Dual Drug Release Electrospun Core-Shell Nanofibers with Tunable Dose in the Second Phase



Abstract
:
1. Introduction
2. Results and Discussion
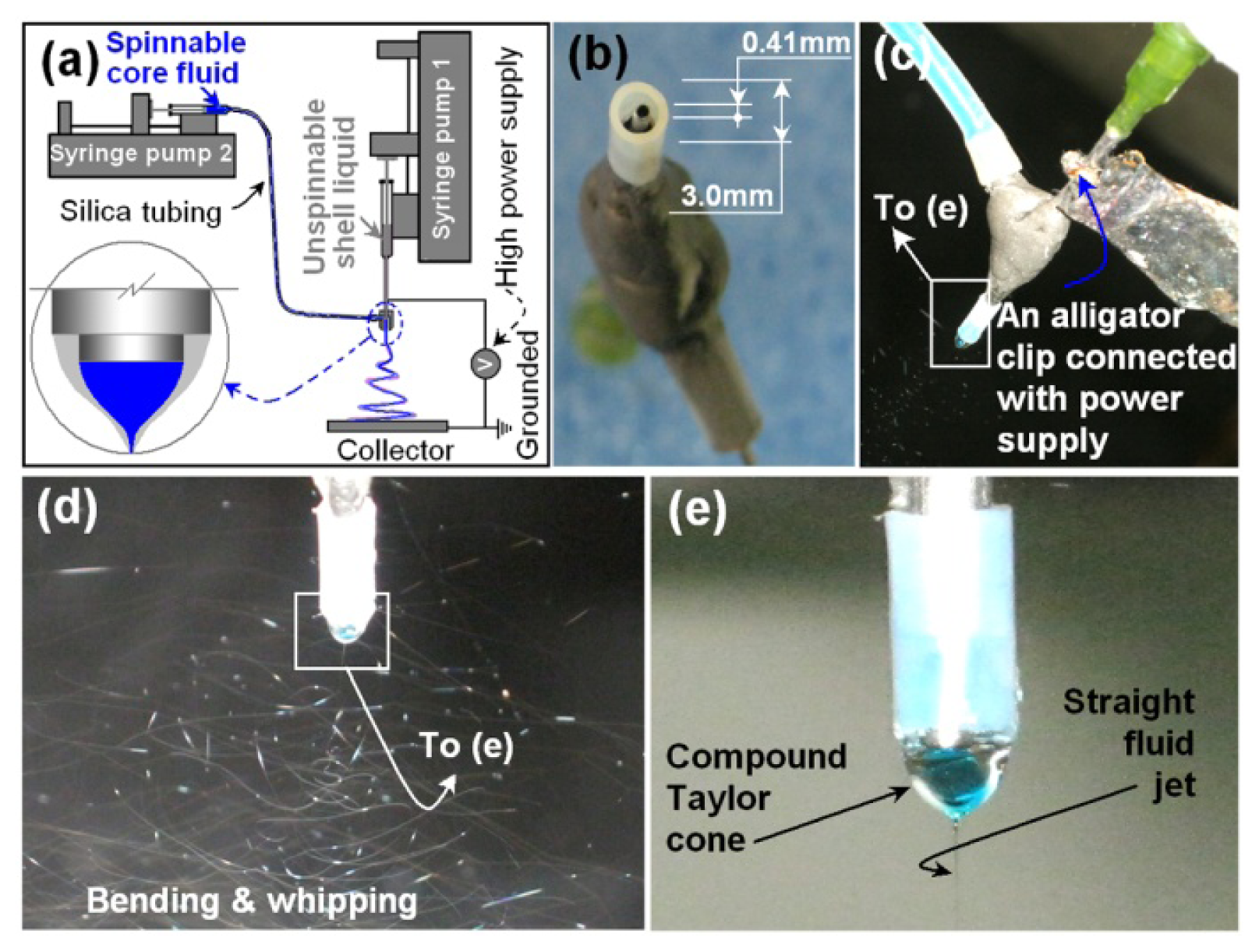
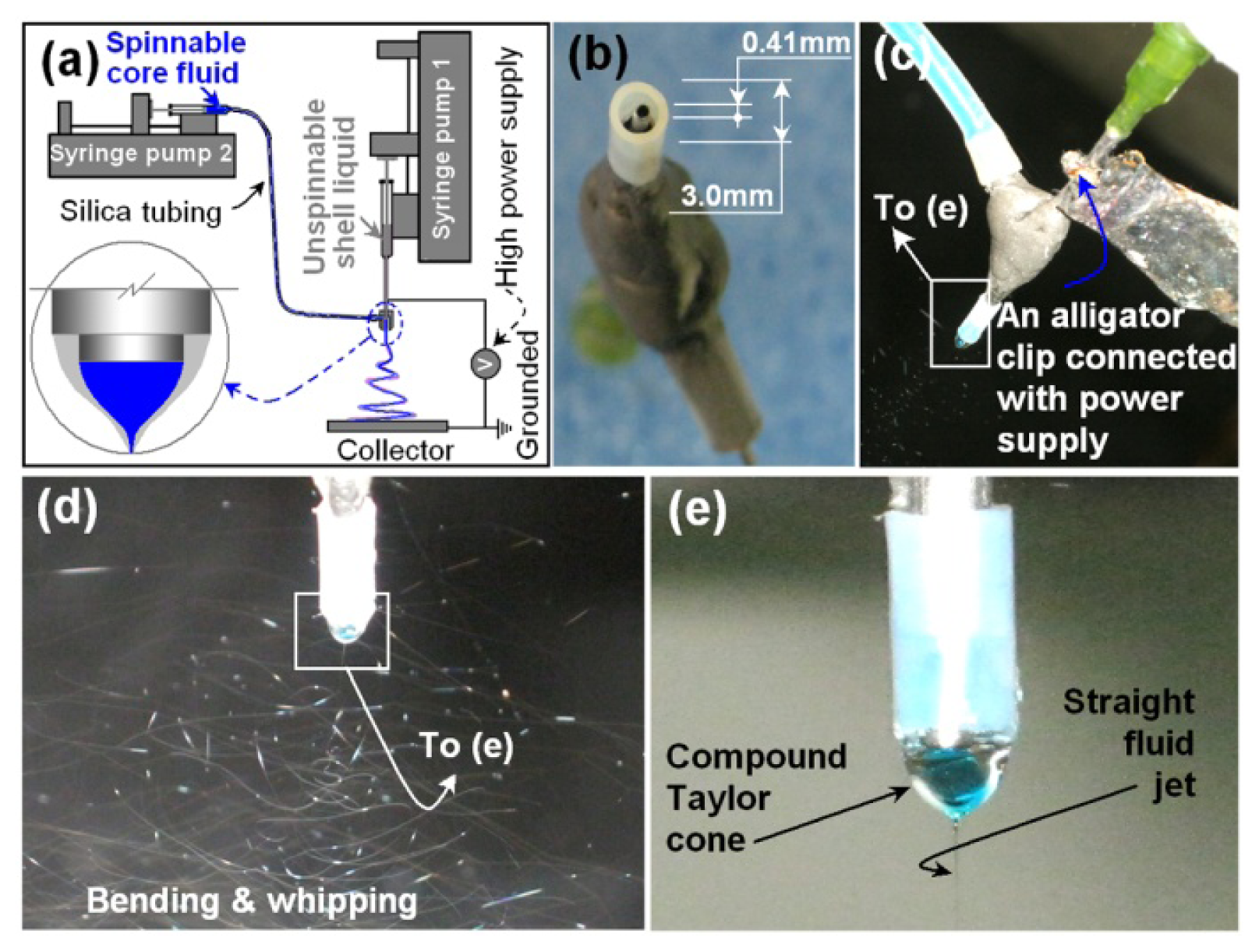
2.1. The Teflon-Coated Concentric Spinneret and the Modified Coaxial Electrospinning
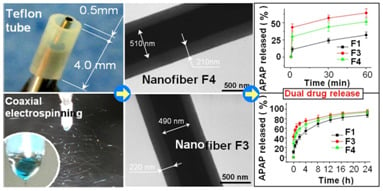
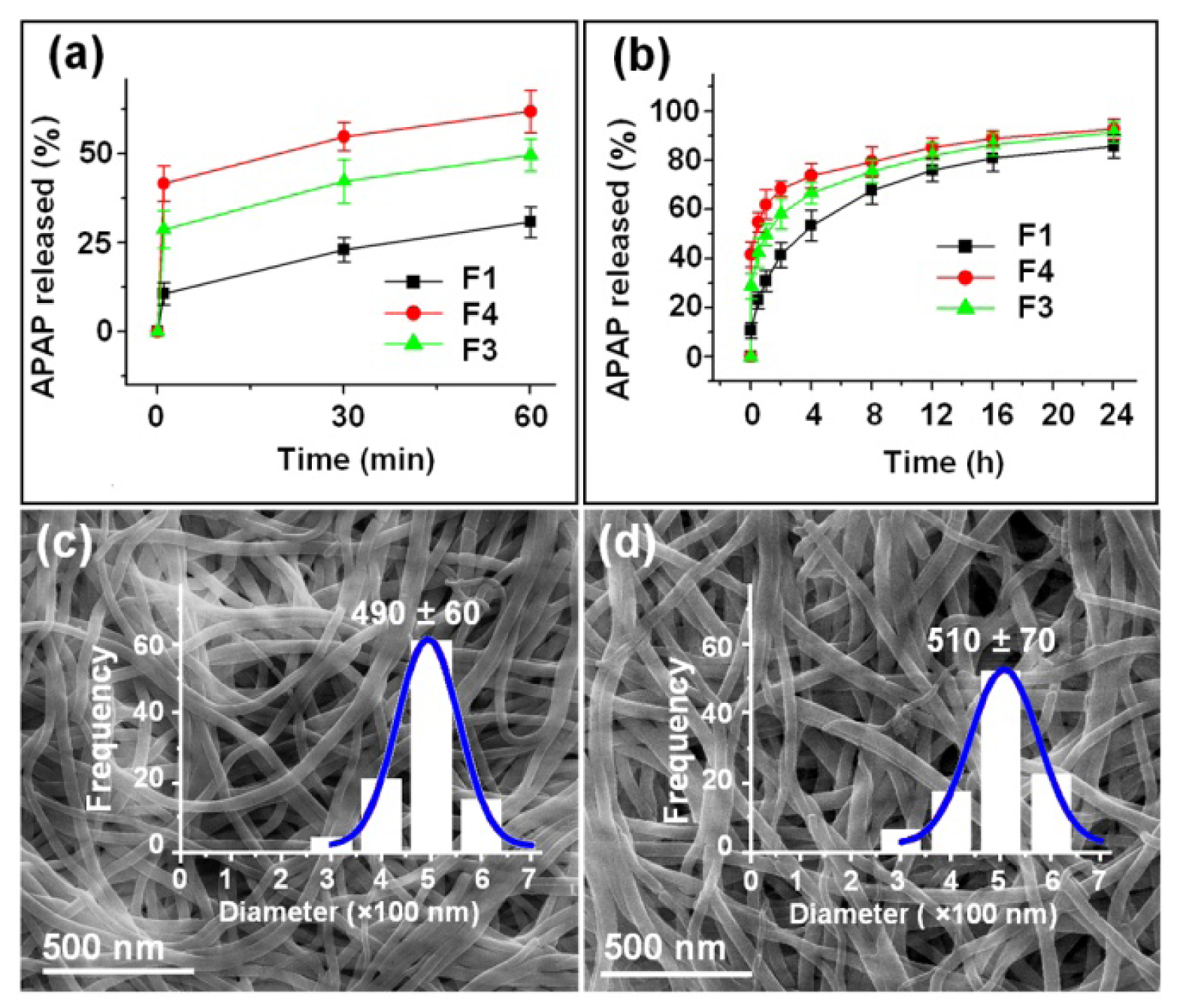
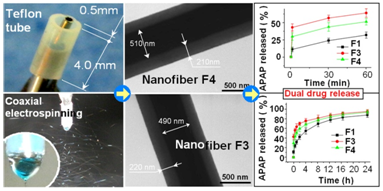
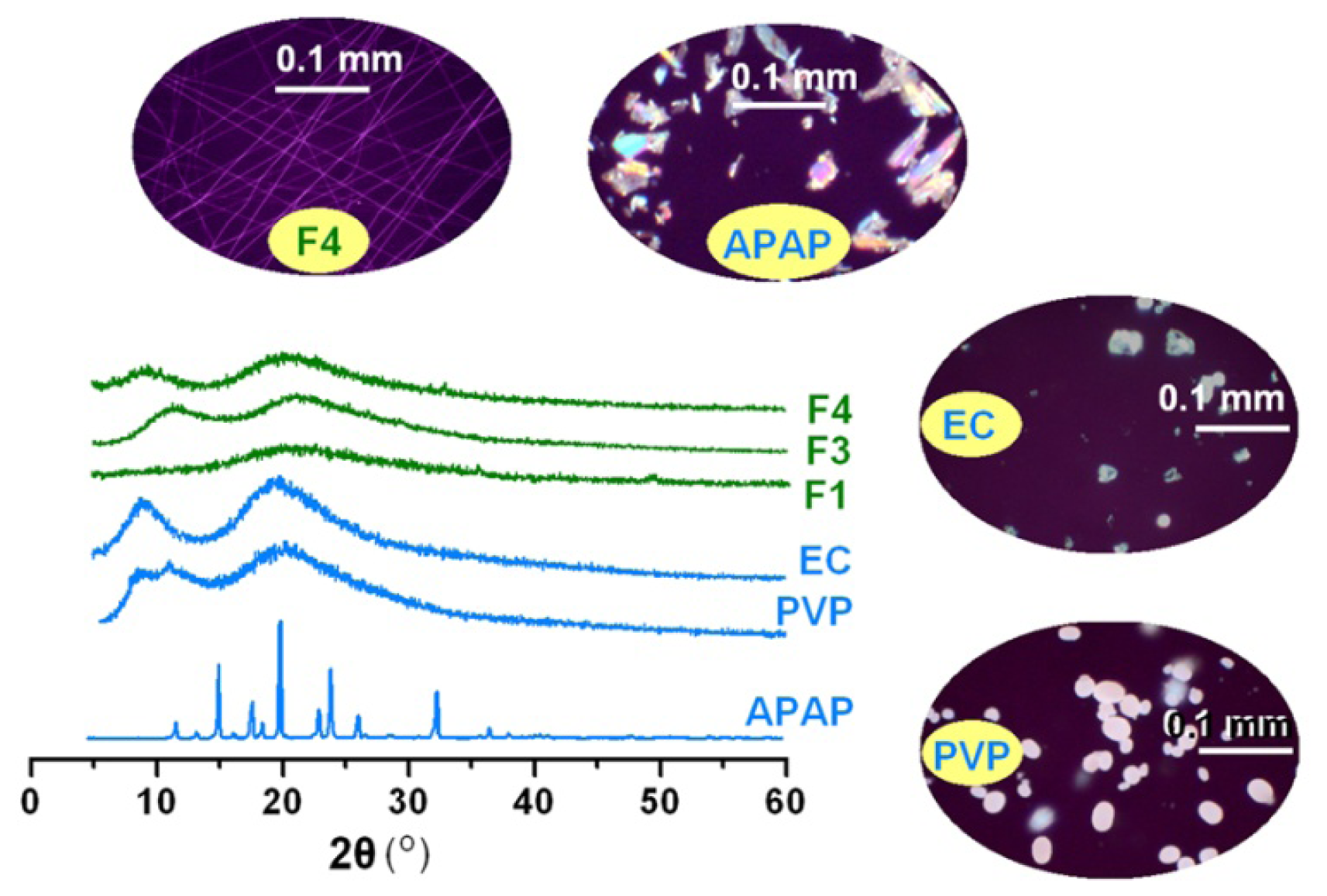
2.2. Morphology and Structure of Nanofibers
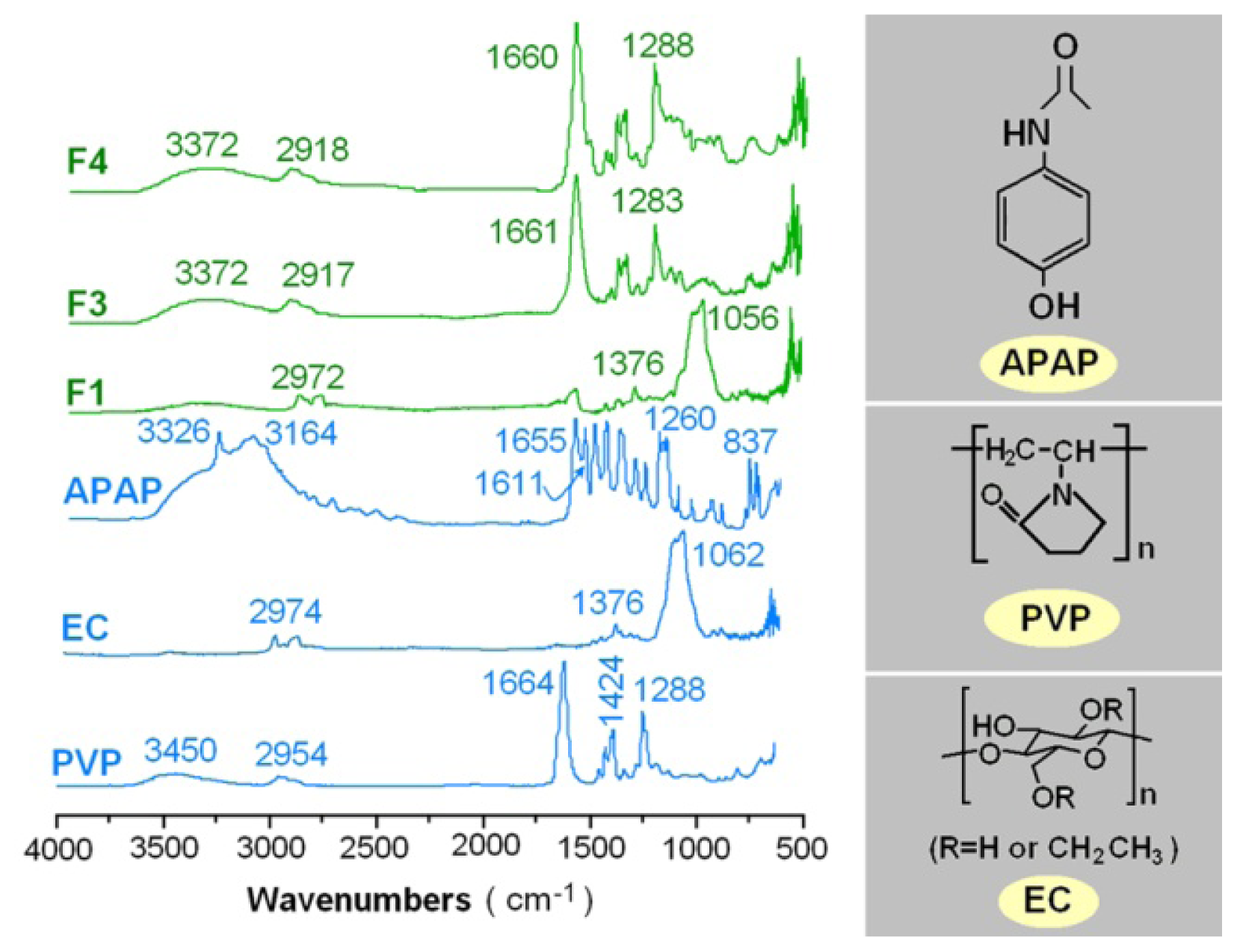
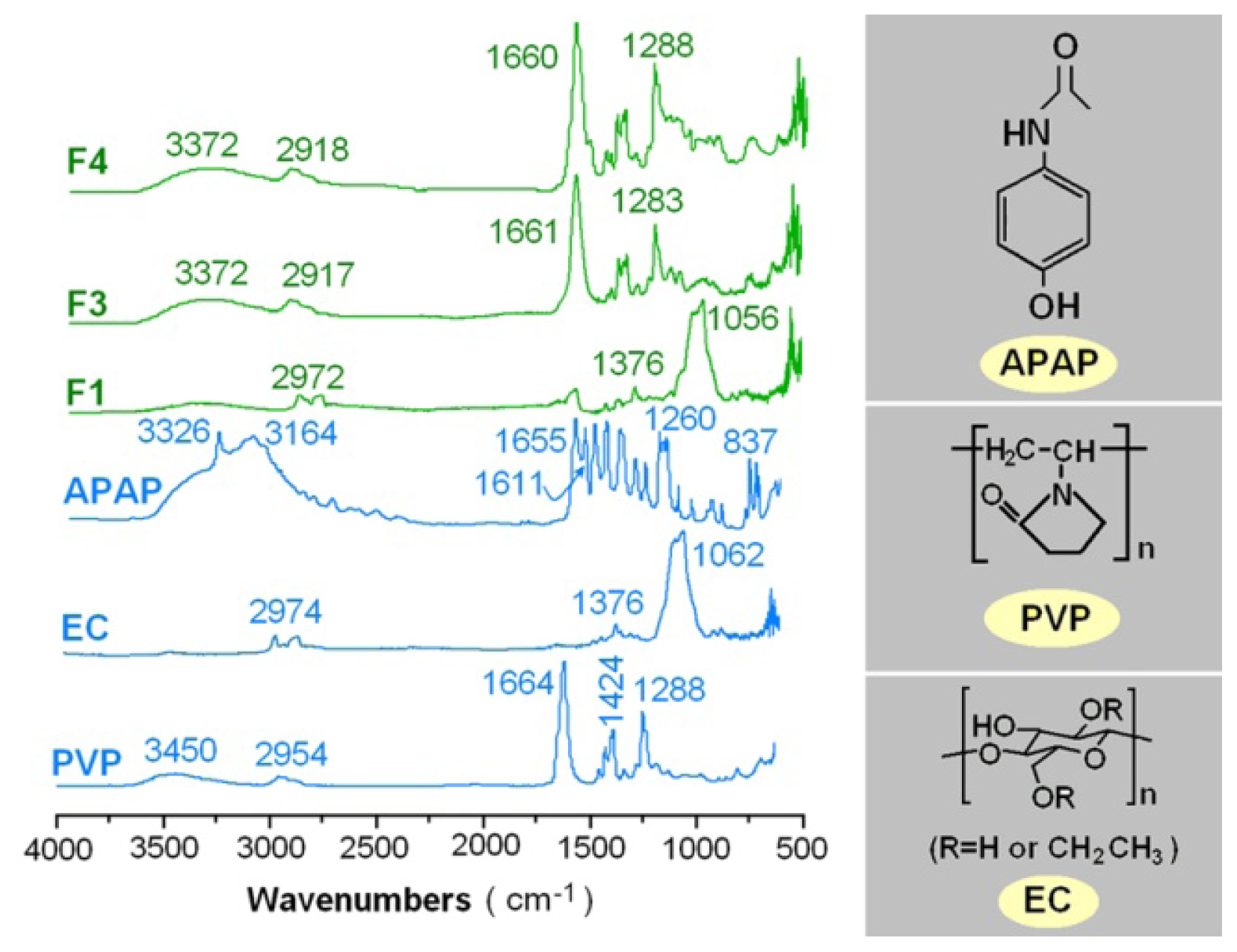
2.3. Physical Status and Compatibility of Components
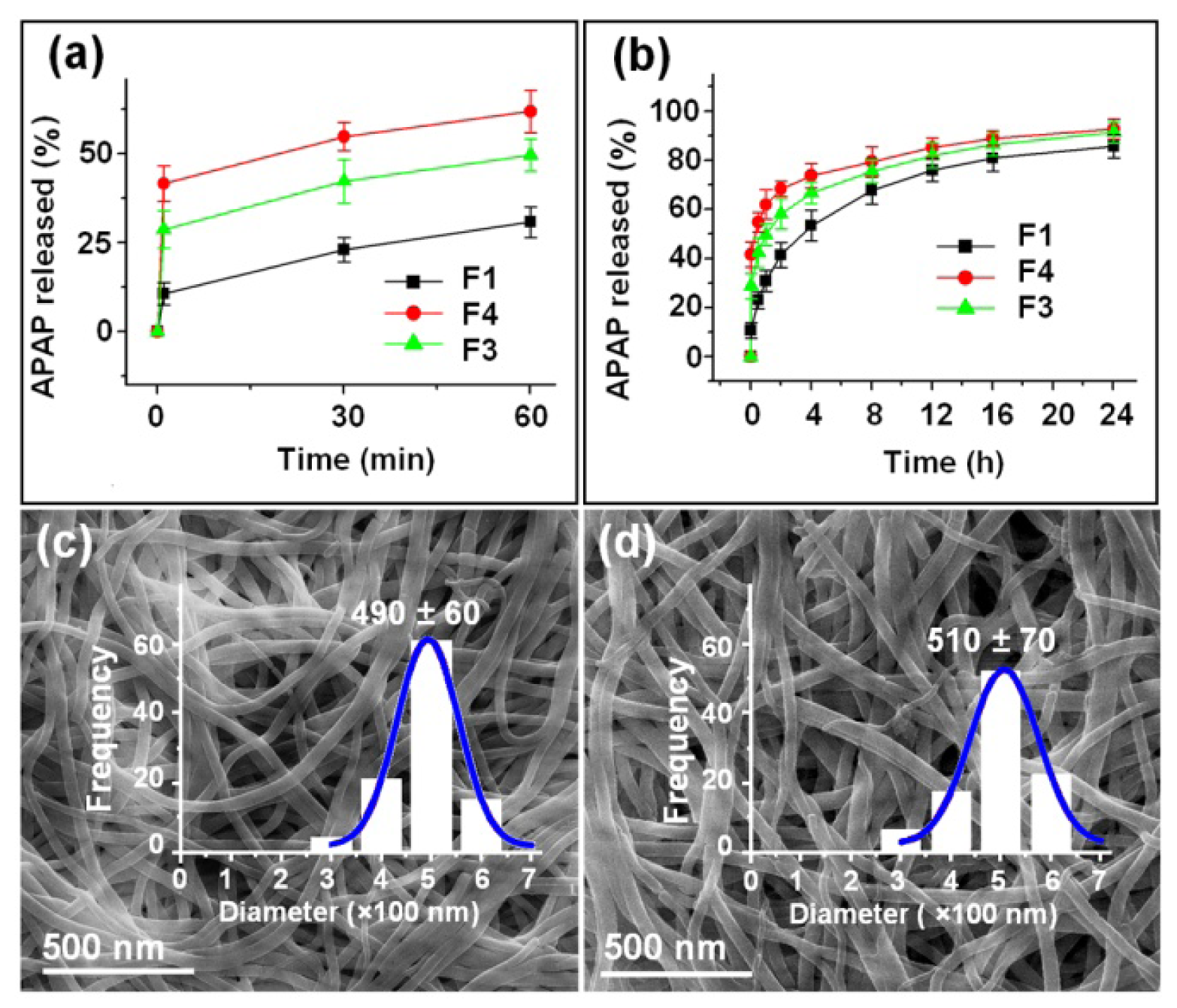
2.4. Dual Release Profiles with Tunable Dose in the Second Phase
3. Experimental Section
3.1. Materials
3.2. Modified Coaxial Electrospinning
3.3. Characterization
3.3.1. Morphology
3.3.2. Physical Status and Compatibility
3.3.3. In Vitro Dissolution Tests
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Agarwal, S.; Greiner, A.; Wendorff, J.H. Functional materials by electrospinning of polymers. Prog. Polym. Sci 2013, 38, 963–991. [Google Scholar]
