shRNA-Mediated XRCC2 Gene Knockdown Efficiently Sensitizes Colon Tumor Cells to X-ray Irradiation in Vitro and in Vivo



Abstract
:
1. Introduction
2. Results
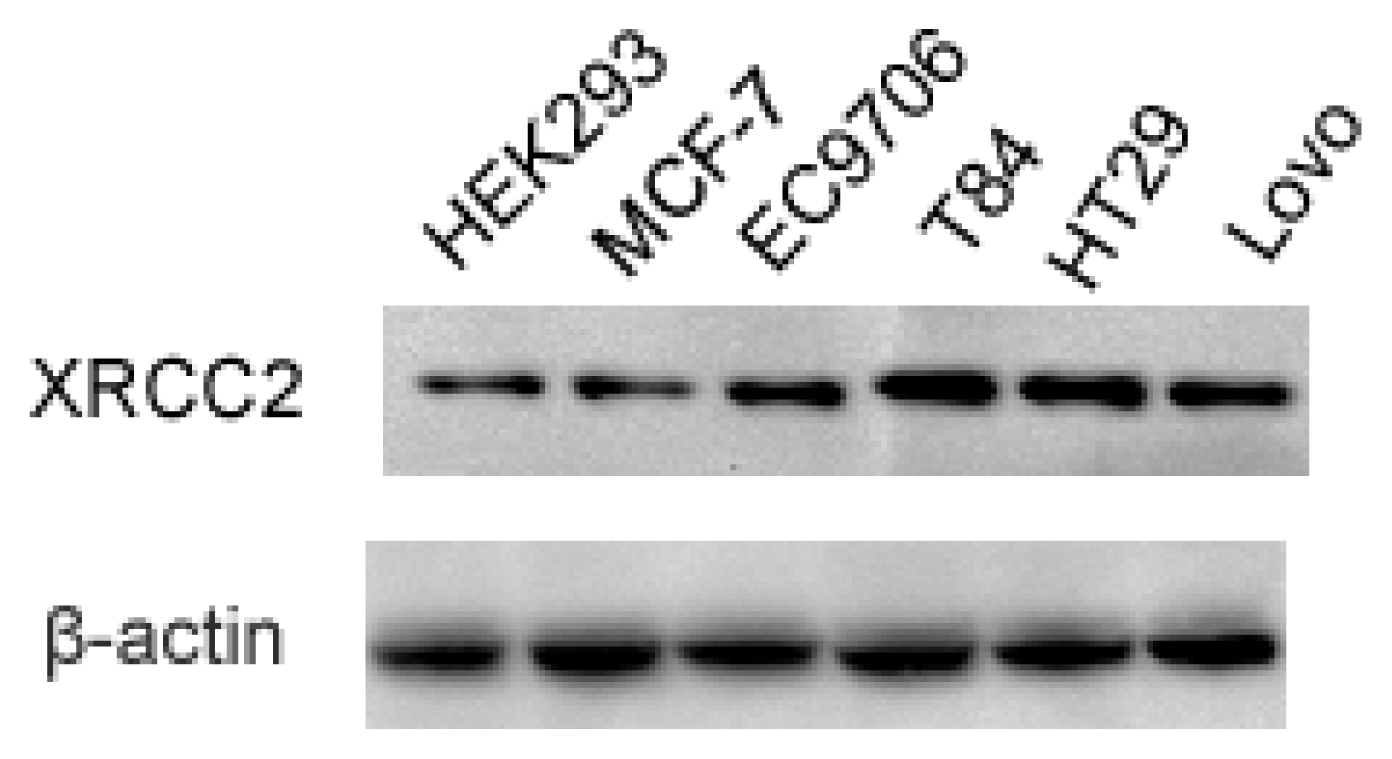
2.1. Expression of XRCC2 in Various Tumor Cell Lines and Normal Cell Line
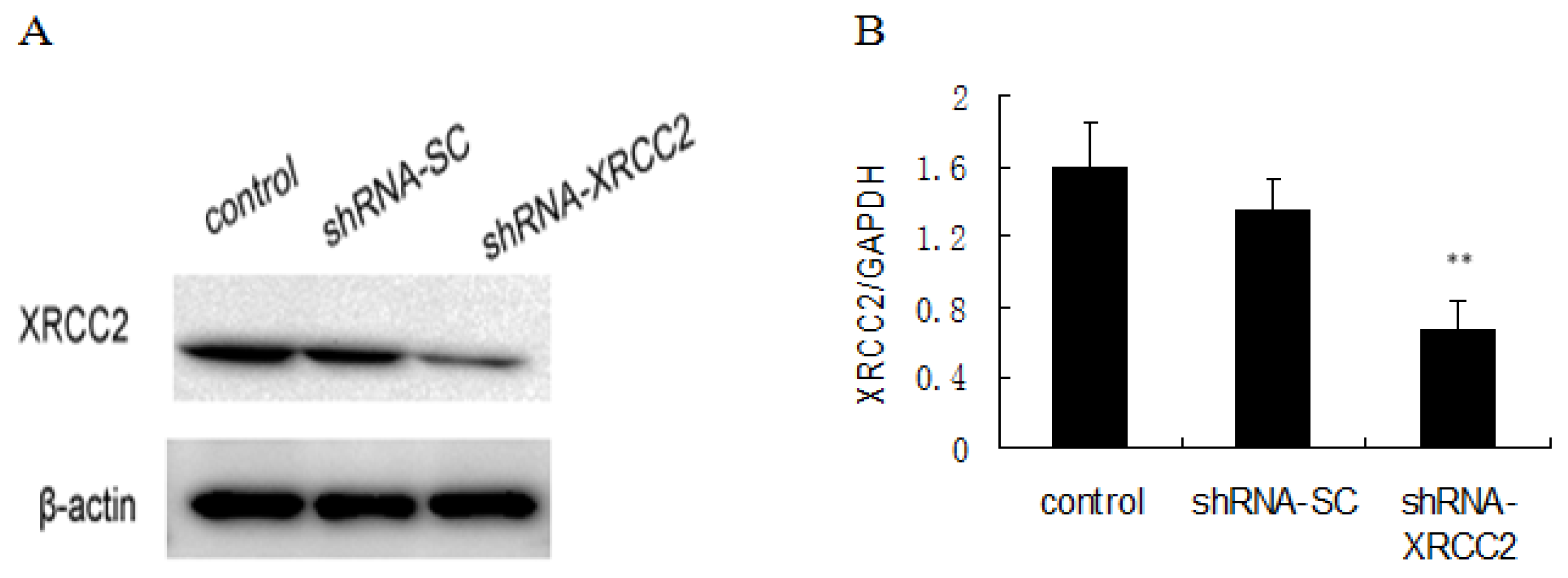
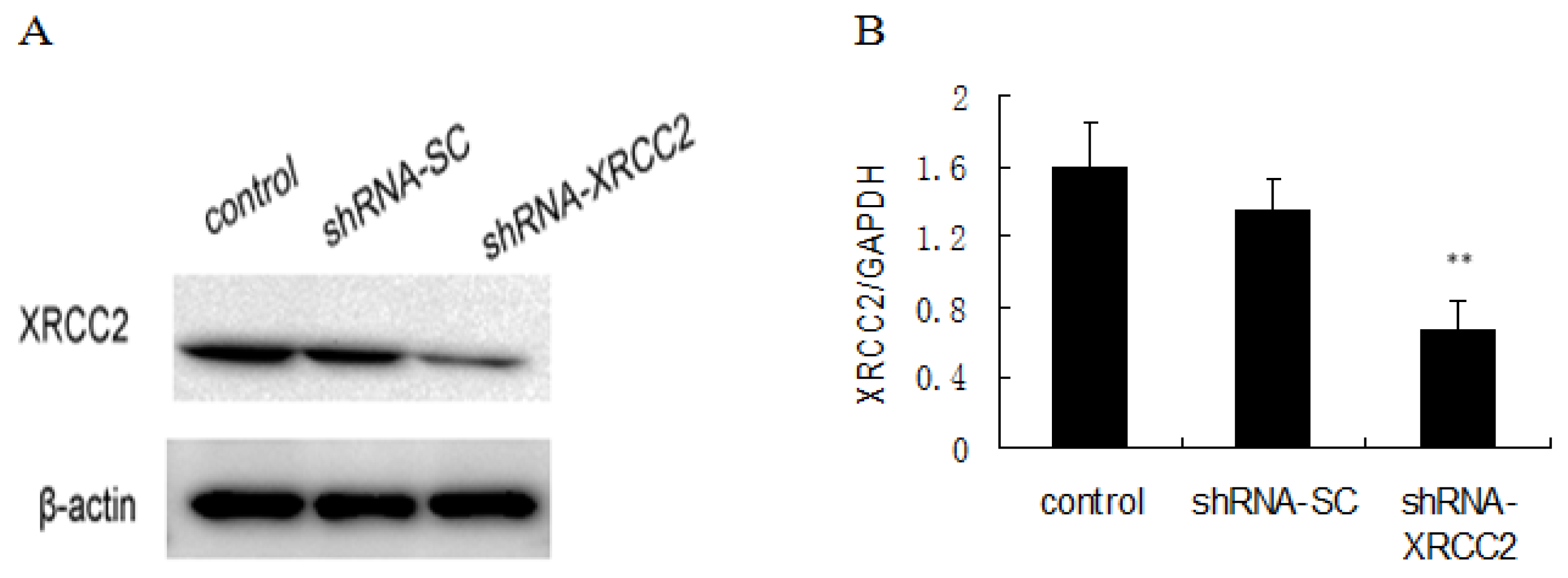
