A Molecular Genetic Linkage Map of Eucommia ulmoides and Quantitative Trait Loci (QTL) Analysis for Growth Traits

Abstract
:1. Introduction
2. Results
2.1. Molecular Markers
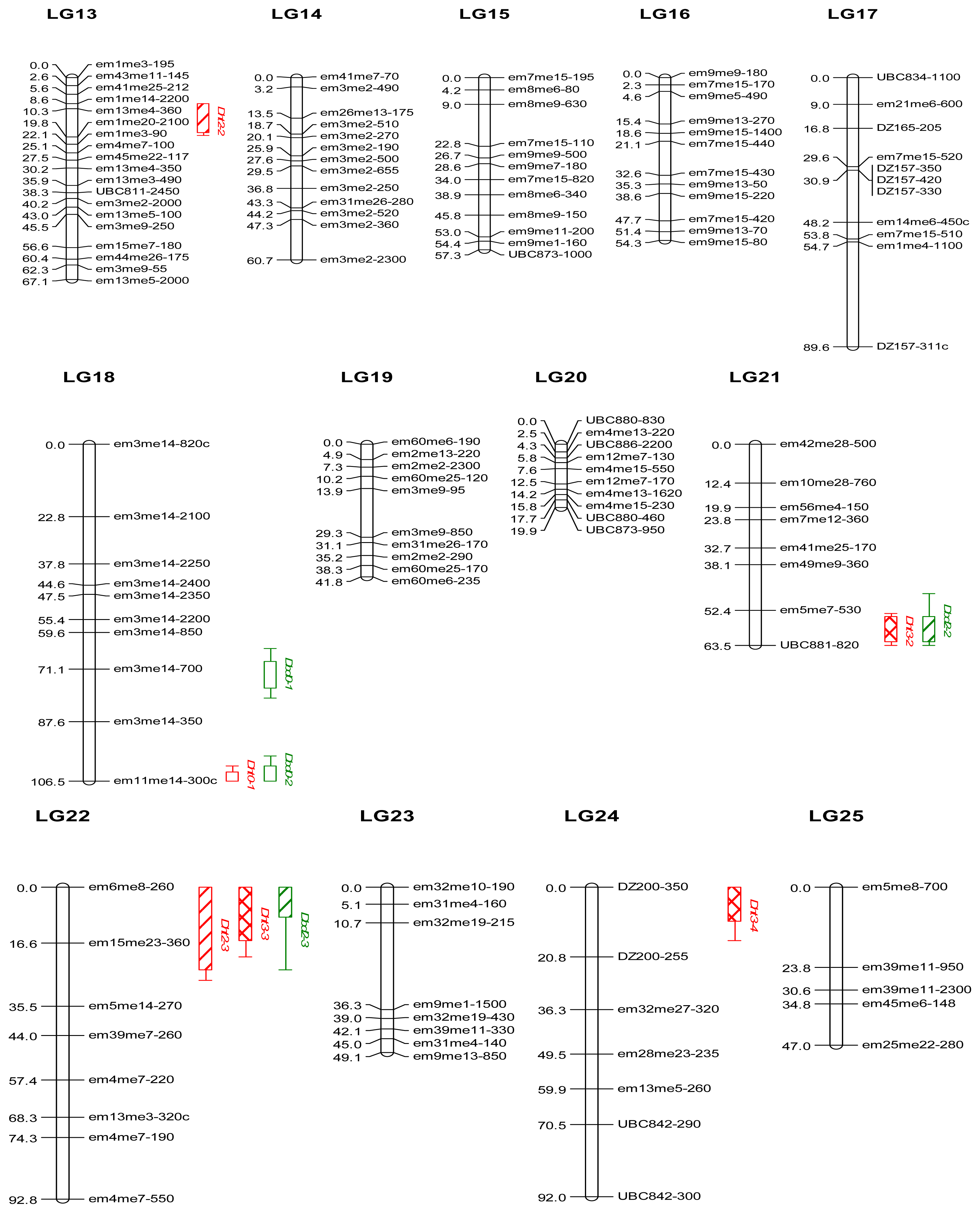
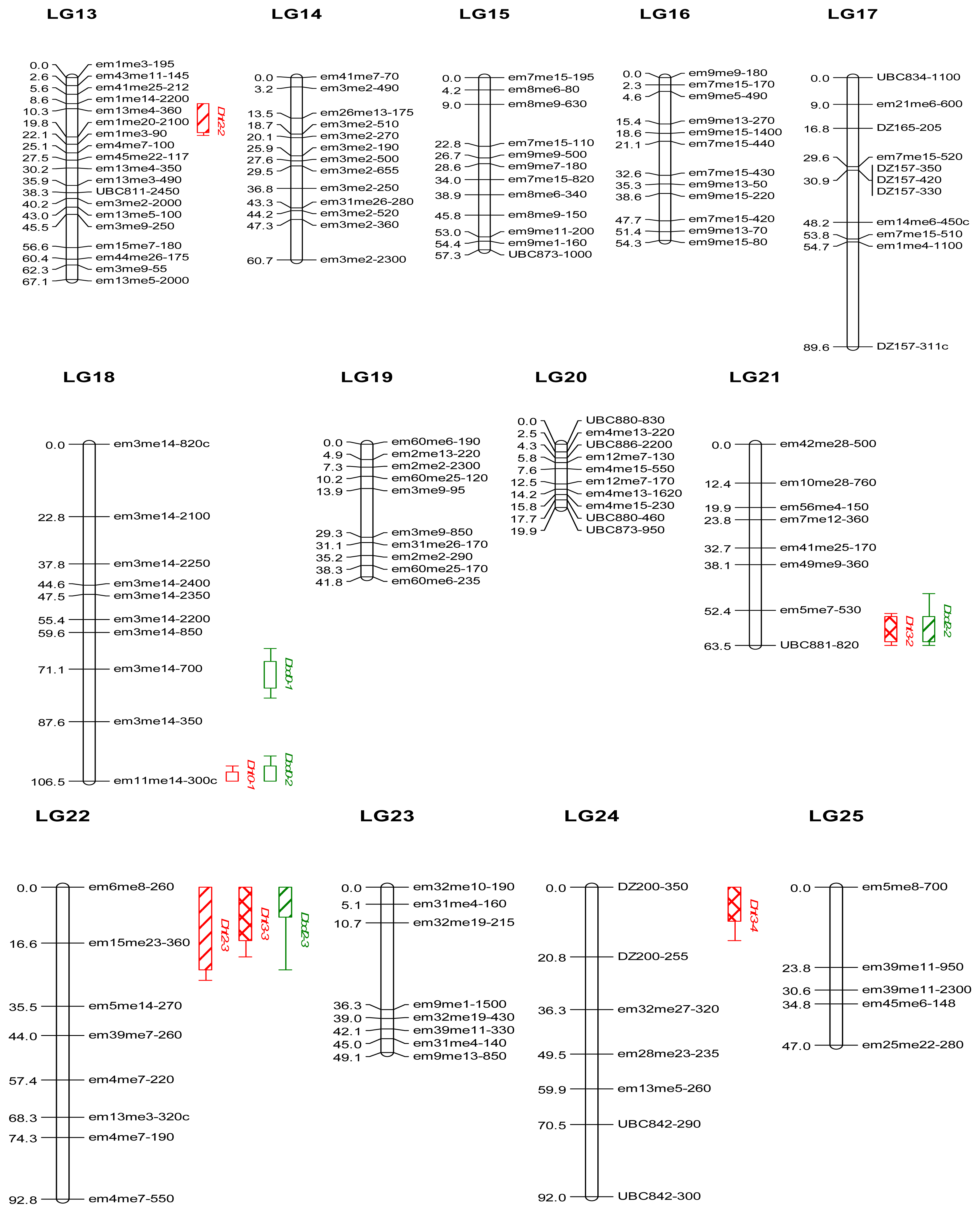
2.2. Genetic Linkage Map
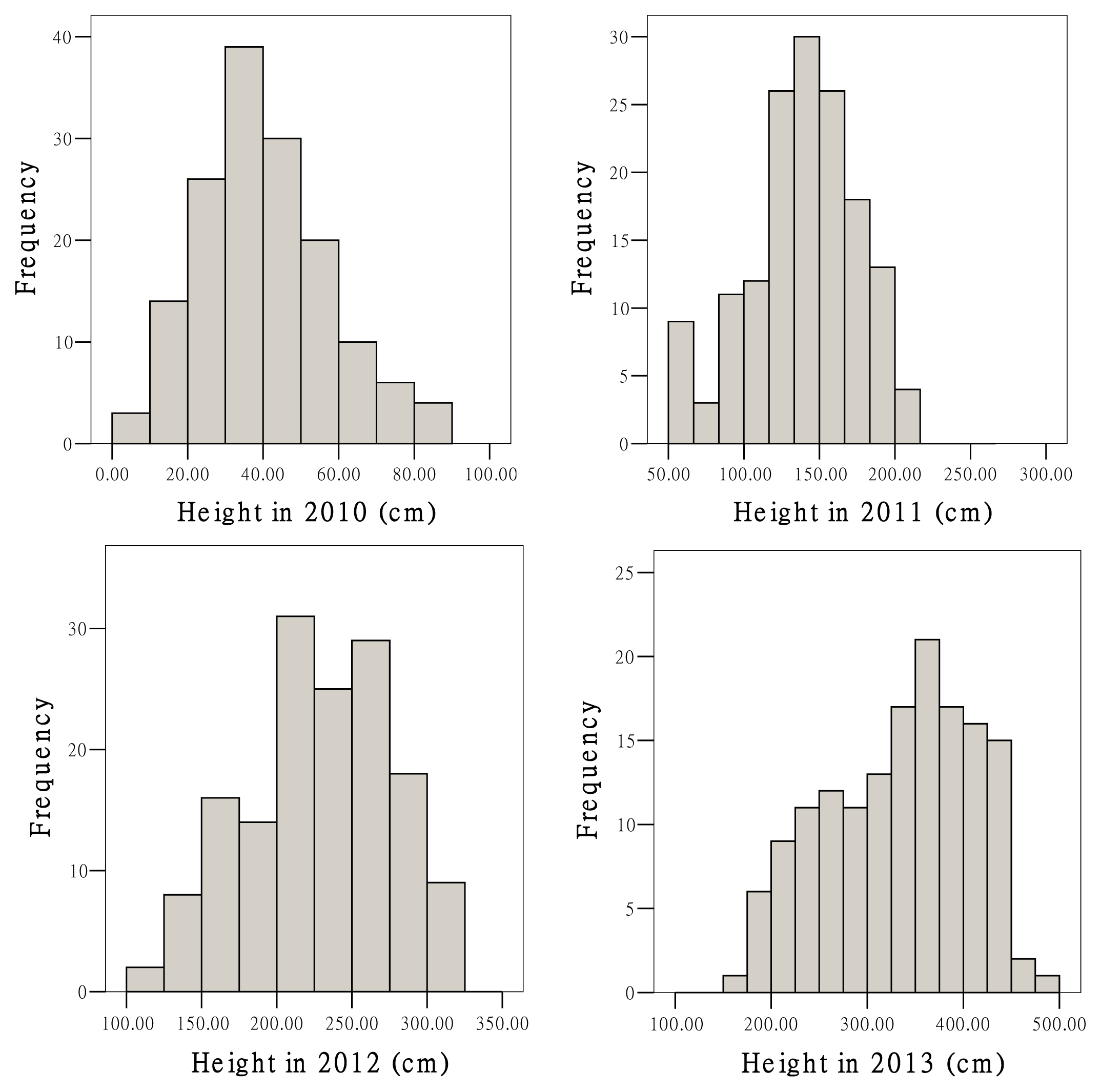
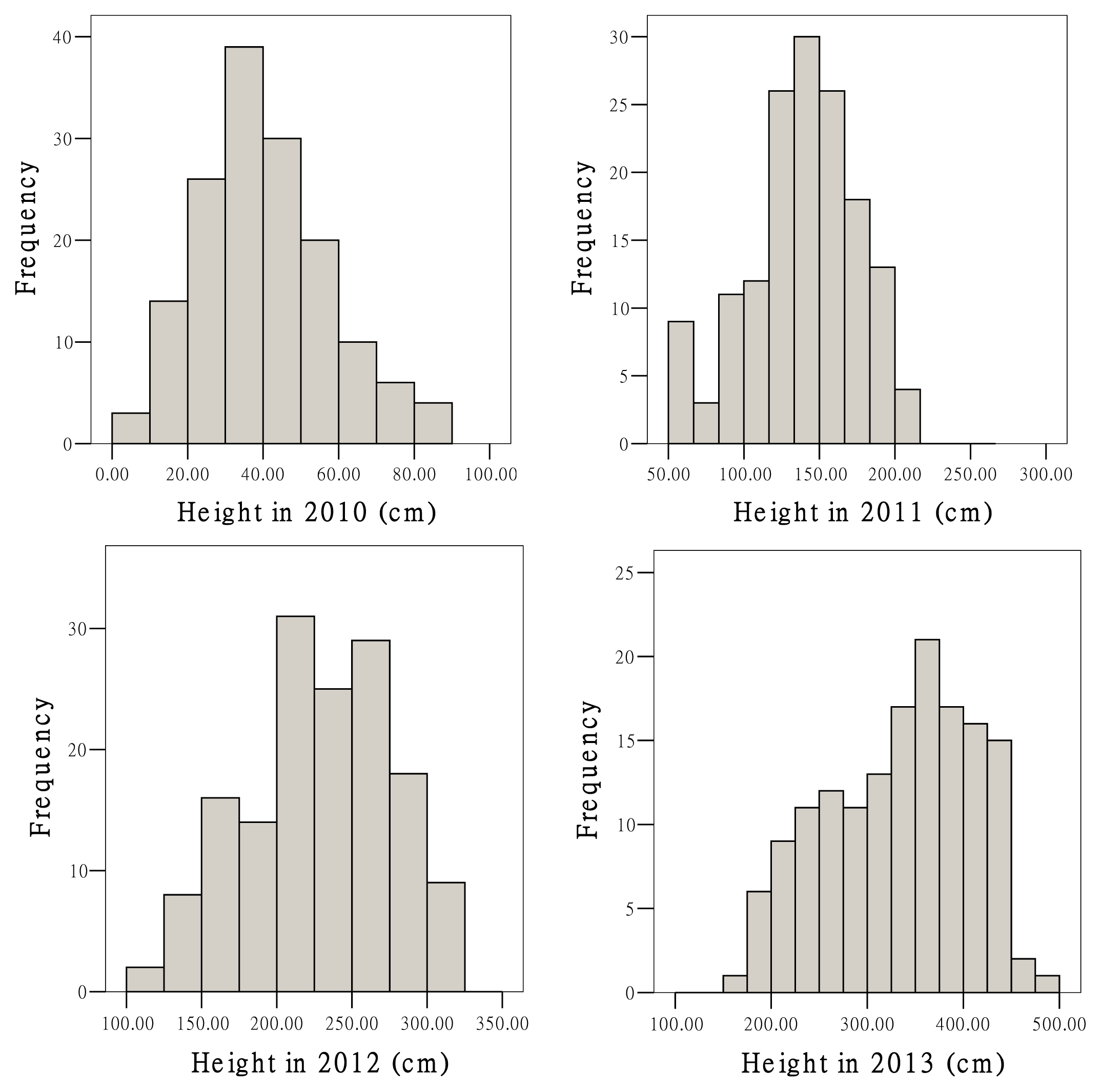
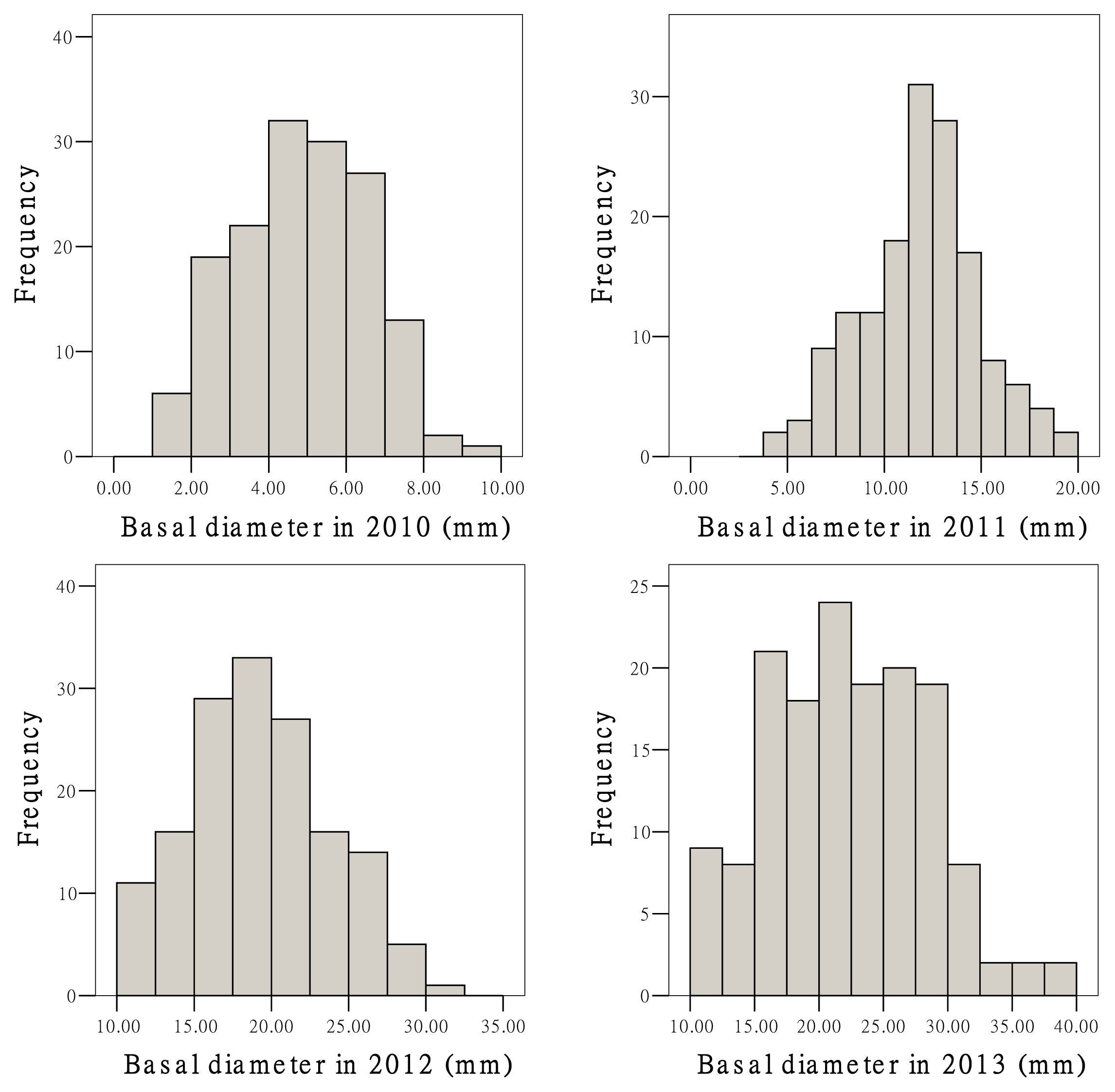
2.3. Growth Traits and QTL Analysis
3. Discussion
3.1. Marker Amplification
3.2. Segregation Distortion
3.3. Genetic Linkage Map
3.4. QTL Analysis
4. Experimental Section
4.1. Plant Material
4.2. DNA Extraction
4.3. SRAP Analysis
4.4. AFLP Analysis
4.5. ISSR Analysis
4.6. SSR Analysis
4.7. Segregation Analysis and Map Construction
4.8. Growth Traits Assessment and QTL Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Tippo, O. The comparative anatomy of the secondary xylem and the phylogeny of the Eucomiaceae. Am. J. Bot 1940, 27, 832–838. [Google Scholar]
- Kwan, C.Y.; Chen, C.X.; Deyama, T.; Nishibe, S. Endothelium-dependent vasorelaxant effects of the aqueous extracts of Eucommia ulmoides Oliv. Leaf and bark: Implications on their anti-hypertensive action. Vascul. Pharmacol 2004, 40, 229–235. [Google Scholar]
- Lee, M.K.; Kim, M.J.; Cho, S.Y.; Park, S.A.; Park, K.K.; Jung, U.J.; Park, H.M.; Choi, M.S. Hypoglycemic effect of Du-zhong (Eucommia ulmoides Oliv.) leaves in streptozotocin-induced diabetic rats. Diabetes Res. Clin. Pract 2005, 67, 22–28. [Google Scholar]
- Hsieh, C.L.; Yen, G.C. Antioxidant actions of Du-zhong (Eucommia ulmoides Oliv.) toward oxidative damage in biomolecules. Life Sci 2000, 66, 1387–1400. [Google Scholar]
- Nakamura, T.; Nakazawa, Y.; Onizuka, S.; Satoh, S.; Chiba, A.; Sekihashi, K.; Miura, A.; Yasugahira, N.; Sasaki, Y.F. Antimutagenicity of Tochu tea (an aqueous extract of Eucommia ulmoides leaves): 1. The clastogen-suppressing effects of Tochu tea in CHO cells and mice. Mutat. Res 1997, 388, 7–20. [Google Scholar]
- Nakazawa, Y.; Bamba, T.; Takeda, T.; Uefuji, H.; Harada, Y.; Li, X.H.; Chen, R.; Inoue, S.; Tutumi, M.; Shimizu, T.; et al. Production of Eucommia-rubber from Eucommia ulmoides Oliv. (Hardy Rubber Tree). Plant Biotechnol 2009, 26, 71–79. [Google Scholar]
- Zhang, K.J.; Su, Y.Q.; Zhang, T. Selection and Breeding of Superior Clones of Chinese Eucommia Ulmoides; Northwest A&F University Press: Yangling, China, 2002; pp. 9–14. [Google Scholar]