- Zhang, K.-H.; Ye, Q.; Yan, Z.-Y. Influence of post-treatment with 75% (v/v) ethanol vapor on the properties of SF/P(LLA-CL) nanofibrous scaffolds. Int. J. Mol. Sci 2012, 13, 2036–2047. [Google Scholar]
- Feng, Q.; Tang, B.; Wei, Q.; Hou, D.; Bi, S.; Wei, A. Preparation of a Cu(II)-PVA/PA6 composite nanofibrous membrane for enzyme immobilization. Int. J. Mol. Sci 2012, 13, 12734–12746. [Google Scholar]
- Luo, C.J.; Stoyanov, S.D.; Stride, E.; Pelan, E.; Edirisinghe, M. Electrospinning versus fibre production methods: From specifics to technological convergence. Chem. Soc. Rev 2012, 41, 4708–4735. [Google Scholar]
- Heunis, T.; Bshena, O.; Klumperman, B.; Dicks, L. Release of bacteriocins from nanofibers prepared with combinations of poly(d,l-lactide) (PDLLA) and poly(Ethylene Oxide) (PEO). Int. J. Mol. Sci 2011, 12, 2158–2173. [Google Scholar]
- Zhang, K.-H.; Yu, Q.-Z.; Mo, X.-M. Fabrication and intermolecular interactions of silk fibroin/hydroxybutyl chitosan blended nanofibers. Int. J. Mol. Sci 2011, 12, 2187–2199. [Google Scholar]