2.2. Knockdown of XRCC2 Using Vector-Based shRNA in T84 Cells
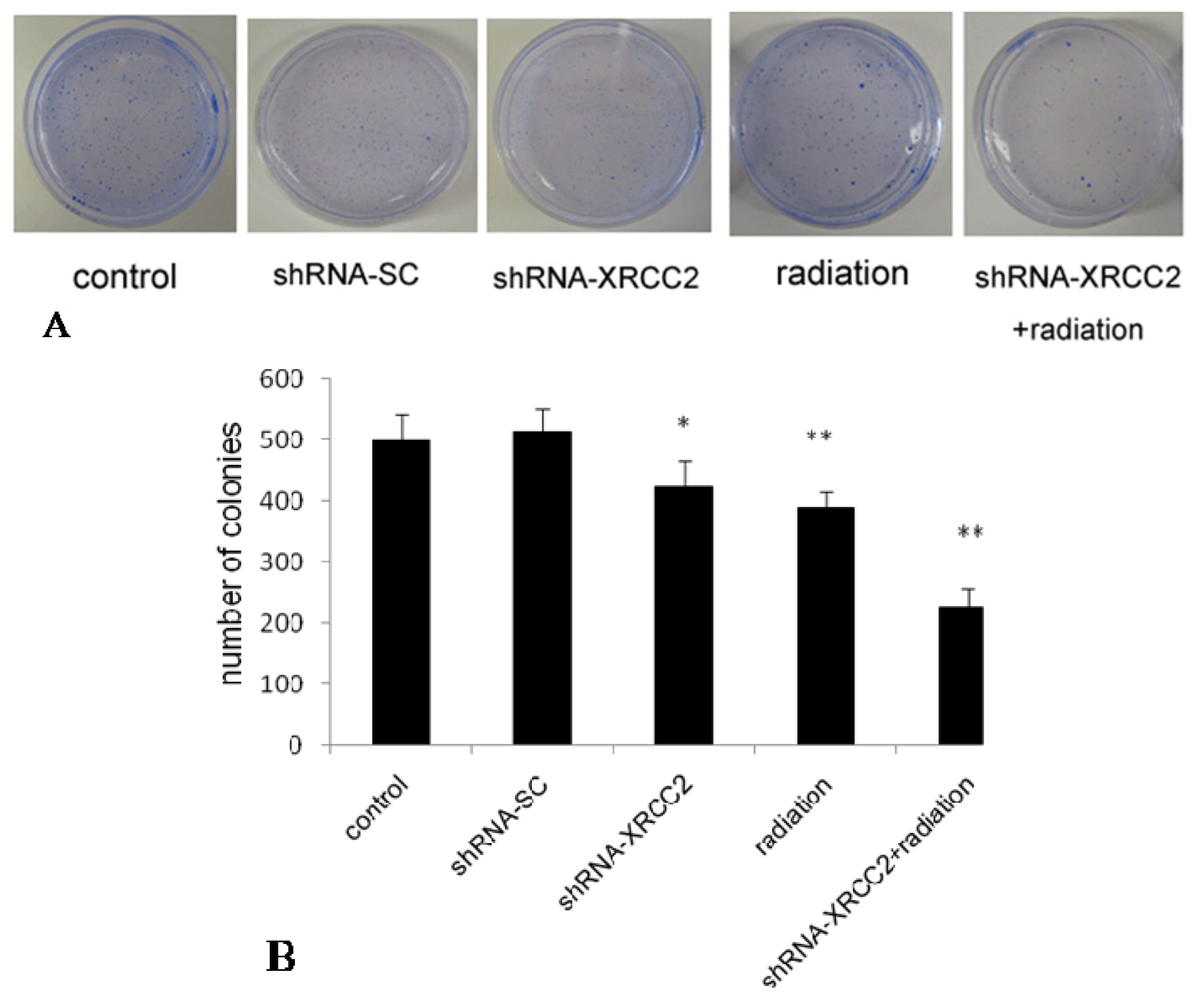
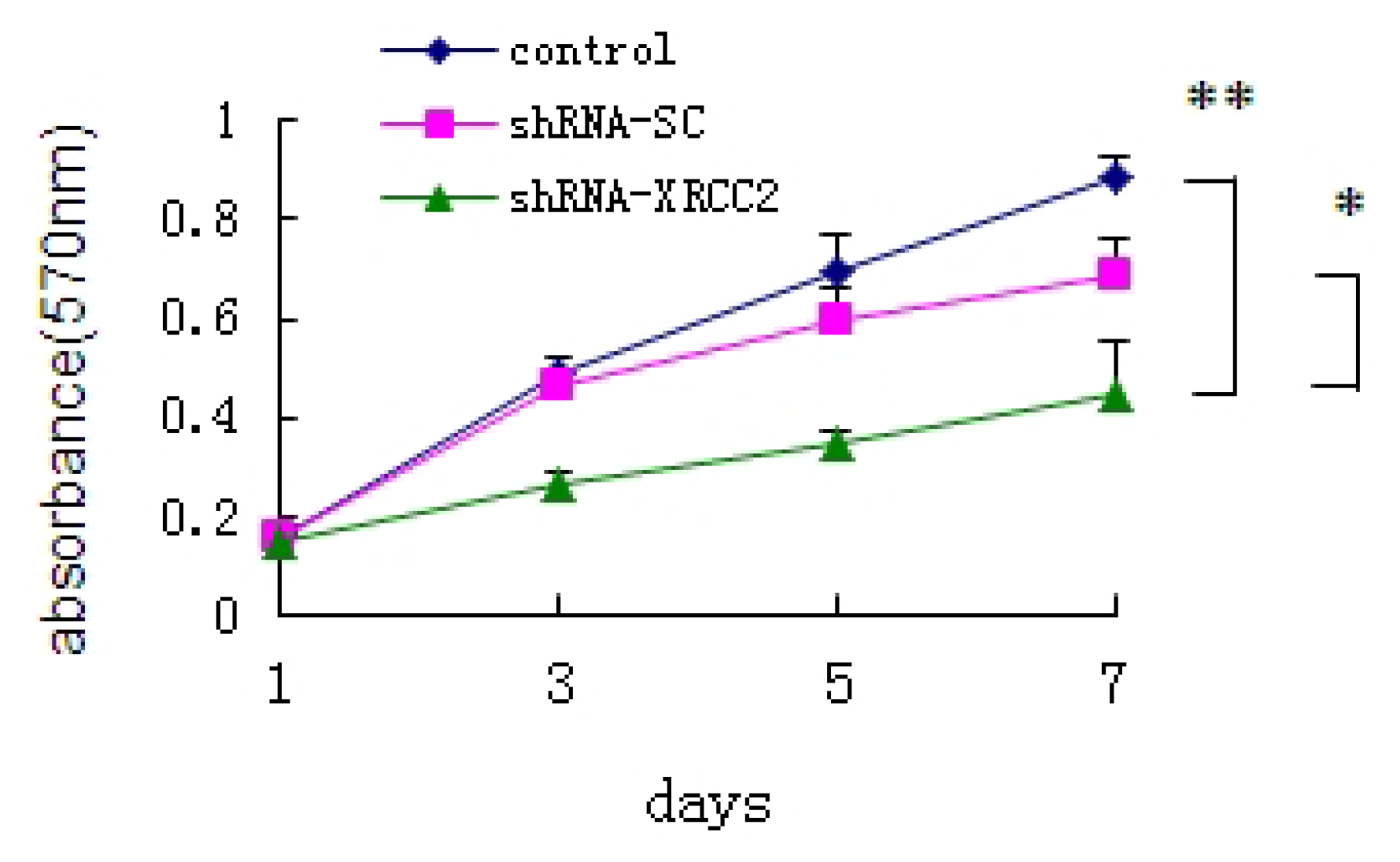
2.3. Effect of XRCC2 Knockdown on Cell Growth of T84 Cells
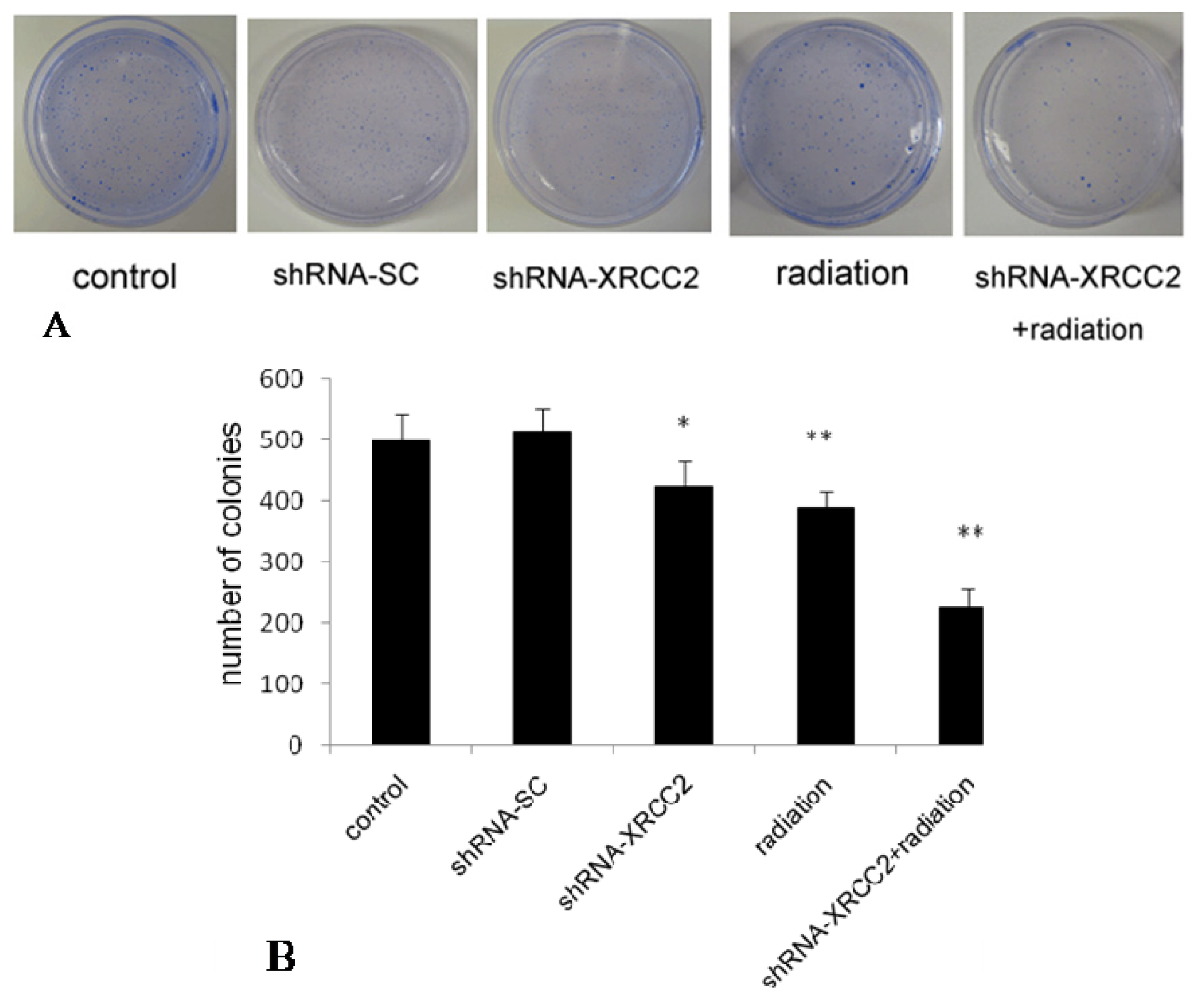
2.4. Effect of XRCC2 Knockdown on the Response of T84 Cells to X-Radiation