- Tsarouhas, V.; Gullberg, U.; Lagercrantz, U. An AFLP and RFLP linkage map and quantitative trait locus (QTL) analysis of growth traits in Salix. Theor. Appl. Genet 2002, 105, 277–288. [Google Scholar]
- Doucleff, M.; Jin, Y.; Gao, F.; Riaz, S.; Krivanek, A.F.; Walker, M.A. A genetic linkage map of grape, utilizing Vitis rupestris and Vitis arizonica. Theor. Appl. Genet 2004, 109, 1178–1187. [Google Scholar]
- Venkateswarlu, M.; Urs, S.R.; Nath, B.S.; Shashidhar, H.E.; Maheswaran, M.; Veeraiah, T.M.; Sabitha, M.G. A first genetic linkage map of mulberry (Morus spp.) using RAPD, ISSR, and SSR markers and pseudotestcross mapping strategy. Tree Genet. Genomes 2006, 3, 15–24. [Google Scholar]
- Pakull, B.; Groppe, K.; Meyer, M.; Markussen, T.; Fladung, M. Genetic linkage mapping in aspen (Populus tremula L. and Populus tremuloides Michx.). Tree Genet. Genomes 2009, 5, 505–515. [Google Scholar]
- Gulsen, O.; Uzun, A.; Canan, I.; Seday, U.; Canihos, E. A new citrus linkage map based on SRAP, SSR, ISSR, POGP, RGA and RAPD markers. Euphytica 2010, 173, 265–277. [Google Scholar]
- Wang, Y.; Sun, X.; Tan, B.; Zhang, B.; Xu, L.A.; Huang, M.; Wang, M. A genetic linkage map of Populus adenopoda Maxim. × P. alba L. hybrid based on SSR and SRAP markers. Euphytica 2010, 173, 193–205. [Google Scholar]
- Fernandez-Fernandez, F.; Antanaviciute, L.; van Dyk, M.M.; Tobutt, K.R.; Evans, K.M.; Rees, D.J.G.; Dunwell, J.M.; Sargent, D.J. A genetic linkage map of an apple rootstock progeny anchored to the Malus genome sequence. Tree Genet. Genomes 2012, 8, 991–1002. [Google Scholar]
- Souza, L.M.; Gazaffi, R.; Mantello, C.C.; Silva, C.C.; Garcia, D.; le Guen, V.; Cardoso, S.E.A.; Garcia, A.A.F.; Souza, A.P. QTL mapping of growth-related traits in a full-Sib family of rubber tree (Hevea brasiliensis) evaluated in a sub-tropical climate. PLoS One 2013, 8, e61238. [Google Scholar]
- Garcia, M.R.; Asins, M.J.; Carbonell, E.A. QTL analysis of yield and seed number in Citrus. Theor. Appl. Genet 2000, 101, 487–493. [Google Scholar]
- Scalfi, M.; Troggio, M.; Piovani, P.; Leonardi, S.; Magnaschi, G.; Vendramin, G.G.; Menozzi, P. A RAPD, AFLP and SSR linkage map, and QTL analysis in European beech (Fagus sylvatica L.). Theor. Appl. Genet 2004, 108, 433–441. [Google Scholar]
- Freeman, J.S.; Whittock, S.P.; Potts, B.M.; Vaillancourt, R.E. QTL influencing growth and wood properties in Eucalyptus globulus. Tree Genet. Genomes 2009, 5, 713–722. [Google Scholar]
