Luteolin Reduces Alzheimer’s Disease Pathologies Induced by Traumatic Brain Injury


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
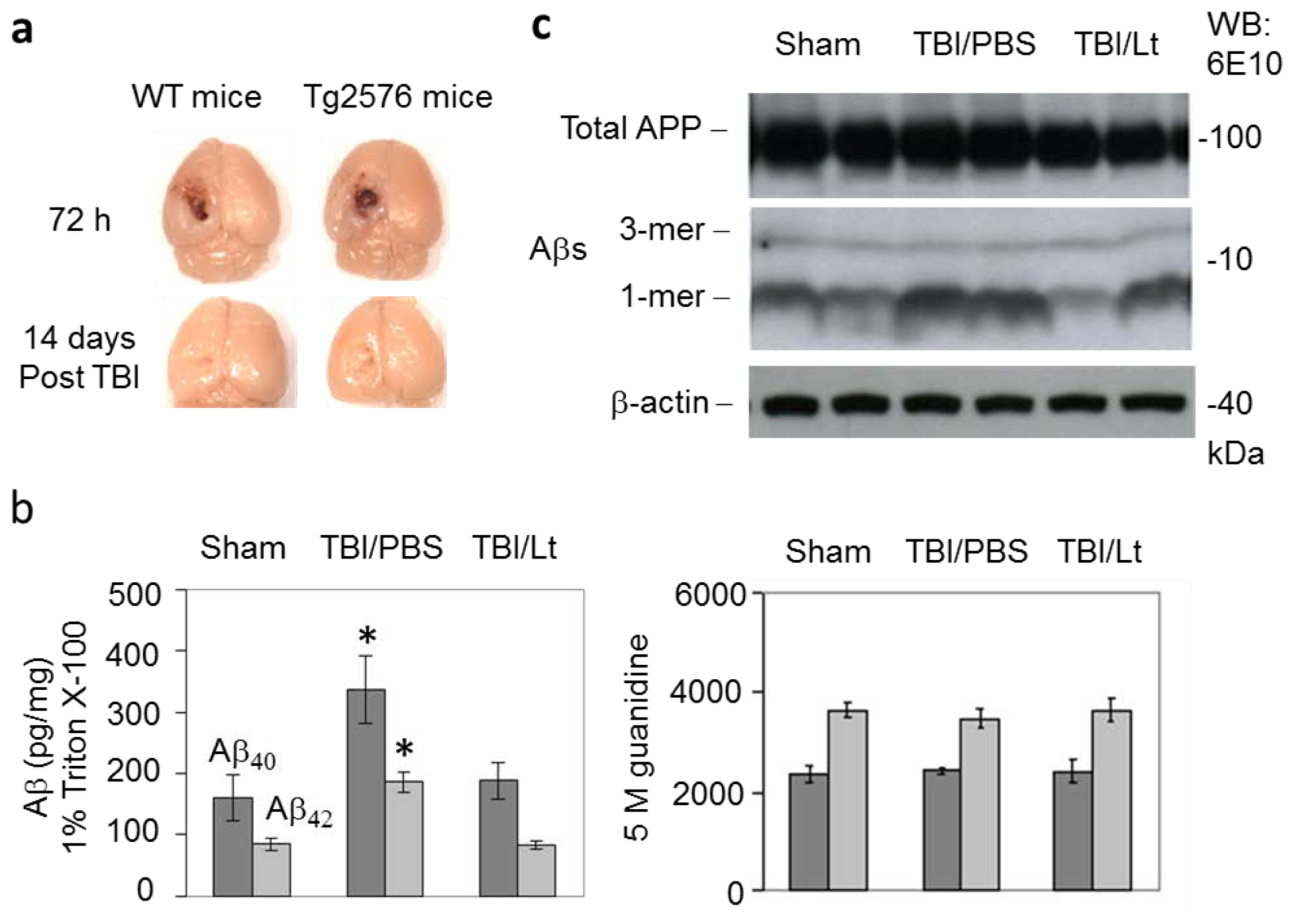
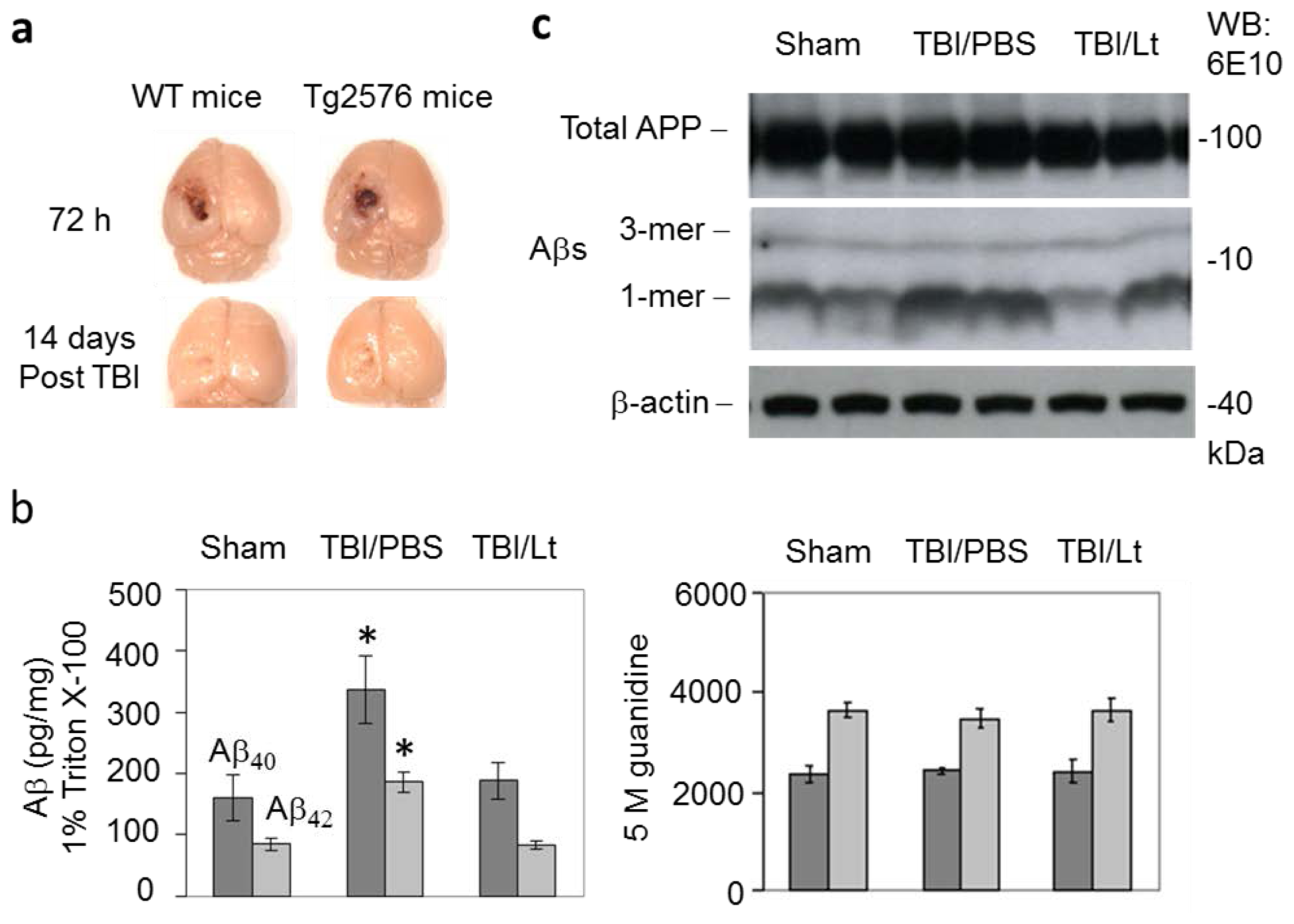
2.1. Luteolin Significantly Reduces Amyloid Pathology Elicited by TBI in Tg2576 Mice (Figure 1a–c)
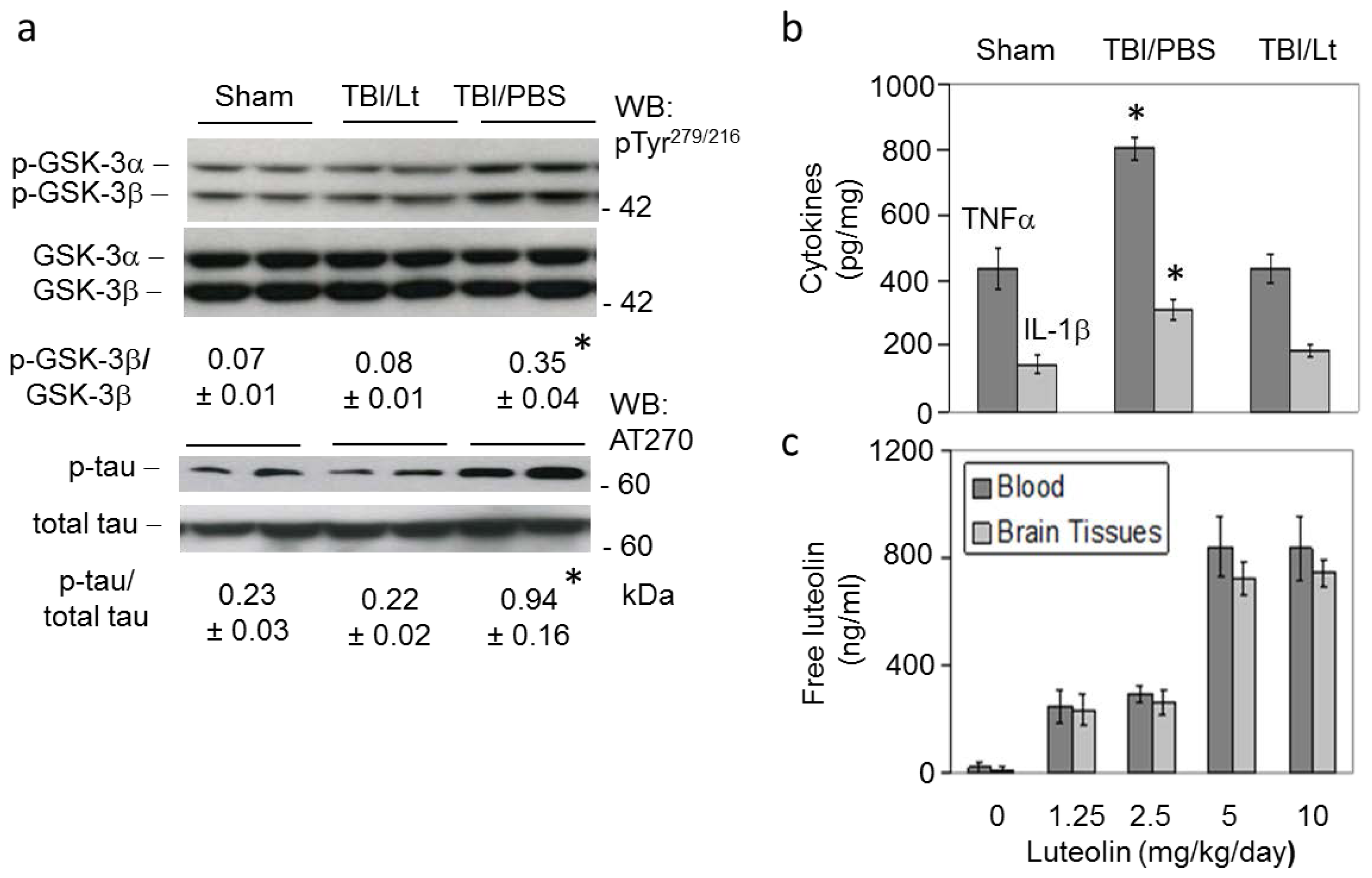
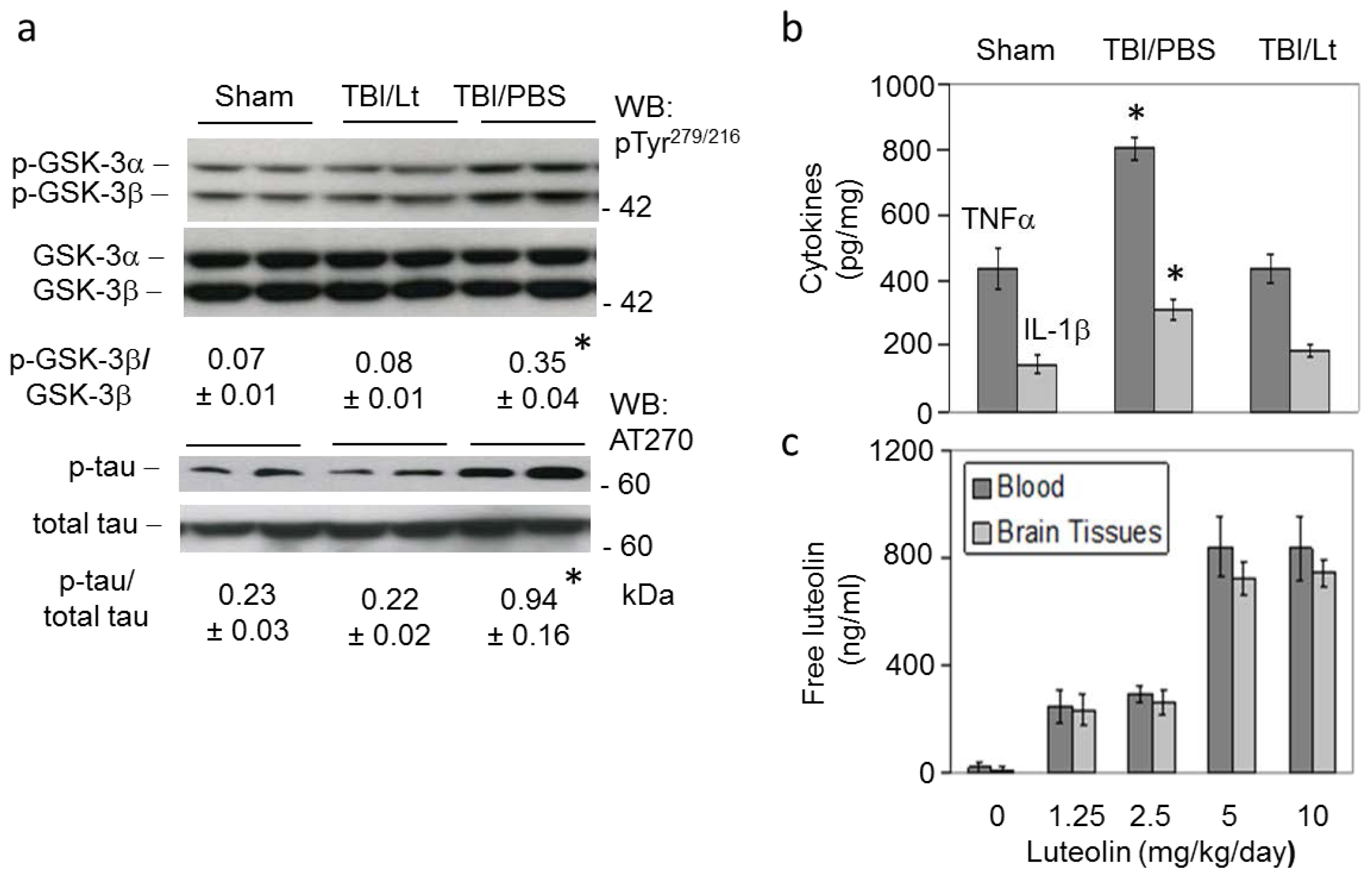
2.2. Luteolin Significantly Reduces GSK Activation, Tau Phosphorylation and Microglial-Induced Release of Inflammatory Cytokines Elicited by TBI in Tg2576 Mice (Figure 2a,b)
2.3. Luteolin Is Brain Permeable (Figure 2c)
3. Experimental Section
3.1. TBI Procedure
3.2. Luteolin Administration
3.3. ELISA
3.4. Western Blot Analysis
3.5. Brain Biodistribution Study
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- O’Meara, E.S.; Kukull, W.A.; Sheppard, L.; Bowen, J.D.; McCormick, W.C.; Teri, L.; Pfanschmidt, M.; Thompson, J.D.; Schellenberg, G.D.; Larson, E.B. Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. Am. J. Epidemiol 1997, 146, 373–384. [Google Scholar]
- Guo, Z.; Cupples, L.A.; Kurz, A.; Auerbach, S.H.; Volicer, L.; Chui, H.; Green, R.C.; Sadovnick, A.D.; Duara, R.; DeCarli, C.; et al. Head injury and the risk of AD in the MIRAGE study. Neurology 2000, 54, 1316–1323. [Google Scholar]