- Bedford, N.M.; Dickerson, M.B.; Drummy, L.F.; Koerner, H.; Singh, K.M.; Vasudev, M.C.; Durstock, M.F.; Naik, R.R.; Steckl, A.J. Nanofiber-based bulk-heterojunction organic solar cells using coaxial electrospinning. Adv. Energy Mater 2012, 2, 1136–1144. [Google Scholar]
- Dzenis, Y. Spinning continuous fibers for nanotechnology. Science 2004, 304, 1917–1919. [Google Scholar]
- Li, X.Y.; Li, Y.C.; Yu, D.G.; Liao, Y.Z.; Wang, X. Fast disintegrating quercetin-Loaded drug delivery systems fabricated using coaxial electrospinning. Int. J. Mol. Sci 2013, 14, 21647–21659. [Google Scholar]
- Moghe, A.K.; Gupta, B.S. Co-axial electrospinning for nanofiber structures: Preparation and applications. Polym. Rev 2008, 48, 353–377. [Google Scholar]
- Yarin, A.L. Coaxial electrospinning and emulsion electrospinning of core–shell fibers. Polym. Adv. Technol 2011, 22, 310–317. [Google Scholar]
- Wang, S.-G.; Jiang, X.; Chen, P.-C.; Yu, A.-G.; Huang, X.-J. Preparation of coaxial-Electrospun Poly[bis(p-methylphenoxy)] phosphazene nanofiber membrane for enzyme immobilization. Int. J. Mol. Sci 2012, 13, 14136–14148. [Google Scholar]
