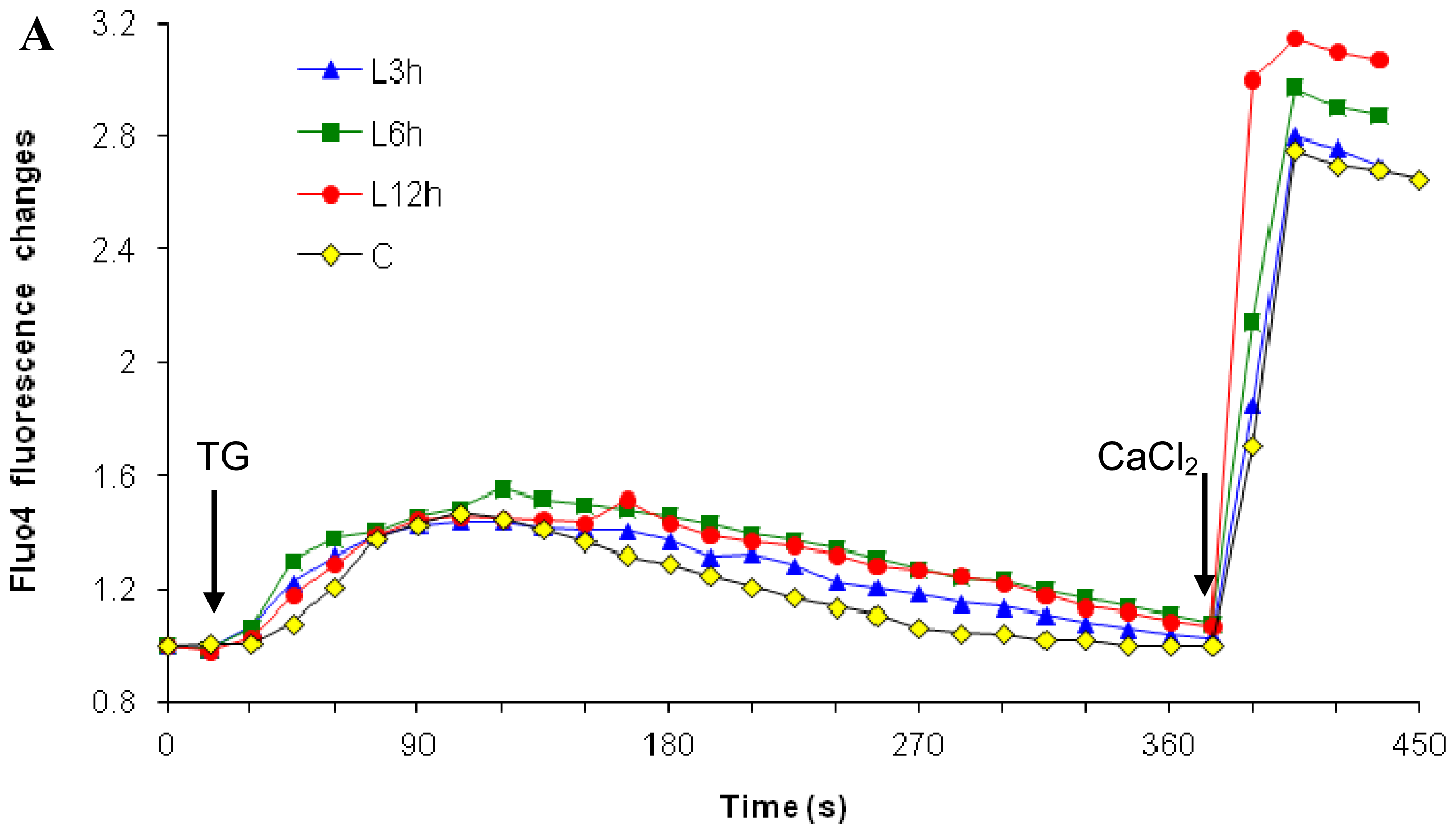
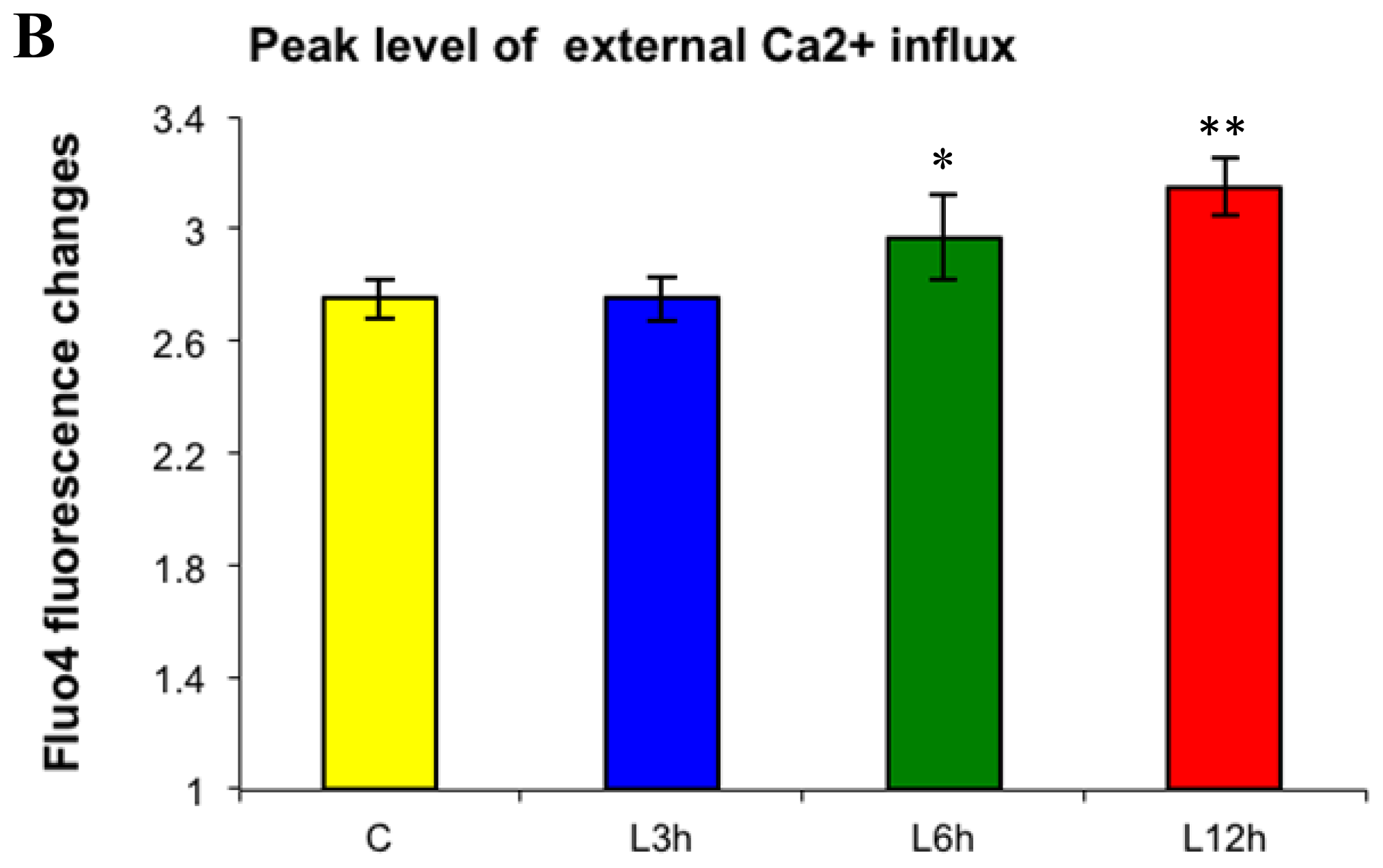
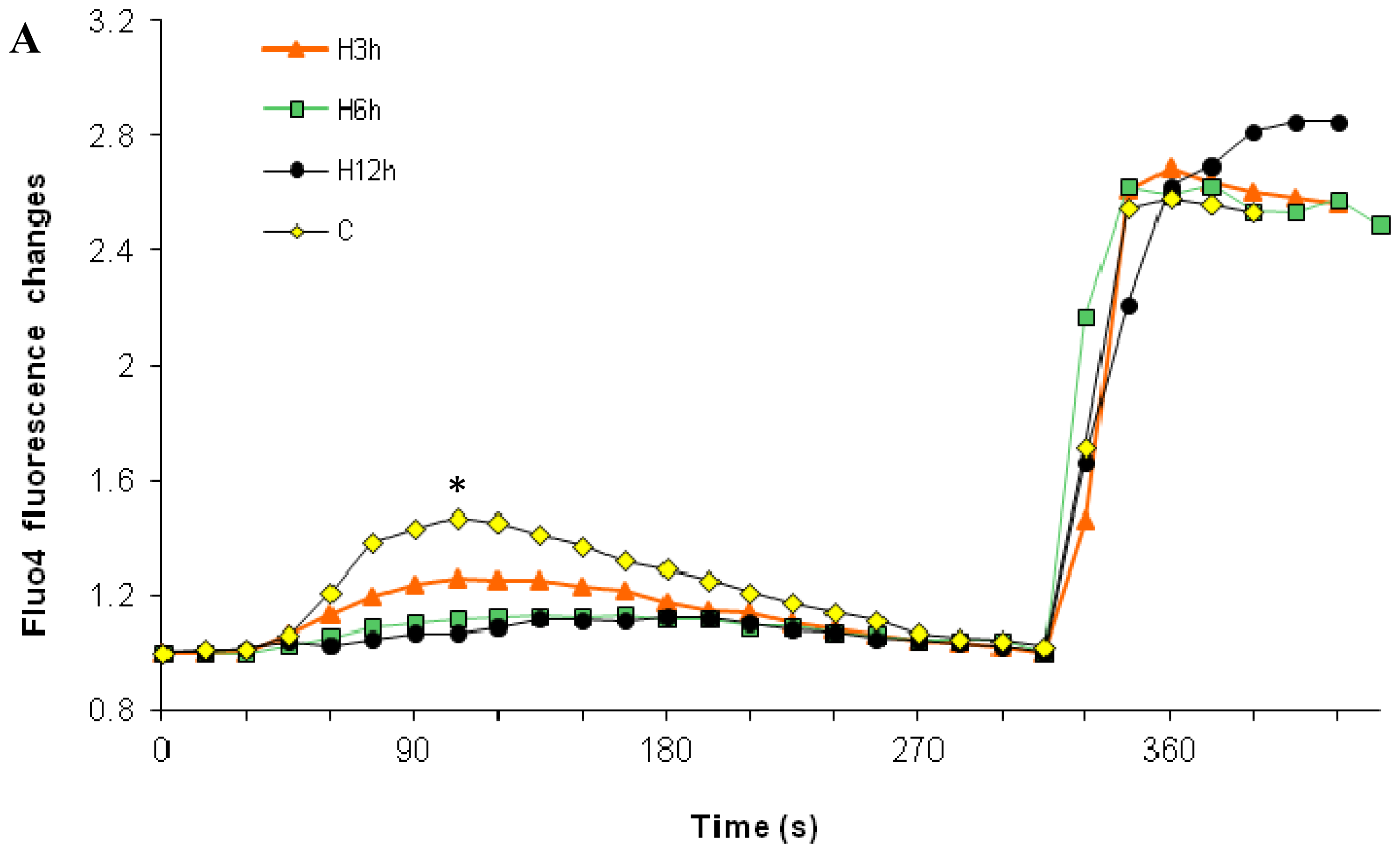
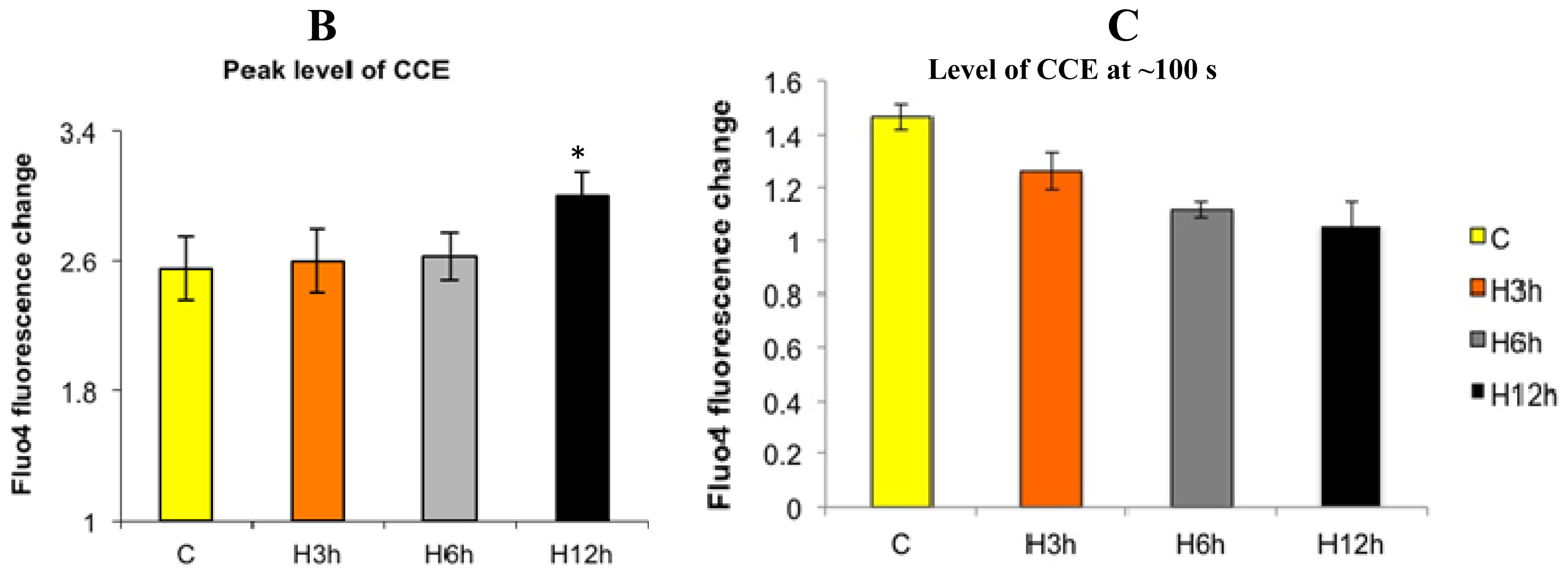
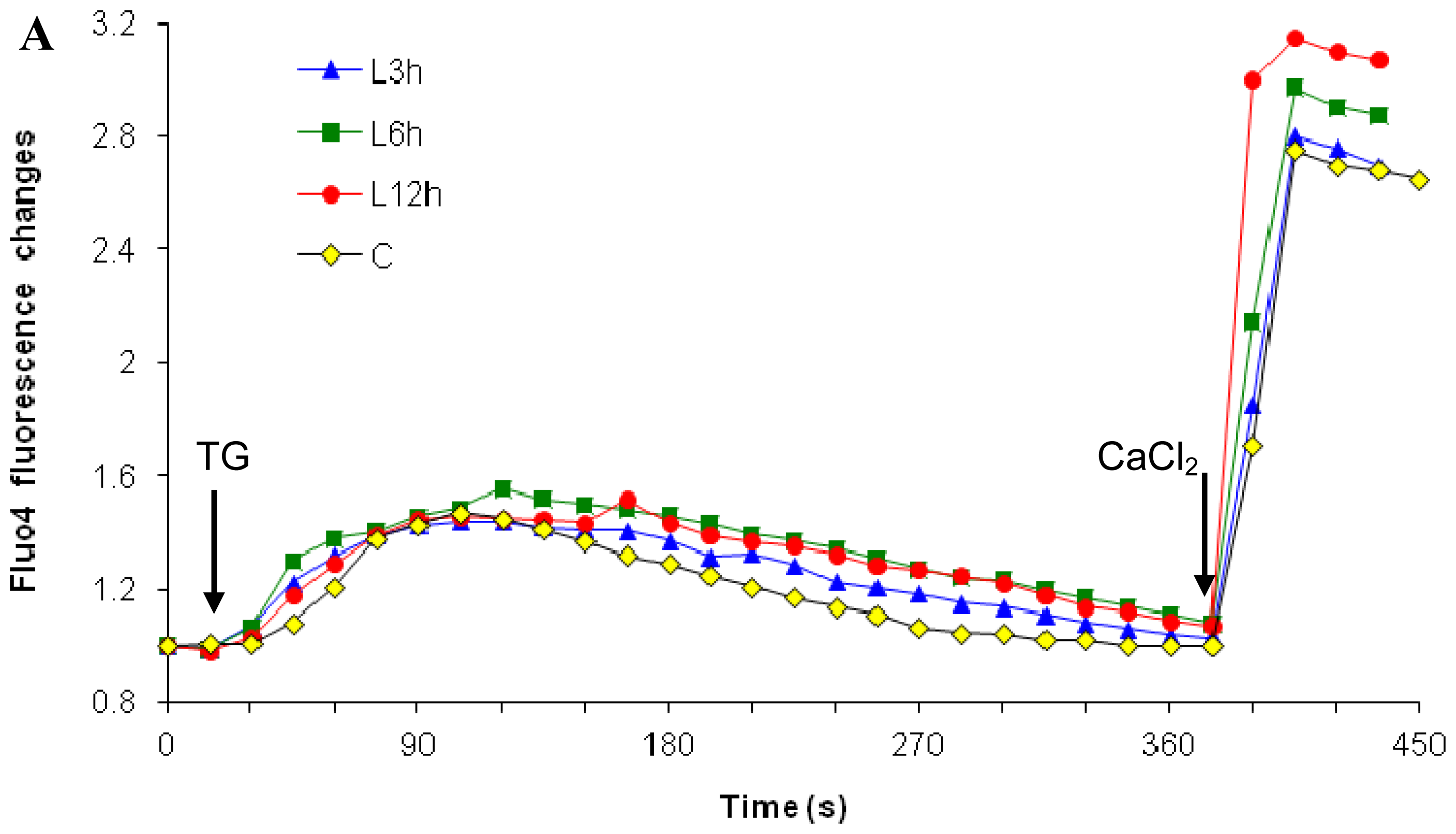
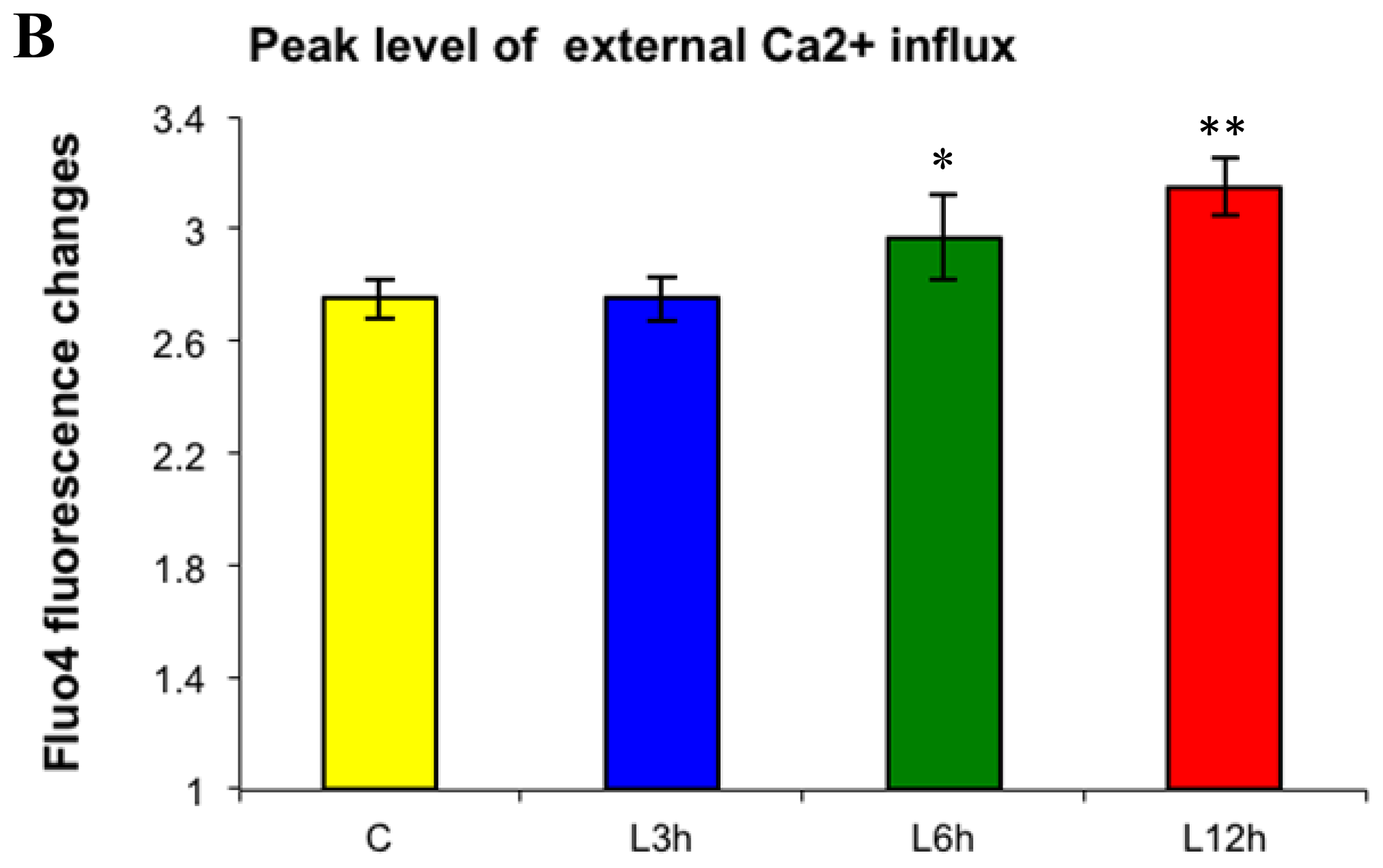
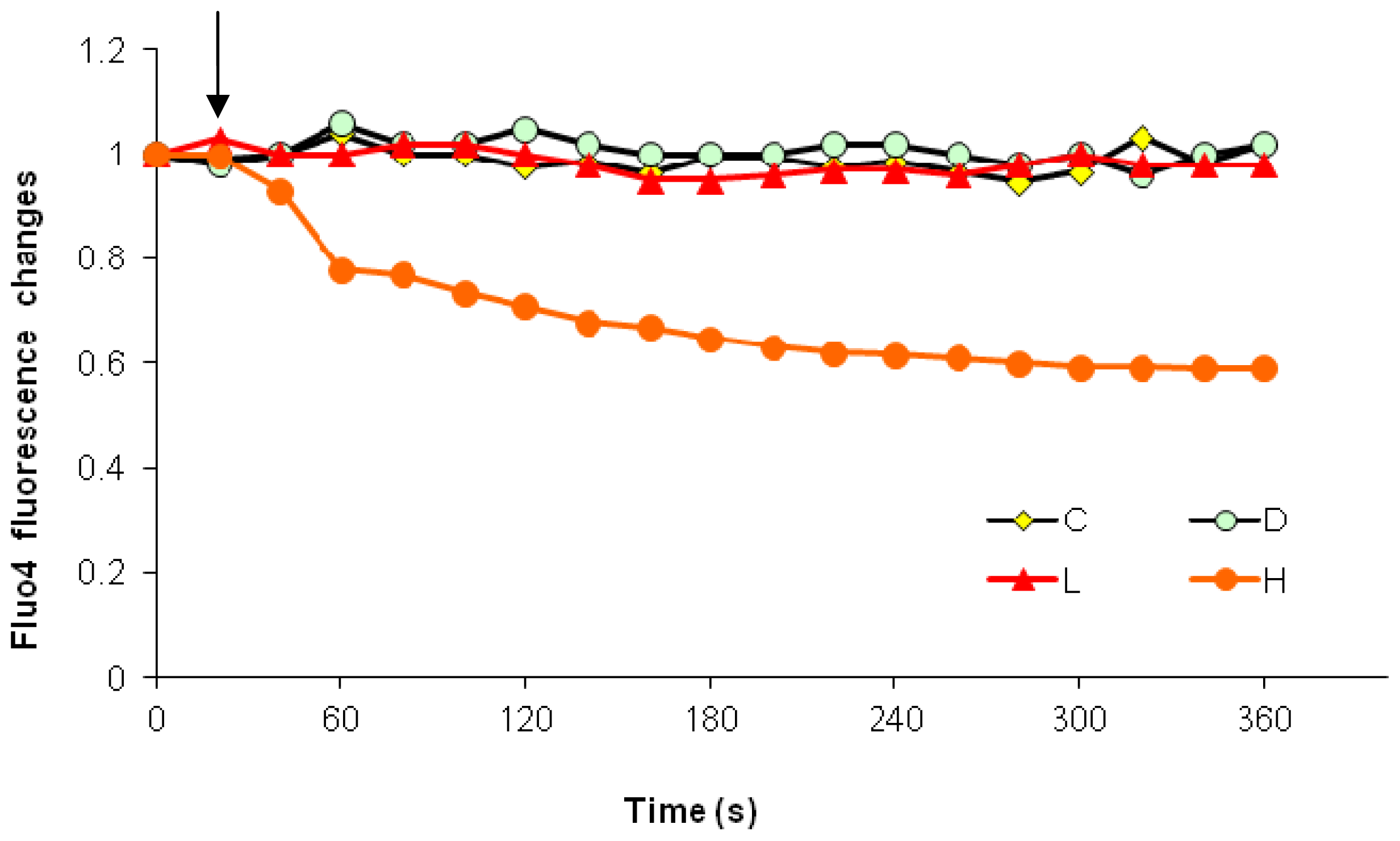
2.1. Effects of Paclitaxel on the CCE of the Cell Depends on Dosage
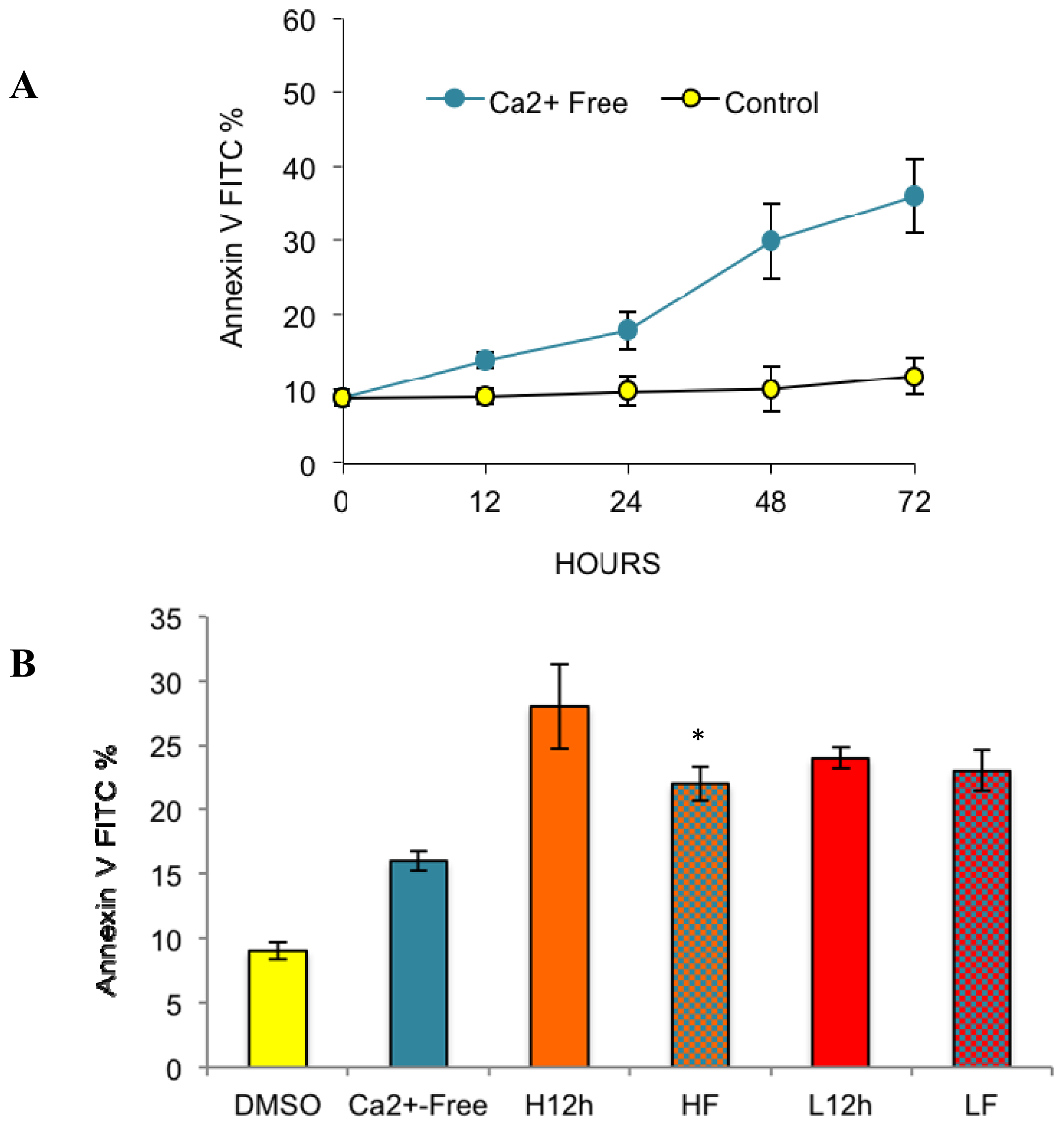
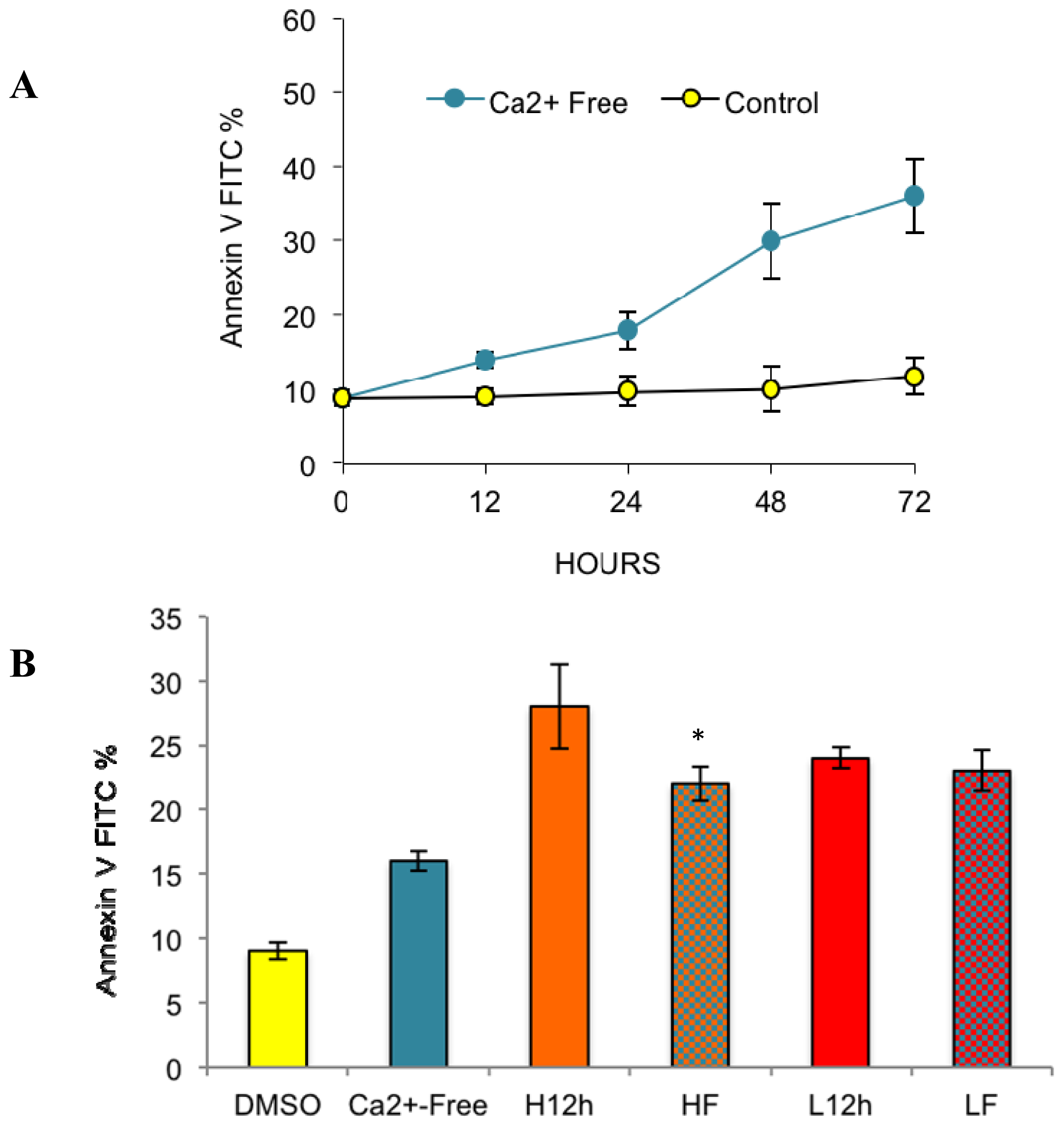
2.2. Effects of Removing External Calcium Influx on Subsequent Apoptosis Using Two Different Methods—The External Calcium Chelator BAPTA, and Calcium-Free Medium
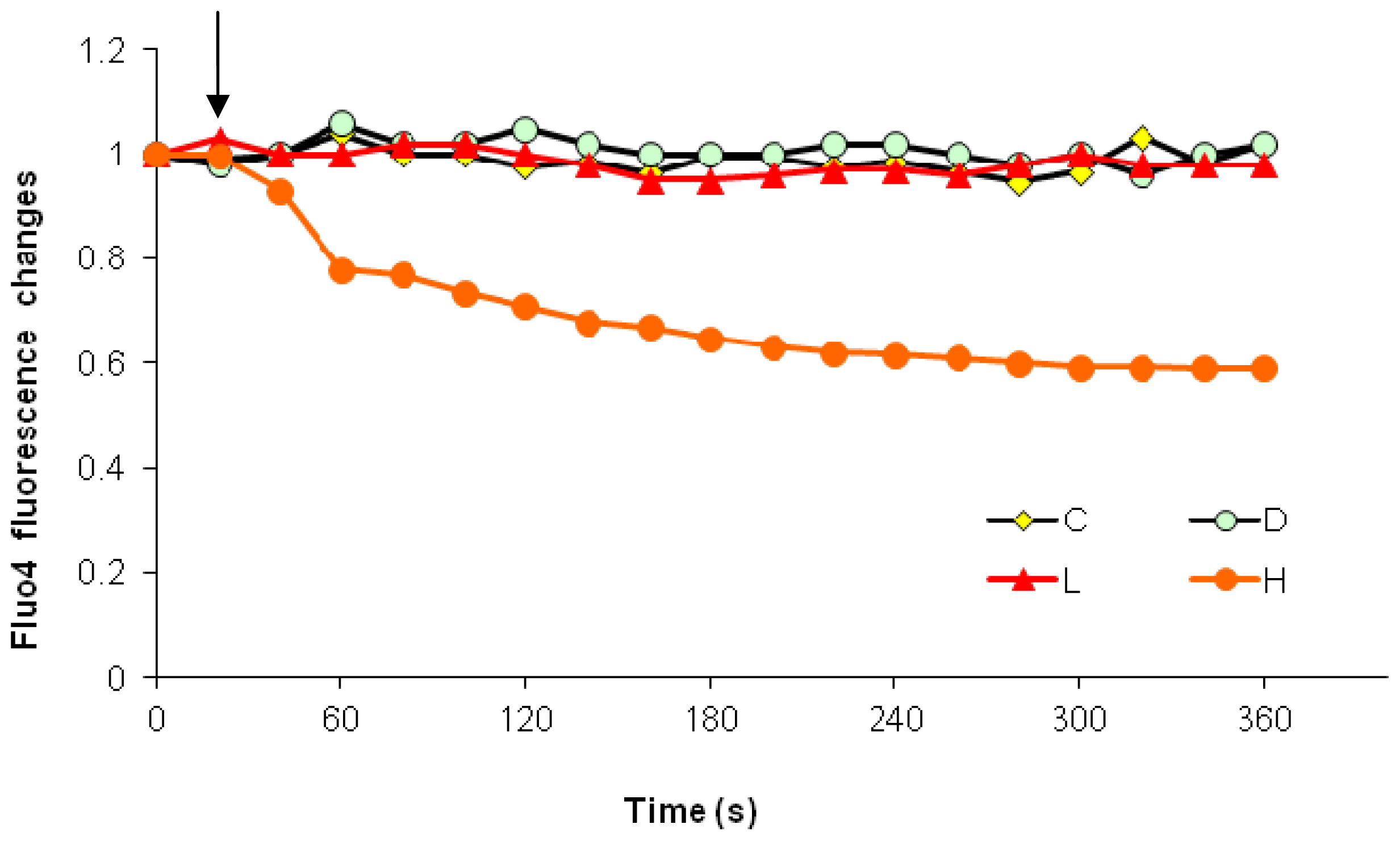
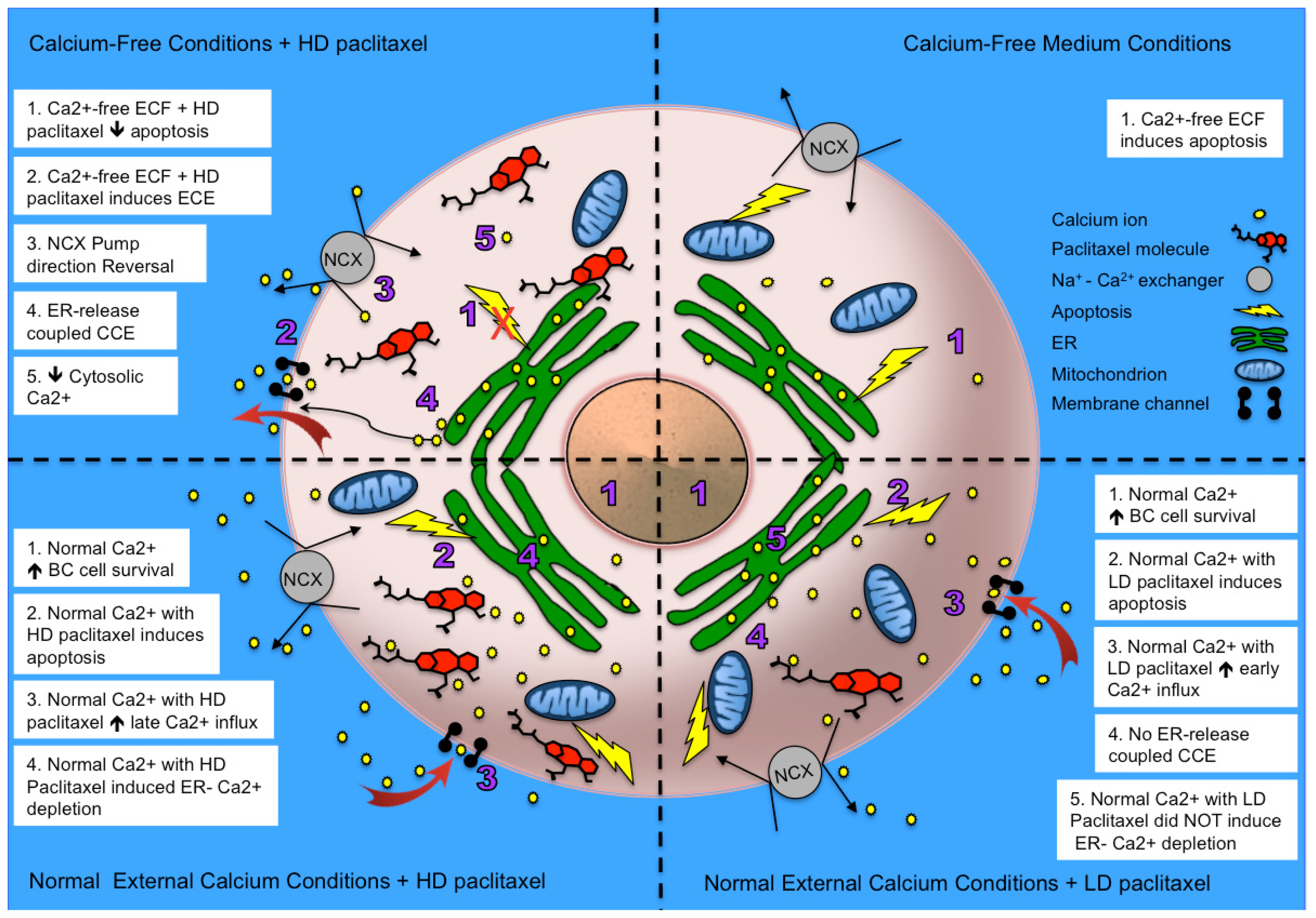
2.2.1. Paclitaxel Has a Different Calcium-Regulating Action in the Absence of External Calcium
2.3. Effects of the Absence of External Calcium on Paclitaxel-Induced Cytosolic Calcium Responses
2.4. Paclitaxel’s Different Calcium-Regulating Actions Affect Its Efficacy to Induce Apoptosis
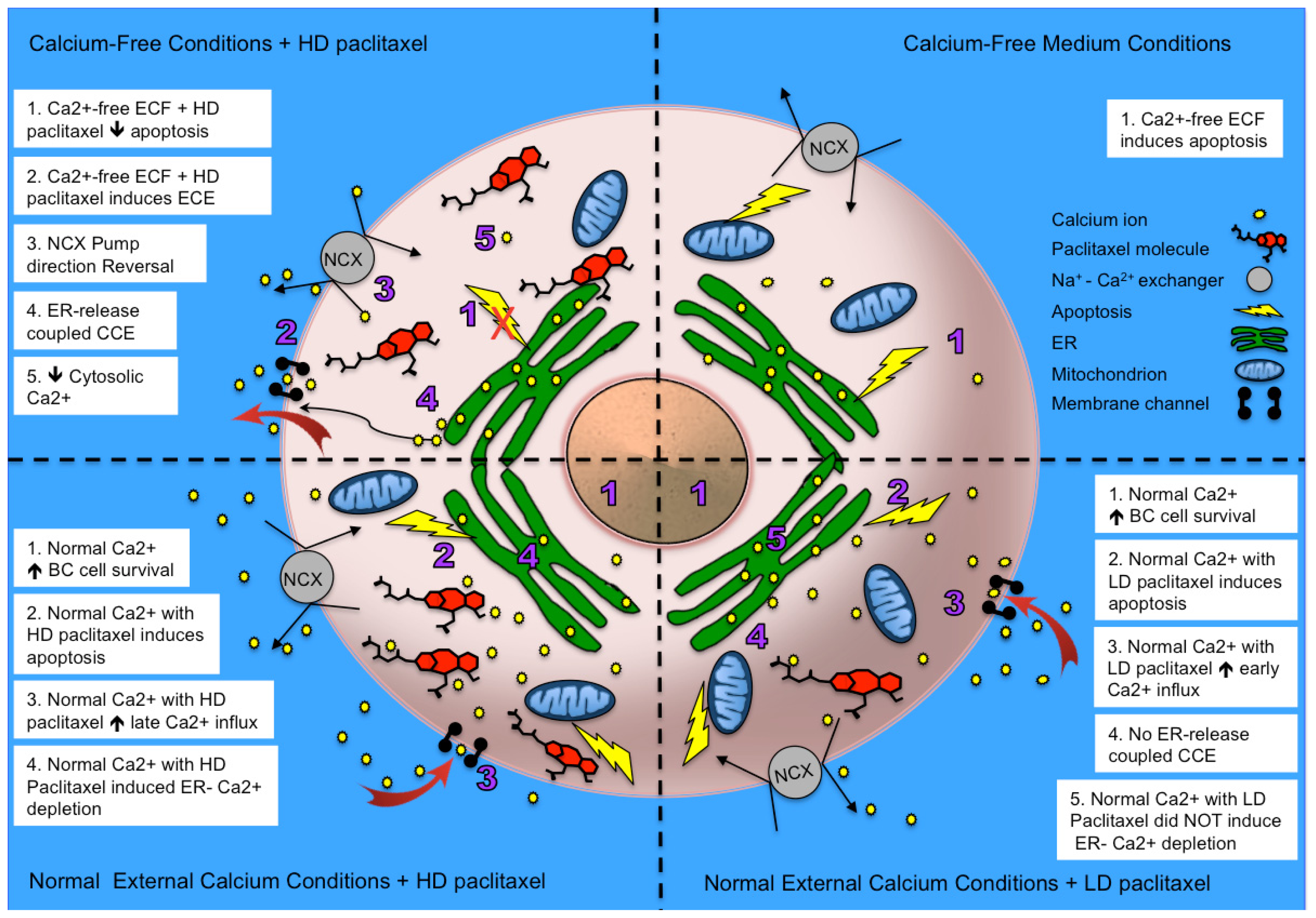
2.5. Alternative Explanations for Paclitaxel’s Different Calcium-Regulating Actions
3. Experimental Section
3.1. Cell Culture and Reagents
3.2. Paclitaxel Treatment
3.3. Apoptosis Measurements
3.4. Cytosolic Calcium Measurements
3.5. Capacitative Calcium Entry Measurements
3.6. Statistical Analyses
3.7. Source Code for Paclitaxel Docking Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Arnaudeau, S.; Kelley, W.L.; Walsh, J.V., Jr.; Demaurex, N. Mitochondria recycle Ca2+ to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J. Biol. Chem 2001, 276, 29430–29439. [Google Scholar]