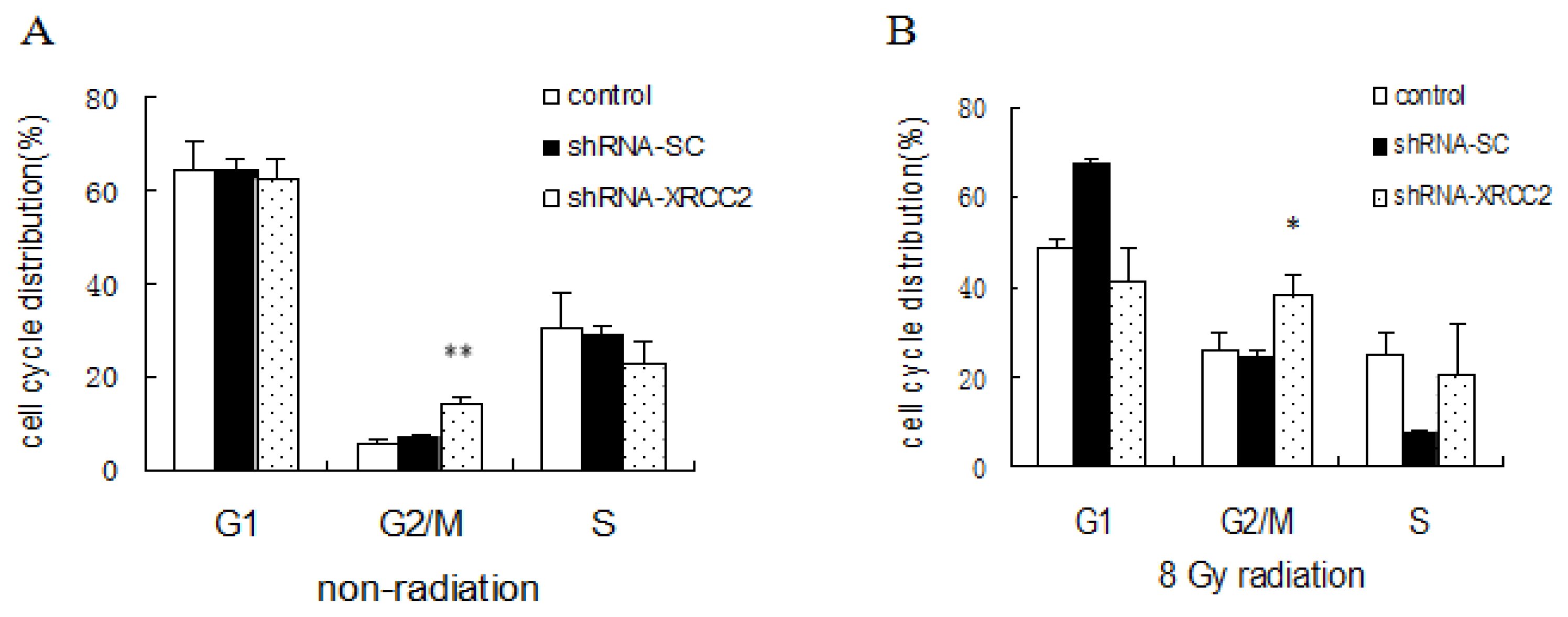
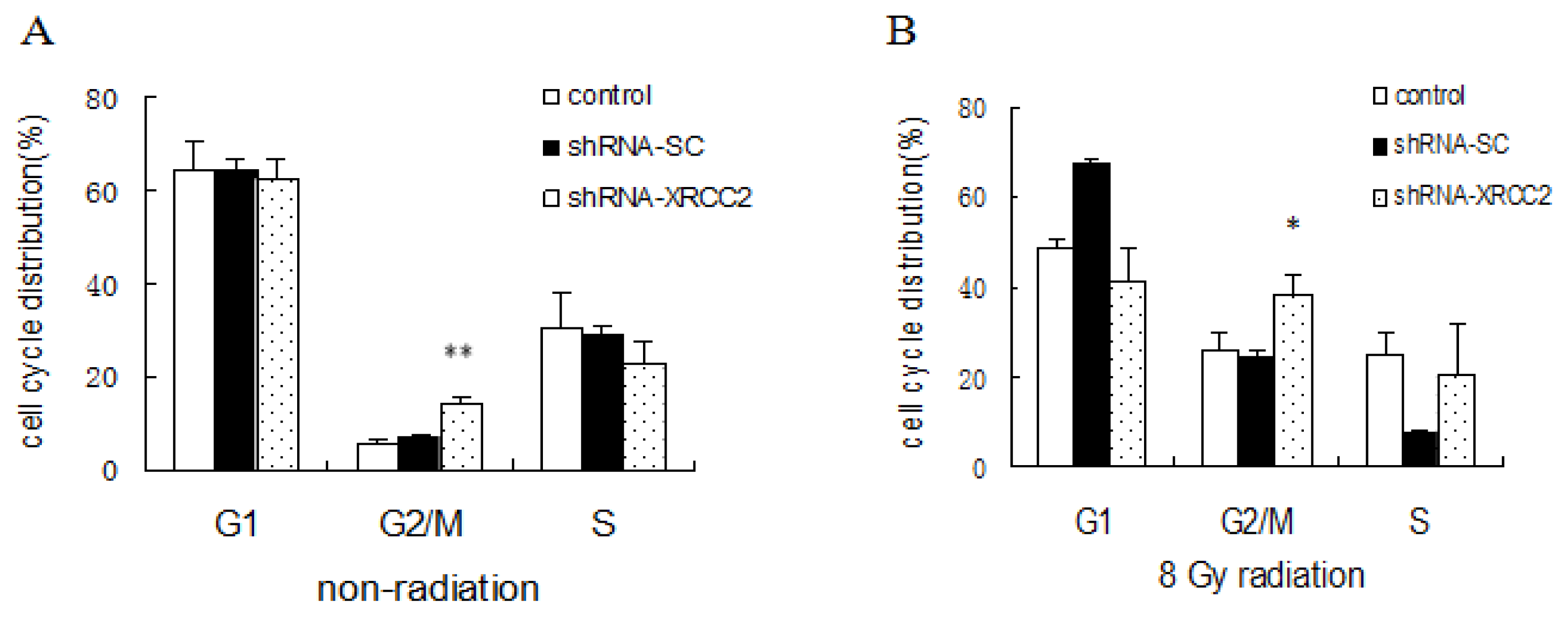
2.5. Effect of XRCC2 Knockdown on Cell Cycle Distribution Induced by Radiation
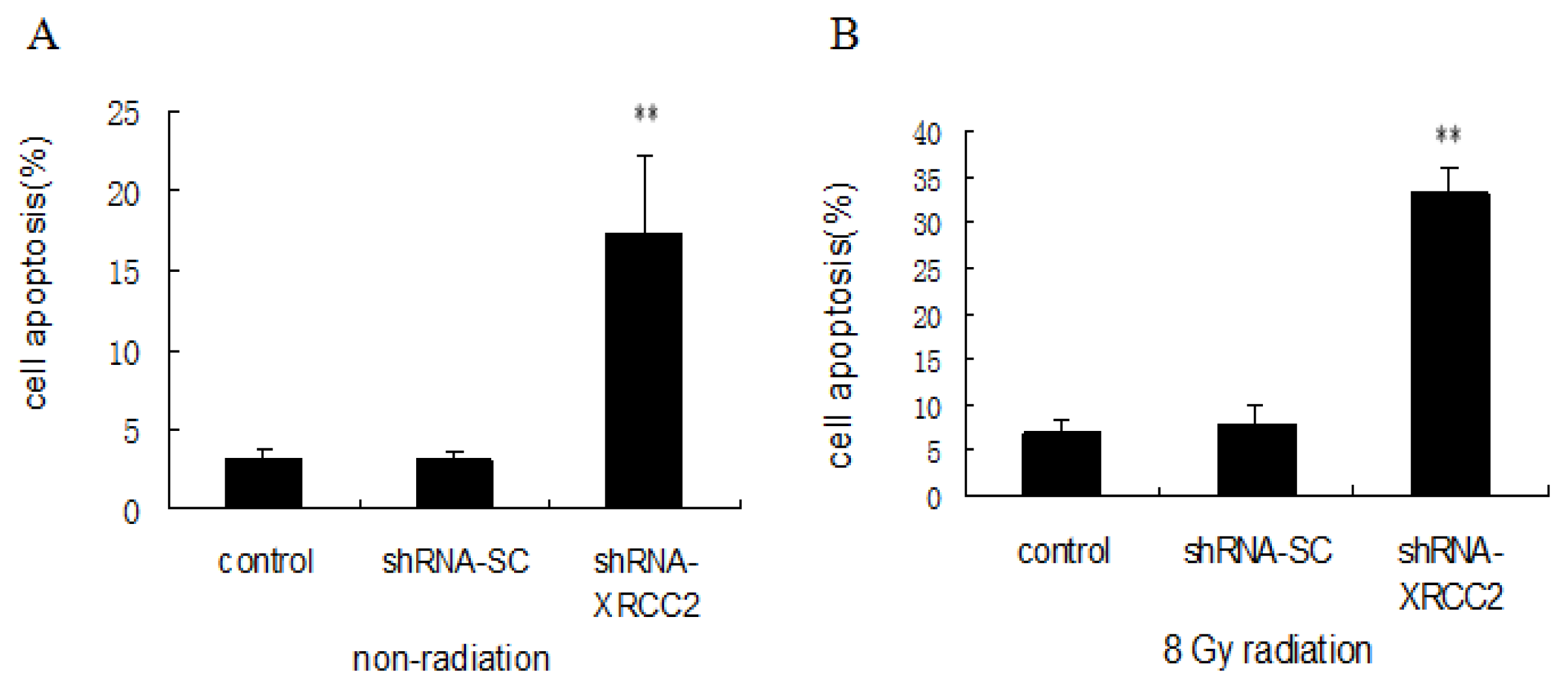
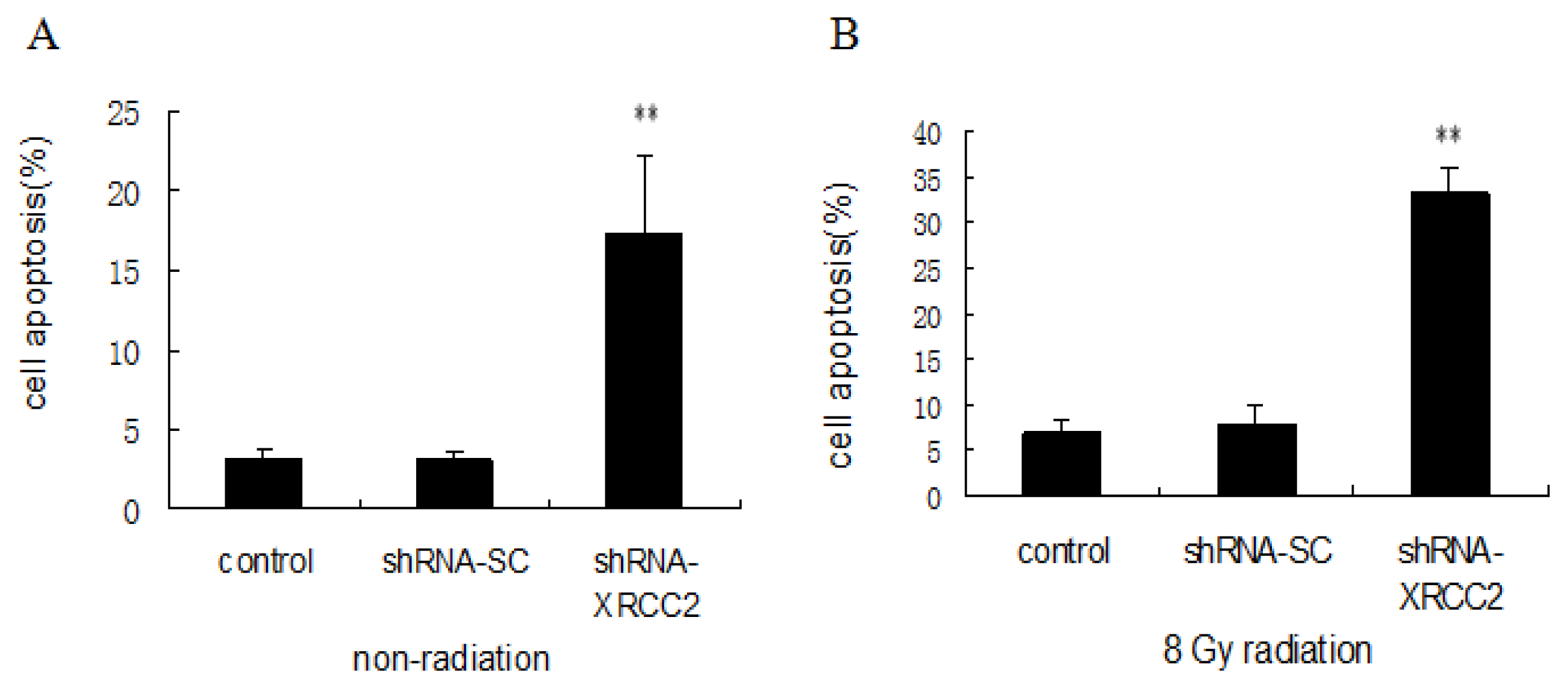
2.6. Effect of XRCC2 Knockdown on Cell Apoptosis Induced by Radiation
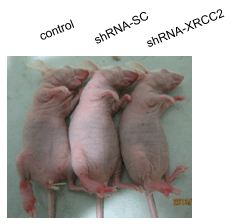