- Thumma, B.R.; Baltunis, B.S.; Bell, J.C.; Emebiri, L.C.; Moran, G.F.; Southerton, S.G. Quantitative trait locus (QTL) analysis of growth and vegetative propagation traits in Eucalyptus nitens full-sib families. Tree Genet. Genomes 2010, 6, 877–889. [Google Scholar]
- Samils, B.; Ronnberg-Wastljung, A.C.; Stenlid, J. QTL mapping of resistance to leaf rust in Salix. Tree Genet. Genomes 2011, 7, 1219–1235. [Google Scholar]
- Sadok, I.B.; Celton, J.M.; Essalouh, L.; El Aabidine, A.Z.; Garcia, G.; Martinez, S.; Grati-Kamoun, N.; Rebai, A.; Costes, E.; Khadari, B. QTL mapping of flowering and fruiting traits in olive. PLoS One 2013, 8, e62831. [Google Scholar]
- Dong, J.; Ma, X.H.; Wei, Q.; Peng, S.B.; Zhang, S.C. Effects of growing location on the contents of secondary metabolites in the leaves of four selected superior clones of Eucommia ulmoides. Ind. Crop. Prod 2011, 34, 1607–1614. [Google Scholar]
- Li, G.; Quiros, C.F. Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: Its application to mapping and gene tagging in Brassica. Theor. Appl. Genet 2001, 103, 455–461. [Google Scholar]
- Grando, M.S.; Bellin, D.; Edwards, K.J.; Pozzi, C.; Stefanini, M.; Velasco, R. Molecular linkage maps of Vitis vinifera L. and Vitis riparia Mchx. Theor. Appl. Genet 2003, 106, 1213–1224. [Google Scholar]
- Zhang, R.; Wu, J.; Li, X.G.; Khan, M.A.; Chen, H.; Korban, S.S.; Zhang, S.L.; An, A.F.L.P. SRAP, and SSR genetic linkage map and identification of QTLs for fruit traits in pear (Pyrus L.). Plant Mol. Biol. Report 2013, 31, 678–687. [Google Scholar]
- Sanchez-Robles, J.M.; Balao, F.; Garcia-Castano, J.L.; Terrab, A.; Navarro-Sampedro, L.; Talavera, S. Nuclear microsatellite primers for the endangered relict fir, Abies pinsapo (pinaceae) and cross-amplification in related mediterranean species. Int. J. Mol. Sci 2012, 13, 14243–14250. [Google Scholar]
- Wang, H.; Chen, N.F.; Zheng, J.Y.; Wang, W.C.; Pei, Y.Y.; Zhu, G.P. Isolation and characterization of eleven polymorphic microsatellite loci for the valuable medicinal plant Dendrobium huoshanense and cross-species amplification. Int. J. Mol. Sci 2012, 13, 16779–16784. [Google Scholar]
- Kang, B.Y.; Major, J.E.; Rajora, O.P. A high-density genetic linkage map of a black spruce (Picea mariana) × red spruce (Picea rubens) interspecific hybrid. Genome 2011, 54, 128–143. [Google Scholar]