- Loscertales, I.G.; Barrero, A.; Guerrero, I.; Cortijo, R.; Marquez, M.; Canan-Calvo, A.M. Micro/nano encapsulation via electrified coaxial liquid jets. Science 2002, 295, 1695–1698. [Google Scholar]
- Su, Y.; Su, Q.; Liu, W.; Lim, M.; Venugopal, J.R.; Mo, X.; Ramakrishna, S.; Al-Deyab, S.S.; El-Newehy, M. Controlled release of bone morphogenetic protein and dexamethasone loaded in core–shell PLLACL–collagen fibers for use in bone tissue engineering. Acta Biomater 2012, 8, 763–771. [Google Scholar]
- Han, D.; Steckl, A. Superhydrophobic and oleophobic fibers by coaxial electrospinning. Langmuir 2009, 25, 9454–9462. [Google Scholar]
- Yu, D.G.; Branford-White, C.; Bligh, S.W.A.; White, K.; Chatterton, N.P.; Zhu, L.M. Improving polymer nanofiber quality using a modified co-axial electrospinning process. Macromol. Rapid Commun 2011, 32, 744–750. [Google Scholar]
- Yu, D.G.; Williams, G.R.; Wang, X.; Liu, X.K.; Li, H.L.; Bligh, S.W.A. Dual drug release nanocomposites prepared using a combination of electrospraying and electrospinning. RSC Adv 2013, 3, 4652–4658. [Google Scholar]