- Cerella, C.; D’Alessio, M.; de Nicola, M.; Magrini, A.; Bergamaschi, A.; Ghibelli, L. Cytosolic and endoplasmic reticulum Ca2+ concentrations determine the extent and the morphological type of apoptosis, respectively. Ann. N. Y. Acad. Sci 2003, 1010, 74–77. [Google Scholar]
- Jackisch, C.; Hahm, H.A.; Tombal, B.; McCloskey, D.; Butash, K.; Davidson, N.E.; Denmeade, S.R. Delayed micromolar elevation in intracellular calcium precedes induction of apoptosis in thapsigargin-treated breast cancer cells. Clin. Cancer Res 2000, 6, 2844–2850. [Google Scholar]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol 2003, 4, 552–565. [Google Scholar]
- Rekasi, Z.; Czompoly, T.; Schally, A.V.; Boldizsar, F.; Varga, J.L.; Zarandi, M.; Berki, T.; Horvath, R.A.; Nemeth, P. Antagonist of growth hormone-releasing hormone induces apoptosis in LNCaP human prostate cancer cells through a Ca2+-dependent pathway. Proc. Nat. Acad. Sci. USA 2005, 102, 3435–3440. [Google Scholar]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar]
- Schmitt, C.A.; Lowe, S.W. Apoptosis is critical for drug response in vivo. Drug Resist. Updates 2001, 4, 132–134. [Google Scholar]
- Sledge, G.W., Jr.; Miller, K.D. Exploiting the hallmarks of cancer: The future conquest of breast cancer. Eur. J. Cancer 2003, 39, 1668–1675. [Google Scholar]
- Baffy, G.; Miyashita, T.; Williamson, J.R.; Reed, J.C. Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J. Biol. Chem 1993, 268, 6511–6519. [Google Scholar]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol 2003, 4, 517–529. [Google Scholar]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar]