- Lorieux, M.; Goffinet, B.; Perrier, X.; Gonzalez de Leon, D.; Lanaud, C. Maximum-likelihood models for mapping genetic markers showing segregation distortion. 1. Backcross populations. Theor. Appl. Genet 1995, 90, 73–80. [Google Scholar]
- Sauge, M.H.; Lambert, P.; Pascal, T. Co-localisation of host plant resistance QTLs affecting the performance and feeding behaviour of the aphid Myzus persicae in the peach tree. Heredity 2012, 108, 292–301. [Google Scholar]
- Jenczewski, E.; Gherardi, M.; Bonnin, I.; Prosperi, J.M.; Olivieri, I.; Huguet, T. Insight on segregation distortions in two intraspecific crosses between annual species of Medicago (Leguminosae). Theor. Appl. Genet 1997, 94, 682–691. [Google Scholar]
- Kasha, K.J.; Kao, K.N. High frequency haploid production in barley (Hordeum vulgare). Nature 1970, 225, 874–876. [Google Scholar]
- Bradshaw, H.D.; Stettler, R.F. Molecular genetics of growth and development in Populus. II. Segregation distortion due to genetic load. Theor. Appl. Genet 1994, 89, 551–558. [Google Scholar]
- Fishman, L.; Willis, J.H. A novel meiotic drive locus almost completely distorts segregation in Mimulus (monkeyflower) hybrids. Genetics 2005, 169, 347–353. [Google Scholar]
- Fishman, L.; Kelly, A.J.; Morgan, E.; Willis, J.H. A genetic map in the Mimulus guttatus species complex reveals transmission ratio distortion due to heterospecific interactions. Genetics 2001, 159, 1701–1716. [Google Scholar]
- Fishman, L.; Aagaard, J.; Tuthill, J.C. Toward the evolutionary genomics of gametophytic divergence: Patterns of transmission ratio distortion in monkey flower (Mimulus) hybrids reveal a complex genetic basis for conspecific pollen precedence. Evolution 2008, 62, 2958–2970. [Google Scholar]
- Bradshaw, H.D.; Stettler, R.F. Molecular genetics of growth and development in Popolus. IV. Mapping QTLs with large effects on growth, form and phenology traits in a forest tree. Genetics 1995, 139, 963–973. [Google Scholar]
- Zhang, D.; Zhang, Z.; Yang, K. QTL analysis of growth and wood chemical content traits in an interspecific backcross family of white poplar (Populus tomentosa × P. bolleana) × P. tomentosa. Can. J. For. Res 2006, 36, 2015–2023. [Google Scholar]
- Ronnberg-Wastljung, A.C.; Glynn, C.; Weih, M. QTL analyses of drought tolerance and growth for a Salix dasyclados × Salix viminalis hybrid in contrasting water regimes. Theor. Appl. Genet 2005, 110, 537–549. [Google Scholar]