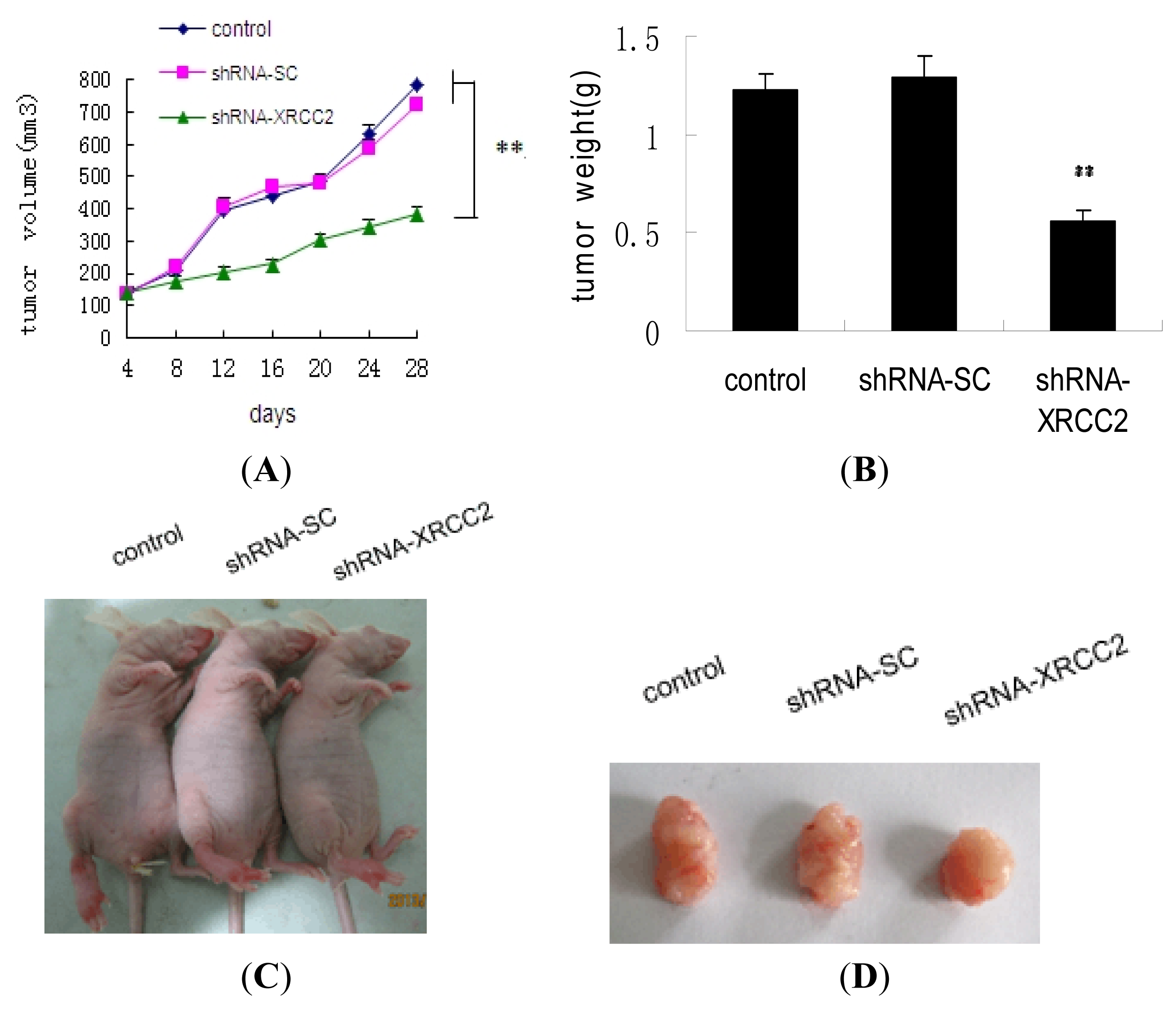
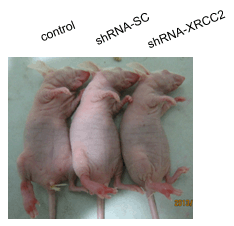
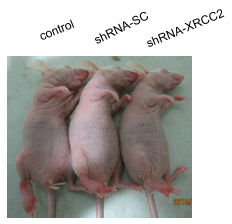
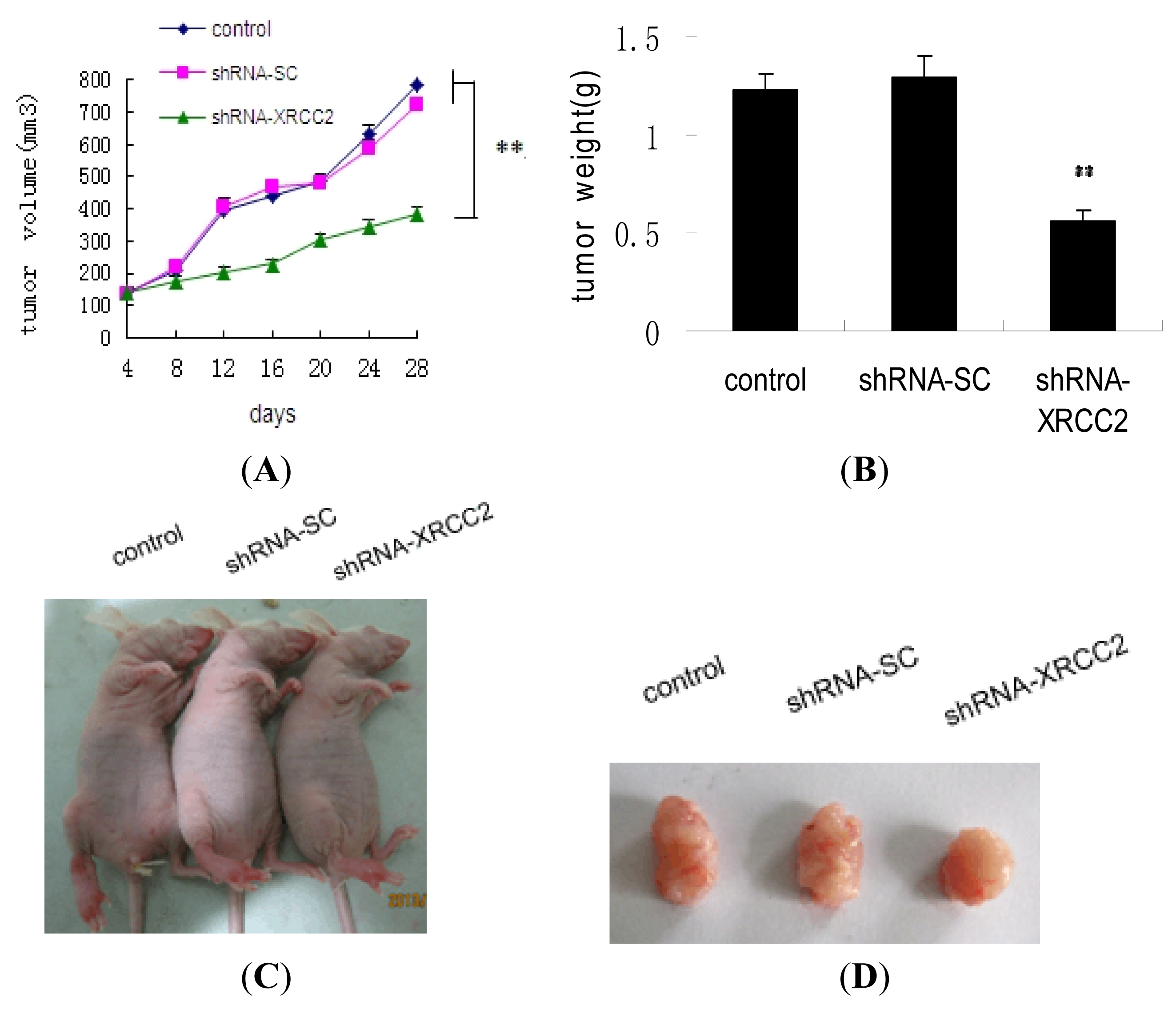
2.7. Effect of XRCC2 Knockdown on Tumorigenicity in Nude Mice
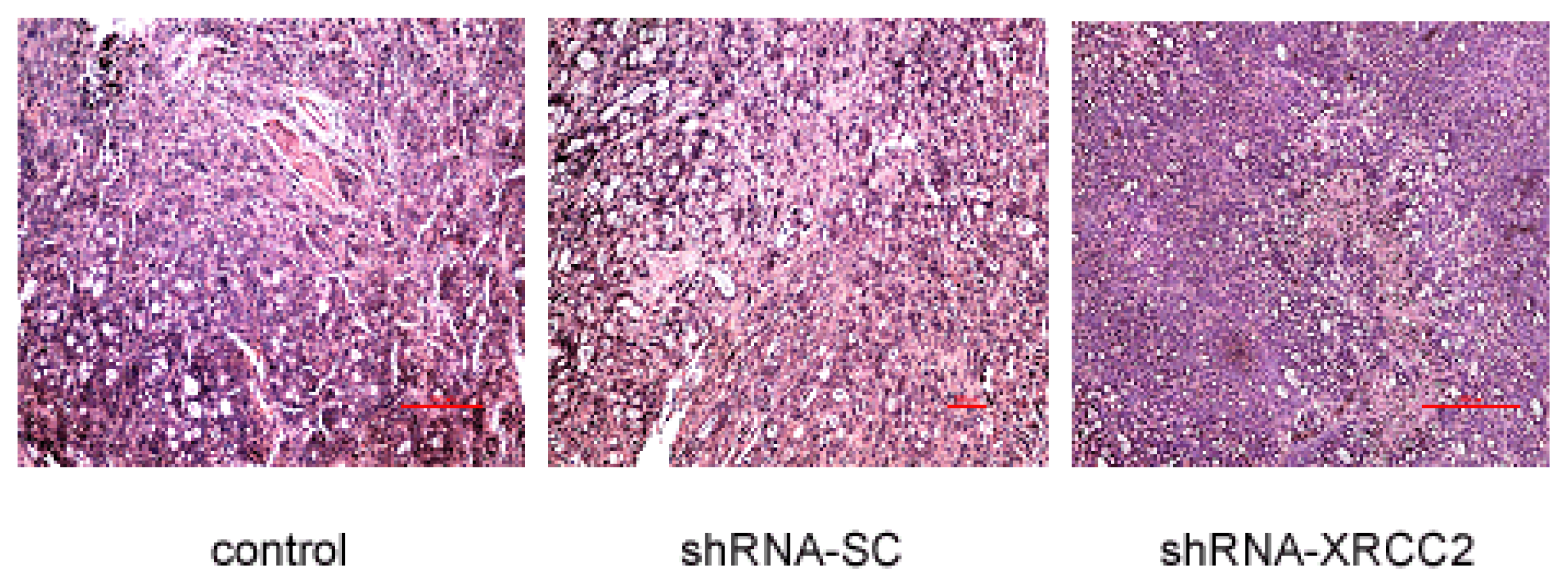
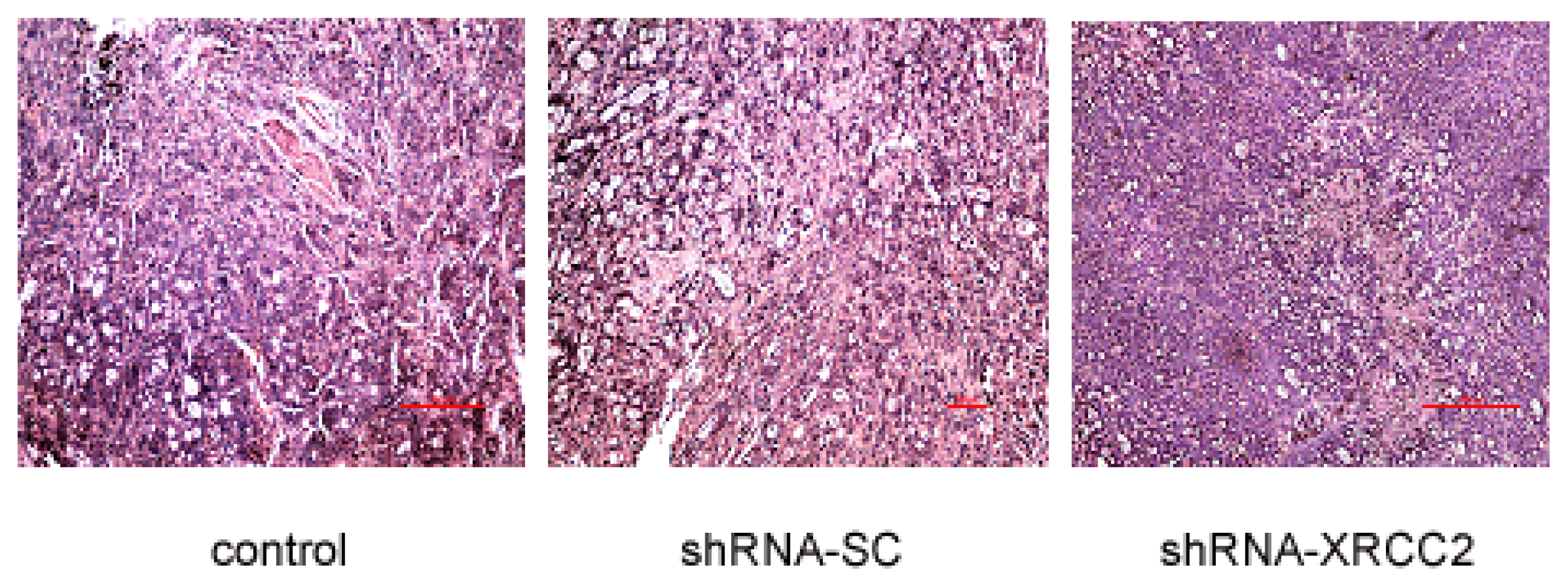
2.8. Effect of XRCC2 Knockdown on Pathology in Nude Mice
3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. Ionizing Radiation
4.3. shRNA Plasmid Stable Transfection
4.4. MTT Assay
4.5. Colony Formation Assay
4.6. Western Blot Analysis
4.7. Quantitative Real-Time PCR
4.8. Cell Cycle
4.9. Cell Apoptosis
4.10. Tumorigenicity in Nude Mice
4.11. Pathological Analysis
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsThe work presented here was carried out in collaboration between all authors. Qin Wang, Feiyue Fan and Qiang Liu conceived and designed the experiments. Qin Wang, Yan Wang and Liqing Du carried out the laboratory experiments, analyzed the data, interpreted the results and wrote the paper. Chang Xu, Yue Fu and Yuanming Sun co-worked on associated data collection and their interpretation. Bing Yang and Lu Cai contributed significantly to analysis and manuscript preparation. Zhijuan Sun and Saijun Fan helped perform the analysis with constructive discussions. All authors have contributed to, seen and approved the manuscript.