- Demaurex, N.; Distelhorst, C. Cell biology. Apoptosis—The calcium connection. Science 2003, 300, 65–67. [Google Scholar]
- Hajnoczky, G.; Davies, E.; Madesh, M. Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun 2003, 304, 445–454. [Google Scholar]
- Kim, M.J.; Jo, D.G.; Hong, G.S.; Kim, B.J.; Lai, M.; Cho, D.H.; Kim, K.W.; Bandyopadhyay, A.; Hong, Y.M.; Kim, D.H.; et al. Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc. Nat. Acad. Sci. USA 2002, 99, 9870–9875. [Google Scholar]
- Mattson, M.P.; Chan, S.L. Calcium orchestrates apoptosis. Nat. Cell Biol 2003, 5, 1041–1043. [Google Scholar]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar]
- Pineiro, D.; Martin, M.E.; Guerra, N.; Salinas, M.; Gonzalez, V.M. Calpain inhibition stimulates caspase-dependent apoptosis induced by taxol in NIH3T3 cells. Exp. Cell Res 2007, 313, 369–379. [Google Scholar]
- Rizzuto, R.; Pinton, P.; Ferrari, D.; Chami, M.; Szabadkai, G.; Magalhaes, P.J.; di Virgilio, F.; Pozzan, T. Calcium and apoptosis: Facts and hypotheses. Oncogene 2003, 22, 8619–8627. [Google Scholar]
- Szabadkai, G.; Rizzuto, R. Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: More than just neighborhood? FEBS Lett 2004, 567, 111–115. [Google Scholar]
- Pan, Z.; Gollahon, L. Taxol directly induces endoplasmic reticulum-associated calcium changes that promote apoptosis in breast cancer cells. Breast J 2011, 17, 56–70. [Google Scholar]
- Blajeski, A.L.; Kottke, T.J.; Kaufmann, S.H. A multistep model for paclitaxel-induced apoptosis in human breast cancer cell lines. Exp. Cell Res 2001, 270, 277–288. [Google Scholar]