- Jha, M.K.; Rahman, M.H.; Rahman, M.M. Biphasic oral solid drug delivery system: A review. Int. J. Pharm. Sci. Res 2011, 2, 1108–1115. [Google Scholar]
- Bandari, S.; Eaga, C.M.; Thadishetty, A.; Yamsani, M.R. Formulation and evaluation of multiple tablets as a biphasic gastroretentive floating drug delivery system for fenoverine. Acta Pharm 2010, 60, 89–97. [Google Scholar]
- Ofori-Kwakye, K.; Fell, J.T. Leaching of pectin from mixed films containing pectin, chitosan and HPMC intended for biphasic drug delivery. Int. J. Pharm 2003, 250, 251–257. [Google Scholar]
- Loo, S.C.J.; Tan, Z.Y.S.; Chow, Y.J.; Lin, S.L.I. Drug release from irradiated PLGA and PLLA multi-layered films. J. Pharm. Sci 2010, 99, 3060–3071. [Google Scholar]
- Monkhouse, D.C.; Yao, J.; Sherwood, M.J.; Cima, M.J.; Bornancini, E. Dosage Forms Exhibiting Multi-Phasic Release Kinetics and Methods of Manufacture thereof. U.S. Patent 6280771, 28 August 2001. [Google Scholar]