- Verhaegen, D.; Plomion, C.; Gion, J.M.; Poitel, M.; Costa, P.; Kremer, A. Quantitative trait dissection analysis in Eucalyptus using RAPD markers. 1. Detection of QTL in interspecific hybrid progeny, stability of QTL expression across different ages. Theor. Appl. Genet 1997, 95, 597–608. [Google Scholar]
- Kenis, K.; Keulemans, J. Study of tree architecture of apple (Malus × domestica Borkh.) by QTL analysis of growth traits. Mol. Breed 2007, 19, 193–208. [Google Scholar]
- Emebiri, L.C.; Devey, M.E.; Matheson, A.C.; Slee, M.U. Age-related changes in the expression of QTLs for growth in radiata pine seedlings. Theor. Appl. Genet 1998, 97, 1053–1061. [Google Scholar]
- Stoeckli, S.; Mody, K.; Gessler, C.; Patocchi, A.; Jermini, M.; Dorn, S. QTL analysis for aphid resistance and growth traits in apple. Tree Genet. Genomes 2008, 4, 833–847. [Google Scholar]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep 1997, 15, 8–15. [Google Scholar]
- Vos, P.; Hoger, R.; Blecker, M. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res 1995, 23, 4407–4414. [Google Scholar]
- Wang, D.W.; Li, Y.; Li, Z.Q. Identification of a male-specific amplified fragment length polymorphism (AFLP) and a sequence characterized amplified region (SCAR) Marker in Eucommia ulmoides Oliv. Int. J. Mol. Sci 2011, 12, 857–864. [Google Scholar]
- Zietkiewicz, E.; Rafalski, A.; Labuda, D. Genome fingerprinting by simple sequence repeats (SSR)-anchored PCR amplification. Genomics 1994, 20, 176–183. [Google Scholar]
- Deng, J.Y.; Liu, Y.F.; Huang, H.W. Development and characterization of microsatellite markers in Eucommia ulmoides Oliver (Eucommiaceae). Mol. Ecol. Notes 2006, 6, 496–498. [Google Scholar]
- Van Ooijen, J.W. Joinmap 4, Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Plant Research International B.V. and Kyazma B.V.: Wageningen, The Netherlands, 2006. [Google Scholar]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered 2002, 93, 77–78. [Google Scholar]
- Chakravarti, A.; Lasher, L.K.; Reefer, J.E. A maximum-likelihood method for estimating genome length using genetic linkage data. Genetics 1991, 128, 175–182. [Google Scholar]
- Van Ooijen, J.W. Mapqtl 5, Software for the Mapping of Quantitative Trait Loci in Experimental Populations; Plant Research International B.V. and Kyazma B.V.: Wageningen, The Netherlands, 2004. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward primer | Reverse primer | ||||
|---|---|---|---|---|---|