- Ofir, R.; Seidman, R.; Rabinski, T.; Krup, M.; Yavelsky, V.; Weinstein, Y.; Wolfson, M. Taxol-induced apoptosis in human SKOV3 ovarian and MCF7 breast carcinoma cells is caspase-3 and caspase-9 independent. Cell Death Differ 2002, 9, 636–642. [Google Scholar]
- Varbiro, G.; Veres, B.; Gallyas, F., Jr.; Sumegi, B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic. Biol. Med 2001, 31, 548–558. [Google Scholar]
- Kidd, J.F.; Pilkington, M.F.; Schell, M.J.; Fogarty, K.E.; Skepper, J.N.; Taylor, C.W.; Thorn, P. Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J. Biol. Chem 2002, 277, 6504–6510. [Google Scholar]
- Mironov, S.L.; Ivannikov, M.V.; Johansson, M. (Ca2+)i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules. From mitochondrial permeability transition pore to Ca2+-induced Ca2+ release. J. Biol. Chem 2005, 280, 715–721. [Google Scholar]
- Charles, A.G.; Han, T.Y.; Liu, Y.Y.; Hansen, N.; Giuliano, A.E.; Cabot, M.C. Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer Chem. Pharmacol 2001, 47, 444–450. [Google Scholar]
- Colina, C.; Flores, A.; Castillo, C.; del Garrido, M.R.; Israel, A.; DiPolo, R.; Benaim, G. Ceramide-1-P induces Ca2+ mobilization in Jurkat T-cells by elevation of Ins(1,4,5)-P3 and activation of a store-operated calcium channel. Biochem. Biophys. Res. Commun 2005, 336, 54–60. [Google Scholar]