- Plassman, B.L.; Havlik, R.J.; Steffens, D.C.; Helms, M.J.; Newman, T.N.; Drosdick, D.; Phillips, C.; Gau, B.A.; Welsh-Bohmer, K.A.; Burke, J.R.; et al. Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 2000, 55, 1158–1166. [Google Scholar]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread tau and amyloid-Beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012, 22, 142–149. [Google Scholar]
- Ikonomovic, M.D.; Uryu, K.; Abrahamson, E.E.; Ciallella, J.R.; Trojanowski, J.Q.; Lee, V.M.; Clark, R.S.; Marion, D.W.; Wisniewski, S.R.; DeKosky, S.T. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol 2004, 190, 192–203. [Google Scholar]
- Roberts, S.B.; Ripellino, J.A.; Ingalls, K.M.; Robakis, N.K.; Felsenstein, K.M. Non-amyloidogenic cleavage of the beta-amyloid precursor protein by an integral membrane metalloendopeptidase. J. Biol. Chem 1994, 269, 3111–3116. [Google Scholar]
- DeKosky, S.T.; Abrahamson, E.E.; Ciallella, J.R.; Paljug, W.R.; Wisniewski, S.R.; Clark, R.S.; Ikonomovic, M.D. Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch. Neurol 2007, 64, 541–544. [Google Scholar]
- Gentleman, S.M.; Nash, M.J.; Sweeting, C.J.; Graham, D.I.; Roberts, G.W. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci. Lett 1993, 160, 139–144. [Google Scholar]
- Goldstein, L.; Fisher, A.; Tagge, C.; Zhang, X.; Velisek, L.; Sullivan, J.; Upreti, C.; Kracht, J.; Ericsson, M.; Wojnarowicz, M.; et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med 2012, 4, 134ra160. [Google Scholar]