| Name | Sequence | Name | Sequence | Name | Sequence |
| me1 | ATA | me33 | GAA | em1 | AAT |
| me2 | AGC | me34 | GAT | em2 | TGC |
| me3 | AAT | me35 | GAG | em3 | GAC |
| me4 | ACC | me36 | GAC | em4 | TGA |
| me5 | AAG | me37 | GTA | em5 | AAC |
| me6 | ACA | me38 | GTT | em6 | GCA |
| me7 | ACG | me39 | GTG | em7 | CAA |
| me8 | ACT | me40 | GTC | em8 | CAC |
| me9 | AGG | me41 | GGA | em9 | CAG |
| me10 | AAA | me42 | GGT | em10 | CAT |
| me11 | AAC | me43 | GGG | em11 | CTA |
| me12 | AGA | me44 | GGC | em12 | CTC |
| me13 | ATG | me45 | GCA | em13 | CTG |
| me14 | ATC | me46 | GCT | em14 | CTT |
| me15 | ATT | me47 | GCG | em15 | GAT |
| me16 | AGT | me48 | GCC | em16 | GTC |
| me17 | TAA | me49 | CAA | em17 | AAG |
| me18 | TAT | me50 | CAT | em18 | ATC |
| me19 | TAG | me51 | CAG | em19 | AGA |
| me20 | TAC | me52 | CAC | em20 | ACT |
| me21 | TTA | me53 | CTA | em21 | TAC |
| me22 | TTT | me54 | CTT | em22 | TTG |
| me23 | TTG | me55 | CTG | em23 | TGT |
| me24 | TTC | me56 | CTC | em24 | TCG |
| me25 | TGA | me57 | CGA | em25 | GAA |
| me26 | TGT | me58 | CGT | em26 | GTG |
| me27 | TGG | me59 | CGG | em27 | GGA |
| me28 | TGC | me60 | CGC | em28 | GCT |
| me29 | TCA | me61 | CCA | em29 | CGA |
| me30 | TCT | me62 | CCT | em30 | CGT |
| me31 | TCG | me63 | CCG | em31 | CCA |
| me32 | TCC | me64 | CCC | em32 | CCT |
| EcoRI primers | MseI primers | ||
|---|---|---|---|
| Name | Sequence | Name | Sequence |
| E1 | AAC | M1 | CAA |
| E2 | AAG | M2 | CAC |
| E3 | ACA | M3 | CAG |
| E4 | ACT | M4 | CAT |
| E5 | ACC | M5 | CTA |
| E6 | ACG | M6 | CTC |
| E7 | AGC | M7 | CTG |
| E8 | AGG | M8 | CTT |
| Name | Sequence | Annealing temperature |
|---|---|---|
| UBC808 | C(AG)8C | 56.0 |
| UBC811 | (GA)8C | 43.6 |
| UBC830 | (TG)8G | 50.0 |
| UBC834 | (AG)8YT | 56.0 |
| UBC835 | (AG)8YC | 43.6 |
| UBC840 | (GA)8YT | 56.0 |
| UBC842 | (GA)8YG | 54.7 |
| UBC853 | (TC)8RT | 46.3 |
| UBC860 | (TG)8RA | 56.0 |
| UBC866 | CT(CCT)5C | 52.6 |
| UBC867 | (GGC)6 | 41.4 |
| UBC868 | (GAA)6 | 46.3 |
| UBC873 | (GACA)4 | 50.0 |
| UBC880 | (GGAGA)3 | 50.0 |
| UBC881 | (GGGGT)3 | 50.0 |
| UBC886 | VDV(CT)7 | 52.6 |
| Marker type a | No. of primer combinations b | No. of polymorphic markers | Lmxll (1:1) c | Nnxnp (1:1) d | Hkxhk (3:1) e | Hkxhk (1:2:1) f | Egxef (1:1:1:1) g | Abxcd (1:1:1:1) h | Distorted markers (p < 0.05) |
|---|---|---|---|---|---|---|---|---|---|
| SRAP | 131 | 1604 | 305 | 326 | 382 | 18 | 13 | 8 | 552 |