- Song, B.; Wu, C.; Chang, J. Dual drug release from electrospun poly(lactic-co-glycolic acid)/mesoporous silica nanoparticles composite mats with distinct release profiles. Acta Biomater 2012, 8, 1901–1907. [Google Scholar]
- Han, D.; Steckl, A. Triaxial electrospun nanofiber membranes for controlled dual release of functional molecules. ACS Appl. Mater. Interfaces 2013, 5, 8241–8245. [Google Scholar]
- Xiang, Q.; Ma, Y.M.; Yu, D.G.; Jin, M.; Williams, G.R. Electrospinning using a Teflon-coated spinneret. Appl. Surf. Sci 2013, 284, 889–893. [Google Scholar]
- Moodley, K.; Pillay, V.; Choonara, Y.E.; du Toit, L.C.; Ndesendo, V.M.K.; Kumar, P.; Cooppan, S.; Bawa, P. Oral drug delivery systems comprising altered geometric configurations for controlled drug delivery. Int. J. Mol. Sci 2012, 13, 18–43. [Google Scholar]
- De Villiers, M.M.; Wurster, D.E.; Van der Watt, J.G.; Ketkar, A. Xray powder diffraction determination of the relative amount of crystalline acetaminophen in solid dispersions with polyvinylpyrrolidone. Int. J. Pharm 1998, 163, 219–224. [Google Scholar]