- Tajiri, N.; Kellogg, S.L.; Shimizu, T.; Arendash, G.W.; Borlongan, C.V. Traumatic brain injury precipitates cognitive impairment and extracellular aβ aggregation in Alzheimer’s disease transgenic mice. PLoS One 2013, 8, e78851. [Google Scholar]
- Shitaka, Y.; Tran, H.T.; Bennett, R.E.; Sanchez, L.; Levy, M.A.; Dikranian, K.; Brody, D.L. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J. Neuropathol. Exp. Neurol 2011, 70, 551–567. [Google Scholar]
- Tran, H.; LaFerla, F.; Holtzman, D.; Brody, D. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-β accumulation and independently accelerates the development of tau abnormalities. J. Neurosci 2011, 31, 9513–9525. [Google Scholar]
- Rezai-Zadeh, K.; Douglas, S.; Bai, Y.; Tian, J.; Hou, H.; Mori, T.; Zeng, J.; Obregon, D.; Town, T.; Tan, J. Flavonoid-mediated presenilin-1 phosphorylation reduces Alzheimer’s disease beta-amyloid production. J. Cell. Mol. Med 2009, 13, 574–588. [Google Scholar]
- Zhou, F.; Chen, S.; Xiong, J.; Li, Y.; Qu, L. Luteolin reduces Zinc-induced Tau phosphorylation at Ser262/356 in an ROS-dependent manner in SH-SY5Y cells. Biol. Trace Elem. Res 2012, 149, 273–279. [Google Scholar]
- Chen, X.H.; Johnson, V.E.; Uryu, K.; Trojanowski, J.Q.; Smith, D.H. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol 2009, 29, 214–223. [Google Scholar]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar]