| AFLP | 18 | 295 | 141 | 108 | 22 | 0 | 0 | 0 | 24 |
| ISSR | 16 | 111 | 27 | 23 | 31 | 0 | 0 | 0 | 30 |
| SSR | 17 | 132 | 33 | 42 | 26 | 6 | 7 | 1 | 17 |
| Total | 182 | 2142 | 506 | 499 | 461 | 24 | 20 | 9 | 623 |
| Linkage group | Length (cM) | No. of markers | SRAPs | AFLPs | ISSRs | SSRs | Mean distance (cM) |
|---|---|---|---|---|---|---|---|
| LG1 | 153.0 | 106 | 70 | 7 | 0 | 29 | 1.5 |
| LG2 | 194.0 | 77 | 9 | 65 | 0 | 3 | 2.6 |
| LG3 | 189.5 | 76 | 74 | 0 | 2 | 0 | 2.5 |
| LG4 | 96.5 | 65 | 64 | 0 | 0 | 1 | 1.5 |
| LG5 | 60.5 | 49 | 37 | 0 | 5 | 7 | 1.3 |
| LG6 | 123.5 | 45 | 0 | 45 | 0 | 0 | 2.8 |
| LG7 | 82.8 | 37 | 37 | 0 | 0 | 0 | 2.3 |
| LG8 | 72.9 | 26 | 25 | 0 | 0 | 1 | 2.9 |
| LG9 | 88.9 | 25 | 12 | 0 | 0 | 13 | 3.7 |
| LG10 | 66.2 | 25 | 25 | 0 | 0 | 0 | 2.8 |
| LG11 | 70.7 | 21 | 20 | 0 | 1 | 0 | 3.5 |
| LG12 | 92.9 | 21 | 21 | 0 | 0 | 0 | 4.7 |
| LG13 | 67.1 | 19 | 18 | 0 | 1 | 0 | 3.7 |
| LG14 | 60.7 | 13 | 13 | 0 | 0 | 0 | 5.1 |
| LG15 | 57.3 | 12 | 11 | 0 | 1 | 0 | 5.2 |
| LG16 | 54.3 | 12 | 12 | 0 | 0 | 0 | 4.9 |
| LG17 | 89.6 | 11 | 10 | 0 | 1 | 0 | 9.0 |
| LG18 | 106.5 | 10 | 10 | 0 | 0 | 0 | 11.8 |
| LG19 | 41.8 | 10 | 10 | 0 | 0 | 0 | 4.6 |
| LG20 | 19.9 | 10 | 6 | 0 | 4 | 0 | 2.2 |
| LG21 | 63.5 | 8 | 7 | 0 | 1 | 0 | 9.1 |
| LG22 | 92.8 | 8 | 8 | 0 | 0 | 0 | 13.3 |
| LG23 | 49.1 | 8 | 8 | 0 | 0 | 0 | 7.0 |
| LG24 | 92.0 | 7 | 3 | 0 | 2 | 2 | 15.3 |
| LG25 | 47.0 | 5 | 5 | 0 | 0 | 0 | 11.8 |
| Total | 2133.0 | 706 | 515 | 117 | 18 | 56 | 3.1 |
| Trait | Mean | SD | Minimum | Maximum | CV (%) |
|---|---|---|---|---|---|
| Height 2010 (cm) | 39.3 | 16.8 | 9.0 | 85.0 | 42.8 |
| Height 2011 (cm) | 138.7 | 36.6 | 50.0 | 216.0 | 26.4 |
| Height 2012 (cm) | 224.8 | 48.4 | 120.0 | 310.0 | 21.5 |
| Height 2013 (cm) | 332.0 | 74.4 | 170.0 | 480.0 | 22.4 |
| Basal diameter 2010 (mm) | 4.9 | 1.7 | 1.3 | 9.4 | 34.0 |
| Basal diameter 2011 (mm) | 11.9 | 3.1 | 3.9 | 19.6 | 25.8 |
| Basal diameter 2012 (mm) | 19.2 | 4.6 | 10.0 | 30.4 | 24.0 |
| Basal diameter 2013 (mm) | 22.4 | 6.1 | 10.4 | 38.3 | 27.2 |
| Traits | Height 2011 | Height 2012 | Height 2013 | Basal diameter 2010 | Basal diameter 2011 | Basal diameter 2012 | Basal diameter 2013 |
|---|---|---|---|---|---|---|---|
| Height 2010 | 0.52 * | −0.04 | −0.02 | 0.80 * | 0.51 * | 0.02 | 0.03 |
| Height 2011 | 0.04 | 0.02 | 0.51 * | 0.83 * | 0.03 | 0.15 | |
| Height 2012 | 0.70 * | −0.02 | 0.03 | 0.66 * | 0.75 * | ||