- Baggott, R.R.; Mohamed, T.M.; Oceandy, D.; Holton, M.; Blanc, M.C.; Roux-Soro, S.C.; Brown, S.; Brown, J.E.; Cartwright, E.J.; Wang, W.; et al. Disruption of the interaction between PMCA2 and calcineurin triggers apoptosis and enhances paclitaxel-induced cytotoxicity in breast cancer cells. Carcinogenesis 2012, 33, 2362–2368. [Google Scholar]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J 2004, 23, 1207–1216. [Google Scholar]
- Brichese, L.; Barboule, N.; Heliez, C.; Valette, A. Bcl-2 phosphorylation and proteasome-dependent degradation induced by paclitaxel treatment: Consequences on sensitivity of isolated mitochondria to Bid. Exp. Cell Res 2002, 278, 101–111. [Google Scholar]
- Pan, Z.; Gollahon, L. Paclitaxel attenuates Bcl-2 resistance to apoptosis in breast cancer cells through an endoplasmic reticulum-mediated calcium release in a dosage dependent manner. Biochem. Biophys. Res. Commun 2013, 432, 431–437. [Google Scholar]
- Putney, J.W., Jr. Capacitative calcium entry: Sensing the calcium stores. J. Cell Biol 2005, 169, 381–382. [Google Scholar]
- Jayadev, S.; Petranka, J.G.; Cheran, S.K.; Biermann, J.A.; Barrett, J.C.; Murphy, E. Reduced capacitative calcium entry correlates with vesicle accumulation and apoptosis. J. Biol. Chem 1999, 274, 8261–8268. [Google Scholar]
- Mason, R.P. Calcium channel blockers, apoptosis and cancer: Is there a biologic relationship? J. Am. Coll. Cardiol 1999, 34, 1857–1866. [Google Scholar]
- Park, E.K.; Kwon, K.B.; Park, K.I.; Park, B.H.; Jhee, E.C. Role of Ca2+ in diallyl disulfide-induced apoptotic cell death of HCT-15 cells. Exp. Mol. Med 2002, 34, 250–257. [Google Scholar]
- Peppiatt, C.M.; Collins, T.J.; Mackenzie, L.; Conway, S.J.; Holmes, A.B.; Bootman, M.D.; Berridge, M.J.; Seo, J.T.; Roderick, H.L. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 2003, 34, 97–108. [Google Scholar]