- Adams, J.H.; Graham, D.I.; Murray, L.S.; Scott, G. Diffuse axonal injury due to nonmissile head injury in humans: An analysis of 45 cases. Ann. Neurol 1982, 12, 557–563. [Google Scholar]
- Geddes, J.F. What’s new in the diagnosis of head injury. J. Clin. Pathol 1997, 50, 271–274. [Google Scholar]
- Geddes, J.F.; Whitwell, H.L.; Graham, D.I. Traumatic axonal injury: Practical issues for diagnosis in medicolegal cases. Neuropathol. Appl. Neurobiol 2000, 26, 105–116. [Google Scholar]
- Reichard, R.R.; White, C.L., 3rd; Hladik, C.L.; Dolinak, D. Beta-amyloid precursor protein staining of nonaccidental central nervous system injury in pediatric autopsies. J. Neurotrauma 2003, 20, 347–355. [Google Scholar]
- Graham, D.I.; Gentleman, S.M.; Nicoll, J.A.; Royston, M.C.; McKenzie, J.E.; Roberts, G.W.; Griffin, W.S. Altered beta-APP metabolism after head injury and its relationship to the aetiology of Alzheimer’s disease. Acta Neurochir. Suppl 1996, 66, 96–102. [Google Scholar]
- Braak, H.; Braak, E.; Bohl, J. Staging of Alzheimer-related cortical destruction. Eur. Neurol 1993, 33, 403–408. [Google Scholar]
- Jellinger, K.A.; Bancher, C. Neuropathology of Alzheimer’s disease: A critical update. J. Neural. Transm. Suppl 1998, 54, 77–95. [Google Scholar]
- Chauhan, N.B.; Siegel, G.J.; Feinstein, D.L. Propentofylline attenuates tau hyperphosphorylation in Alzheimer’s Swedish mutant model Tg2576. Neuropharmacology 2005, 48, 93–104. [Google Scholar]