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin 2011, 61, 69–90. [Google Scholar]
- Center, M.M.; Jemal, A.; Smith, R.A.; Ward, E. Worldwide variations in colorectal cancer. CA Cancer J. Clin 2009, 59, 366–378. [Google Scholar]
- Ganapathi, S.; Kumar, D.; Katsoulas, N.; Melville, D.; Hodgson, S.; Finlayson, C.; Hagger, R. Colorectal cancer in the young: Trends, charactistics and outcome. Int. J. Color. Dis 2011, 26, 927–934. [Google Scholar]
- Tambini, C.E.; Spink, K.G.; Ross, C.J.; Hill, M.A.; Thacker, J. The importance of XRCC2 in RAD51-related DNA damage repair. DNA Repair 2010, 9, 517–525. [Google Scholar]
- Romanowicz, H.; Smolarz, B.; Baszczyński, J.; Zadrożny, M.; Kulig, A. Genetics polymorphism in DNA repair genes by base excision repair pathway (XRCC1) and homologous recombination (XRCC2 and RAD51) and the risk of breast carcinoma in the Polish population. Pol. J. Pathol 2010, 61, 206–212. [Google Scholar]
- Rajesh, C.; Gruver, A.M.; Basrur, V.; Pittman, D.L. The interaction profile of homologous recombination repair proteins RAD51C, RAD51D and XRCC2 as determined by proteomic analysis. Proteomics 2009, 9, 4071–4086. [Google Scholar]
- Park, S.W.; Yoo, N.J.; Lee, S.H. Mutational analysis of mononucleotide repeats in XRCC2 and XRCC6 in cancers with microsatellite instability. Pathology 2011, 43, 78–79. [Google Scholar]
- Liu, Y.; Shete, S.; Wang, L.E.; El-Zein, R.; Etzel, C.J.; Liang, F.W.; Armstrong, G.; Tsavachidis, S.; Gilbert, M.R.; Aldape, K.D.; et al. γ-Radiation sensitivity and polymorphisms in RAD51L1 modulate glioma risk. Carcinogenesis 2010, 31, 1762–1769. [Google Scholar]
- Shi, W.L.; Li, J.; Chen, P.; Dai, C.H. Effect of X-ray on expression levels of XRCC2 and XRCC3 in lung cancer cell line A549. Acta Univ. Med. Nanjing Nat. Sci 2009, 29, 1517–1520. [Google Scholar]
- Lin, W.Y.; Camp, N.J.; Cannon-Albright, L.A.; Allen-Brady, K.; Balasubramanian, S.; Reed, M.W.; Hopper, J.L.; Apicella, C.; Giles, G.G.; Southey, M.C.; et al. A role for XRCC2 gene polymorphisms in breast cancer risk and survival. Med. Genet 2011, 48, 477–484. [Google Scholar]
- Shamseldin, H.E.; Elfaki, M.; Alkuraya, F.S. Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J. Med. Genet 2012, 49, 184–186. [Google Scholar]
- Silva, S.N.; Tomar, M.; Paulo, C.; Gomes, B.C.; Azevedo, A.P.; Teixeira, V.; Pina, J.E.; Rueff, J.; Gaspar, J.F. Breast cancer risk and common single nucleotide polymorphisms in homologous recombination DNA repair pathway genes. XRCC2, XRCC3, NBS1 and RAD51. Cancer Epidemiol 2010, 34, 85–92. [Google Scholar]
- Bastos, H.N.; Antao, M.R.; Silva, S.N.; Azevedo, A.P.; Manita, I.; Teixeira, V.; Pina, J.E.; Gil, O.M.; Ferreira, T.C.; Limbert, E.; et al. Association of polymorphisms in genes of the homologous recombination DNA repair pathway and thyroid cancer risk. Thyroid 2009, 19, 1067–1075. [Google Scholar]
- Pearce, C.L.; Near, A.M.; van den Berg, D.J.; Ramus, S.J.; Gentry-Maharaj, A.; Menon, U.; Gayther, S.A.; Anderson, A.R.; Edlund, C.K.; Wu, A.H.; et al. Validating genetic risk associations for ovarian cancer through the international ovarian cancer association consortium. Br. J .Cancer 2009, 100, 412–420. [Google Scholar]
- Krupa, R.; Sliwinski, T.; Wisniewska-Jarosinska, M.; Chojnacki, J.; Wasylecka, M.; Dziki, L.; Morawiec, J.; Blasiak, J. Polymorphisms in RAD51, XRCC2 and XRCC3 genes of the homologous recombination repair in colorectal cancer—A case control study. Mol. Biol. Rep 2011, 38, 2849–2854. [Google Scholar]
- Slattery, M.L.; Curtin, K.; Wolff, R.K.; Boucher, K.M.; Sweeney, C.; Edwards, S.; Caan, B.J.; Samowitz, W. A comparison of colon and rectal somatic DNA alterations. Dis. Colon Rectum 2009, 52, 1304–1311. [Google Scholar]
- Curtin, K.; Lin, W.Y.; George, R.; Katory, M.; Shorto, J.; Cannon-Albright, L.A.; Bishop, D.T.; Cox, A.; Camp, N.J. Colorectal Cancer Study Group. Meta association of colorectal cancer confirms risk alleles at 8q24 and 18q21. Cancer Epidemiol. Biomark. Prev 2009, 18, 616–621. [Google Scholar]
- Curtin, K.; Lin, W.Y.; George, R.; Katory, M.; Shorto, J.; Cannon-Albright, L.A.; Smith, G.; Bishop, D.T.; Cox, A.; Camp, N.J.; et al. Genetic variants in XRCC2: New insights into colorectal cancer tumorigenesis. Cancer Epidemiol. Biomark. Prev 2009, 18, 2476–2484. [Google Scholar]
- Reeves, S.G.; Meldrum, C.; Groombridge, C.; Spigelman, A.; Suchy, J.; Kurzawski, G.; Lubinski, J.; Scott, R.J. DNA repair gene polymorphisms and risk of early onset colorectal cancer in Lynch syndrome. Cancer Epidemiol 2012, 36, 183–189. [Google Scholar]
- Gladstone, M.; Su, T.T. Radiation responses and resistance. Int. Rev. Cell Mol. Biol 2012, 299, 235–253. [Google Scholar]
- Bai, J.; Guo, X.G.; Bai, X.P. Epidermal growth factor receptor-related DNA repair and radiation-resistance regulatory mechanisms: A mini-review. Asian Pac. J. Cancer Prev 2012, 13, 4879–4881. [Google Scholar]
- Haines, J.W.; Coster, M.R.; Adam, J.; Cheeseman, M.; Ainsbury, E.A.; Thacker, J.; Bouffler, S.D. Xrcc2 modulates spontaneous and radiation-induced tumorigenesis in Apcmin/+ mice. Mol. Cancer Res 2010, 8, 1227–1233. [Google Scholar]
- Zheng, Z.; Ng, W.L.; Zhang, X.; Olson, J.J.; Hao, C.; Curran, W.J.; Wang, Y. RNAi-mediated targeting of noncoding and coding sequences in DNA repair gene messages efficiently radiosensitizes human tumor cells. Cancer Res 2012, 72, 1221–1228. [Google Scholar]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys 2004, 59, 928–942. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | shRNA-SC | shRNA-XRCC2 | |
|---|---|---|---|
| Tumor margin | + | + | + |
| Architectural atypia | + | + | +/− |
| Cellular atypia | + | + | + |
| Karyokinesis | ++ | ++ | + |
| Necrotic area | +/− | +/− | + |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, Q.; Wang, Y.; Du, L.; Xu, C.; Sun, Y.; Yang, B.; Sun, Z.; Fu, Y.; Cai, L.; Fan, S.; et al. shRNA-Mediated XRCC2 Gene Knockdown Efficiently Sensitizes Colon Tumor Cells to X-ray Irradiation in Vitro and in Vivo . Int. J. Mol. Sci. 2014, 15, 2157-2171. https://doi.org/10.3390/ijms15022157
Wang Q, Wang Y, Du L, Xu C, Sun Y, Yang B, Sun Z, Fu Y, Cai L, Fan S, et al. shRNA-Mediated XRCC2 Gene Knockdown Efficiently Sensitizes Colon Tumor Cells to X-ray Irradiation in Vitro and in Vivo . International Journal of Molecular Sciences. 2014; 15(2):2157-2171. https://doi.org/10.3390/ijms15022157
Chicago/Turabian StyleWang, Qin, Yan Wang, Liqing Du, Chang Xu, Yuanming Sun, Bing Yang, Zhijuan Sun, Yue Fu, Lu Cai, Saijun Fan, and et al. 2014. "shRNA-Mediated XRCC2 Gene Knockdown Efficiently Sensitizes Colon Tumor Cells to X-ray Irradiation in Vitro and in Vivo " International Journal of Molecular Sciences 15, no. 2: 2157-2171. https://doi.org/10.3390/ijms15022157