- Pigozzi, D.; Ducret, T.; Tajeddine, N.; Gala, J.L.; Tombal, B.; Gailly, P. Calcium store contents control the expression of TRPC1, TRPC3 and TRPV6 proteins in LNCaP prostate cancer cell line. Cell Calcium 2006, 39, 401–415. [Google Scholar]
- Turman, M.A.; Bates, C.M.; Mathews, A.; Haun, S.E. Effect of extracellular calcium on survival of human proximal tubular cells exposed to hypoxia. Ren. Fail 1995, 17, 421–435. [Google Scholar]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarden, Y.; Seger, R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 2001, 20, 147–155. [Google Scholar]
- Seidman, R.; Gitelman, I.; Sagi, O.; Horwitz, S.B.; Wolfson, M. The role of ERK 1/2 and p38 MAP-kinase pathways in taxol-induced apoptosis in human ovarian carcinoma cells. Exp. Cell Res 2001, 268, 84–92. [Google Scholar]
- Selimovic, D.; Hassan, M.; Haikel, Y.; Hengge, U.R. Taxol-induced mitochondrial stress in melanoma cells is mediated by activation of c-Jun N-terminal kinase (JNK) and p38 pathways via uncoupling protein 2. Cell Signal 2008, 20, 311–322. [Google Scholar]
- Hassan, M.; Alaoui, A.; Feyen, O.; Mirmohammadsadegh, A.; Essmann, F.; Tannapfel, A.; Gulbins, E.; Schulze-Osthoff, K.; Hengge, U.R. The BH3-only member Noxa causes apoptosis in melanoma cells by multiple pathways. Oncogene 2008, 27, 4557–4568. [Google Scholar]
- Heath-Engel, H.M.; Chang, N.C.; Shore, G.C. The endoplasmic reticulum in apoptosis and autophagy: Role of the BCL-2 protein family. Oncogene 2008, 27, 6419–6433. [Google Scholar]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev 2007, 87, 593–658. [Google Scholar]