- Tan, J.; Town, T.; Paris, D.; Mori, T.; Suo, Z.; Crawford, F.; Mattson, M.P.; Flavell, R.A.; Mullan, M. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science 1999, 17, 2352–2355. [Google Scholar]
- Lee, K.K.; Seow, W.T.; Ng, I. Demographical profiles of adult severe traumatic brain injury patients: Implications for healthcare planning. Singapore Med. J 2006, 47, 31–36. [Google Scholar]
- Molgaard, C.A.; Stanford, E.P.; Morton, D.J.; Ryden, L.A.; Schubert, K.R.; Golbeck, A.L. Epidemiology of head trauma and neurocognitive impairment in a multi-ethnic population. Neuroepidemiology 1990, 9, 233–242. [Google Scholar]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-beta pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci 2010, 11, 361–370. [Google Scholar]
- McKee, A.C.; Stern, R.A.; Nowinski, C.J.; Stein, T.D.; Alvarez, V.E.; Daneshvar, D.H.; Lee, H.S.; Wojtowicz, S.M.; Hall, G.; Baugh, C.M.; et al. The spectrum of disease in chronic traumatic encephalopathy. Brain: J. Neurol 2013, 136, 43–64. [Google Scholar]
- Stern, R.A.; Riley, D.O.; Daneshvar, D.H.; Nowinski, C.J.; Cantu, R.C.; McKee, A.C. Long-term consequences of repetitive brain trauma: Chronic traumatic encephalopathy. Phys. Med. Rehabil 2011, 3, S460–S467. [Google Scholar]
- Gavett, B.; Stern, R.; McKee, A. Chronic traumatic encephalopathy: A potential late effect of sport-related concussive and subconcussive head trauma. Clin. Sports Med 2011, 30, 179–188. [Google Scholar]