| Height 2013 | 0.05 | −0.04 | 0.64 * | 0.72 * | |||
| Basal diameter 2010 | 0.47 * | 0.01 | 0.04 | ||||
| Basal diameter 2011 | 0.10 | 0.10 | |||||
| Basal diameter 2012 | 0.92 * |
| QTL a | Linkage group | Peak position (cM) b | LOD peak c | Marker d | % Var.expl. e | KW f |
|---|---|---|---|---|---|---|
| Height 2010 | ||||||
| Dht0-1 | LG18 | 106.5 | 7.8 ** | em11me14–300c | 17.1 | **** |
| Height 2011 | ||||||
| Dht1-1 | LG10 | 14.9 | 4.4 ** | em39me7–750 | 29.7 | - |
| Dht1-2 | LG10 | 27.1 | 4.4 ** | em39me7–330 | 27.7 | * |
| Dht1-3 | LG12 | 40.1 | 3.1 | em9me7–230 | 22.8 | - |
| Height 2012 | ||||||
| Dht2-1 | LG9 | 62.0 | 3.2 | DZ126–280 | 12.6 | - |
| Dht2-2 | LG13 | 14.3 | 3.1 | em13me4–360 | 12.4 | **** |
| Dht2-3 | LG22 | 14.0 | 3.3 | em15me23–360 | 33.3 | - |
| Height 2013 | ||||||
| Dht3-1 | LG9 | 62.0 | 3.9 | DZ126–280 | 13.5 | - |
| Dht3-2 | LG21 | 57.4 | 3.3 | UBC881–820 | 26.6 | - |
| Dht3-3 | LG22 | 0.0 | 3.8 | em6me8–260 | 25.3 | - |
| Dht3-4 | LG24 | 0.0 | 4.3 | DZ200–350 | 27.1 | - |
| Basal diameter 2010 | ||||||
| Dbd0-1 | LG18 | 72.1 | 3.8 | em3me14–700 | 29.8 | ** |
| Dbd0-2 | LG18 | 106.5 | 4.7 ** | em11me14–300c | 13.4 | **** |
| Basal diameter 2011 | ||||||
| Dbd1-1 | LG12 | 40.1 | 3.0 | em9me7–230 | 20.2 | - |
| Basal diameter 2012 | ||||||
| Dbd2-1 | LG1 | 153.0 | 3.0 | em12me11–300 | 17.7 | - |
| Dbd2-2 | LG21 | 58.4 | 3.2 | em5me7–530 | 25.1 | ** |
| Dbd2-3 | LG22 | 0.0 | 3.6 | em6me8–260 | 21.4 | ** |
| Basal diameter 2013 | ||||||
| Dbd3-1 | LG1 | 153.0 | 3.0 | em12me11–300 | 16.8 | - |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, Y.; Wang, D.; Li, Z.; Wei, J.; Jin, C.; Liu, M. A Molecular Genetic Linkage Map of Eucommia ulmoides and Quantitative Trait Loci (QTL) Analysis for Growth Traits. Int. J. Mol. Sci. 2014, 15, 2053-2074. https://doi.org/10.3390/ijms15022053
Li Y, Wang D, Li Z, Wei J, Jin C, Liu M. A Molecular Genetic Linkage Map of Eucommia ulmoides and Quantitative Trait Loci (QTL) Analysis for Growth Traits. International Journal of Molecular Sciences. 2014; 15(2):2053-2074. https://doi.org/10.3390/ijms15022053
Chicago/Turabian StyleLi, Yu, Dawei Wang, Zhouqi Li, Junkun Wei, Cangfu Jin, and Minhao Liu. 2014. "A Molecular Genetic Linkage Map of Eucommia ulmoides and Quantitative Trait Loci (QTL) Analysis for Growth Traits" International Journal of Molecular Sciences 15, no. 2: 2053-2074. https://doi.org/10.3390/ijms15022053