- Li, C.; Wang, X.; Vais, H.; Thompson, C.B.; Foskett, J.K.; White, C. Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc. Natl. Acad. Sci. USA 2007, 104, 12565–12570. [Google Scholar]
- Xu, L.; Kong, D.; Zhu, L.; Zhu, W.; Andrews, D.W.; Kuo, T.H. Suppression of IP3-mediated calcium release and apoptosis by Bcl-2 involves the participation of protein phosphatase 1. Mol. Cell. Biochem 2007, 295, 153–165. [Google Scholar]
- Li, N.; Lin, P.; Cai, C.; Pan, Z.; Weisleder, N.; Ma, J. The amino-terminal peptide of Bax perturbs intracellular Ca2+ homeostasis to enhance apoptosis in prostate cancer cells. Am. J. Physiol. Cell Physiol 2009, 296, C267–C272. [Google Scholar]
- Ionescu, L.; White, C.; Cheung, K.H.; Shuai, J.; Parker, I.; Pearson, J.E.; Foskett, J.K.; Mak, D.O. Mode switching is the major mechanism of ligand regulation of InsP3 receptor calcium release channels. J. Gen. Physiol 2007, 130, 631–645. [Google Scholar]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov 2008, 7, 1013–1030. [Google Scholar]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol 2001, 21, 1249–1259. [Google Scholar]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar]
- Liao, P.-C.; Tan, S.; Lieu, C.-H.; Jung, H.-K. Crosstalk between Endoplasmic Reticulum and Mitochondria-Dependent Death Pathways in Paclitaxel-Induced Apoptosis. Available online: http://www.ym.edu.tw/rnd/phdconf/reports/2006/%B9%F9%AF%5C%A7g/%B9%F9%AF%5C%A7g%20%A5%FE%A4%E5.pdf (accessed on 12 December 2013).