- Garekani, H.A.; Sadeghi, F.; Ghazi, A. Increasing the aqueous solubility of acetaminophen in the presence of polyvinylpyrrolidone and investigation of the mechanisms involved. Drug Dev. Ind. Pharm 2003, 29, 173–179. [Google Scholar]
- Yu, D.G.; Wang, X.; Li, X.Y.; Chian, W.; Li, Y.; Liao, Y.Z. Electrospun biphasic drug release polyvinylpyrrolidone/ethyl cellulose core/shell nanofibers. Acta Biomater 2013, 9, 5665–5672. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Process | Drug con. (w/v) | Flow rate (mL/h) | Morphology c | Total drug content d | Diameter (nm) | ||
|---|---|---|---|---|---|---|---|---|
| Shell a | Core b | Shell | Core | |||||
| F1 | Single | - | 3% | - | 1.0 | Linear | 11.11% | 570 ± 140 |
| F2 | 5% | - | 1.0 | - | - | - | - | |
| F3 | Coaxial | 5% | 3% | 0.3 | 1.0 | Linear | 13.64% | 930 ± 170 |
| F4 | 5% | 6% | 0.3 | 1.0 | Linear | 20.83% | 910 ± 220 | |
| F5 | 5% | 6% | 0.3 | 0.5 | Mixed | - | - | |
| Nanofiber No. | Release in the 1st phase (1 min) | Release after 24 h b | Release in the 2nd phase | ||
|---|---|---|---|---|---|