- Giunta, B.; Hou, H.; Zhu, Y.; Salemi, J.; Ruscin, A.; Shytle, R.D.; Tan, J. Fish oil enhances anti-amyloidogenic properties of green tea EGCG in Tg 2576 mice. Neurosci. Lett 2010, 471, 134–138. [Google Scholar]
- Smith, A.; Giunta, B.; Bickford, P.C.; Fountain, M.; Tan, J.; Shytle, R.D. Nanolipidic particles improve the bioavailability and alpha-secretase inducing ability of epigallocatechin-3-gallate (EGCG) for the treatment of Alzheimer’s disease. Int. J. Pharm 2010, 389, 207–212. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sawmiller, D.; Li, S.; Shahaduzzaman, M.; Smith, A.J.; Obregon, D.; Giunta, B.; Borlongan, C.V.; Sanberg, P.R.; Tan, J. Luteolin Reduces Alzheimer’s Disease Pathologies Induced by Traumatic Brain Injury. Int. J. Mol. Sci. 2014, 15, 895-904. https://doi.org/10.3390/ijms15010895
Sawmiller D, Li S, Shahaduzzaman M, Smith AJ, Obregon D, Giunta B, Borlongan CV, Sanberg PR, Tan J. Luteolin Reduces Alzheimer’s Disease Pathologies Induced by Traumatic Brain Injury. International Journal of Molecular Sciences. 2014; 15(1):895-904. https://doi.org/10.3390/ijms15010895
Chicago/Turabian StyleSawmiller, Darrell, Song Li, Md Shahaduzzaman, Adam J. Smith, Demian Obregon, Brian Giunta, Cesar V. Borlongan, Paul R. Sanberg, and Jun Tan. 2014. "Luteolin Reduces Alzheimer’s Disease Pathologies Induced by Traumatic Brain Injury" International Journal of Molecular Sciences 15, no. 1: 895-904. https://doi.org/10.3390/ijms15010895