- Liao, P.C.; Tan, S.K.; Lieu, C.H.; Jung, H.K. Involvement of endoplasmic reticulum in paclitaxel-induced apoptosis. J. Cell. Biochem 2008, 104, 1509–1523. [Google Scholar]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; de Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol 2011. [Google Scholar] [CrossRef]
- Venkatachalam, K.; van Rossum, D.B.; Patterson, R.L.; Ma, H.-T.; Gill, D.L. The cellular and molecular basis of store-operated calcium entry. Nat. Cell Biol 2002, 4, E263–E272. [Google Scholar]
- Baldi, C.; Vazquez, G.; Boland, R. Capacitative calcium influx in human epithelial breast cancer and non-tumorigenic cells occurs through Ca2+ entry pathways with different permeabilities to divalent cations. J. Cell. Biochem 2003, 88, 1265–1272. [Google Scholar]
- Mandic, A.; Viktorsson, K.; Strandberg, L.; Heiden, T.; Hansson, J.; Linder, S.; Shoshan, M.C. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: Two separate pathways in cisplatin-induced apoptosis. Mol. Cell. Biol 2002, 22, 3003–3013. [Google Scholar]
- Neville, M.C. Calcium secretion into milk. J. Mammary Gland Biol. Neoplasia 2005, 10, 119–128. [Google Scholar]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev 2005, 85, 757–810. [Google Scholar]
- Carafoli, E.; Santella, L.; Branca, D.; Brini, M. Generation, control, and processing of cellular calcium signals. Crit. Rev. Biochem. Mol. Biol 2001, 36, 107–260. [Google Scholar]
- Wang, N.S.; Unkila, M.T.; Reineks, E.Z.; Distelhorst, C.W. Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J. Biol. Chem 2001, 276, 44117–44128. [Google Scholar]
- Palmer, A.E.; Tsien, R.Y. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Prot 2006, 1, 1057–1065. [Google Scholar]
- Squibb, B.-M. Taxol (Paclitaxel) Injection Product Information. Available online: http://packageinserts.bms.com/pi/pi_taxol.pdf (accessed on 15 August 2011).
- Ball, M.; Andrews, S.P.; Wierschem, F.; Cleator, E.; Smith, M.D.; Ley, S.V. Total synthesis of thapsigargin, a potent SERCA pump inhibitor. Org. Lett 2007, 9, 663–666. [Google Scholar]
- Toyoshima, C.; Nomura, H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 2002, 418, 605–611. [Google Scholar]
- Guerini, D.; Guidi, F.; Carafoli, E. Differential membrane targeting of the SERCA and PMCA calcium pumps: Experiments with recombinant chimeras. FASEB J 2002, 16, 519–528. [Google Scholar]
- Brandt, P.C.; Vanaman, T.C. The plasma membrane calcium pump: Not just another pretty ion translocase. Glycobiology 1996, 6, 665–668. [Google Scholar]
- Treiman, M.; Caspersen, C.; Christensen, S.B. A tool coming of age: Thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol. Sci 1998, 19, 131–135. [Google Scholar]
- Uehara, A.; Iwamoto, T.; Nakamura, Y.; Imanaga, I. Forefront of Na+/Ca2+ exchanger studies: Physiology and molecular biology of monovalent cation sensitivities in Na+/Ca2+ exchangers. J. Pharmacol. Sci 2004, 96, 19–22. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Priority Docking Number | RCSB_ID | Energy (kcal/mol) | Species | Protein |
|---|---|---|---|---|
| 1 | 2ekg | −10.5 | Thermus thermophilus | flavoenzyme proline dehydrogenase |
| 2 | 2h7m | −10.4 | Mycobacterium tuberculosis | enoyl reductase |
| 3 | 2i6x | −10.3 | Porphyromonas gingivalis | hydrolase |
| 4 | 3ek2 | −10.3 | Burkholderia pseudomallei | eonyl reductase |
| 5 | 3g7w | −9.9 | Homo sapiens | Islet Amyloid Polypeptide |
| 6 | 2xn5 | −9.9 | Homo sapiens | Thyroxine-Binding Globulin |
| 7 | 3i6i | −9.9 | Vitis vinifera | leucoanthocyanidin reductase |
| 8 | 1hvb | −9.7 | Streptomyces | DD-Peptidase |
| 9 | 3i6o | −9.6 | HIV-1 | HIV-1 protease |
| 10 | 1hv8 | −9.6 | Hyperthermophile Methanococcus Jannaschii | Dead Box Protein |
| 11 | 2prb | −9.6 | Salmonella typhimurium | coenzyme A |
| 12 | 1ekk | −9.6 | Bacillus subtilis | Hydroxyethylthiazole Kinase |
| 13 | 1xpj | −9.5 | Vibrio cholerae | MCSG Target APC26283 |
| 14 | 3h2h | −9.4 | Xanthomonas oryzae | esterase LipA |
| 15 | 1xfs | −9.6 | Nitrosomonas europaea | Protein NE0264 |
| 16 | 1set | −9.5 | Thermus thermophilus | Seryl-TRNA Synthase |
| 17 | 2h7x | −9.3 | Streptomyces venezuelae | Pikromycin Thioesterase |
| 18 | 3hv5 | −9.3 | Homo sapiens | p38 MAP Kinase |
| 19 | 2xff | −9.3 | Hordeum vulgare | Beta-Amylase |
| 20 | 1yp3 | −9.7 | Solanum tuberosum | ADP-glucose pyrophosphorylase |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pan, Z.; Avila, A.; Gollahon, L. Paclitaxel Induces Apoptosis in Breast Cancer Cells through Different Calcium—Regulating Mechanisms Depending on External Calcium Conditions. Int. J. Mol. Sci. 2014, 15, 2672-2694. https://doi.org/10.3390/ijms15022672
Pan Z, Avila A, Gollahon L. Paclitaxel Induces Apoptosis in Breast Cancer Cells through Different Calcium—Regulating Mechanisms Depending on External Calcium Conditions. International Journal of Molecular Sciences. 2014; 15(2):2672-2694. https://doi.org/10.3390/ijms15022672
Chicago/Turabian StylePan, Zhi, Andrew Avila, and Lauren Gollahon. 2014. "Paclitaxel Induces Apoptosis in Breast Cancer Cells through Different Calcium—Regulating Mechanisms Depending on External Calcium Conditions" International Journal of Molecular Sciences 15, no. 2: 2672-2694. https://doi.org/10.3390/ijms15022672