| Theoretical a | Experimental b | Theoretical a | Experimental b | ||
| F1 | - | 10.7% (2.1) | 85.4% (17.1) | - | 74.7% (14.9) |
| F3 | 33.3% (6.7) | 41.6% (8.3) | 92.7% (18.5) | 66.7% (13.3) | 51.1% (10.2) |
| F4 | 20.0% (4.0) | 28.7% (5.7) | 91.4% (18.3) | 80.0% (16.0) | 62.7% (12.5) |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qian, W.; Yu, D.-G.; Li, Y.; Liao, Y.-Z.; Wang, X.; Wang, L. Dual Drug Release Electrospun Core-Shell Nanofibers with Tunable Dose in the Second Phase. Int. J. Mol. Sci. 2014, 15, 774-786. https://doi.org/10.3390/ijms15010774
Qian W, Yu D-G, Li Y, Liao Y-Z, Wang X, Wang L. Dual Drug Release Electrospun Core-Shell Nanofibers with Tunable Dose in the Second Phase. International Journal of Molecular Sciences. 2014; 15(1):774-786. https://doi.org/10.3390/ijms15010774
Chicago/Turabian StyleQian, Wei, Deng-Guang Yu, Ying Li, Yao-Zu Liao, Xia Wang, and Lu Wang. 2014. "Dual Drug Release Electrospun Core-Shell Nanofibers with Tunable Dose in the Second Phase" International Journal of Molecular Sciences 15, no. 1: 774-786. https://doi.org/10.3390/ijms15010774