Efficacy and Feasibility of the Epithelial Cell Adhesion Molecule (EpCAM) Immunomagnetic Cell Sorter for Studies of DNA Methylation in Colorectal Cancer


 ,
, 
Abstract
:1. Introduction
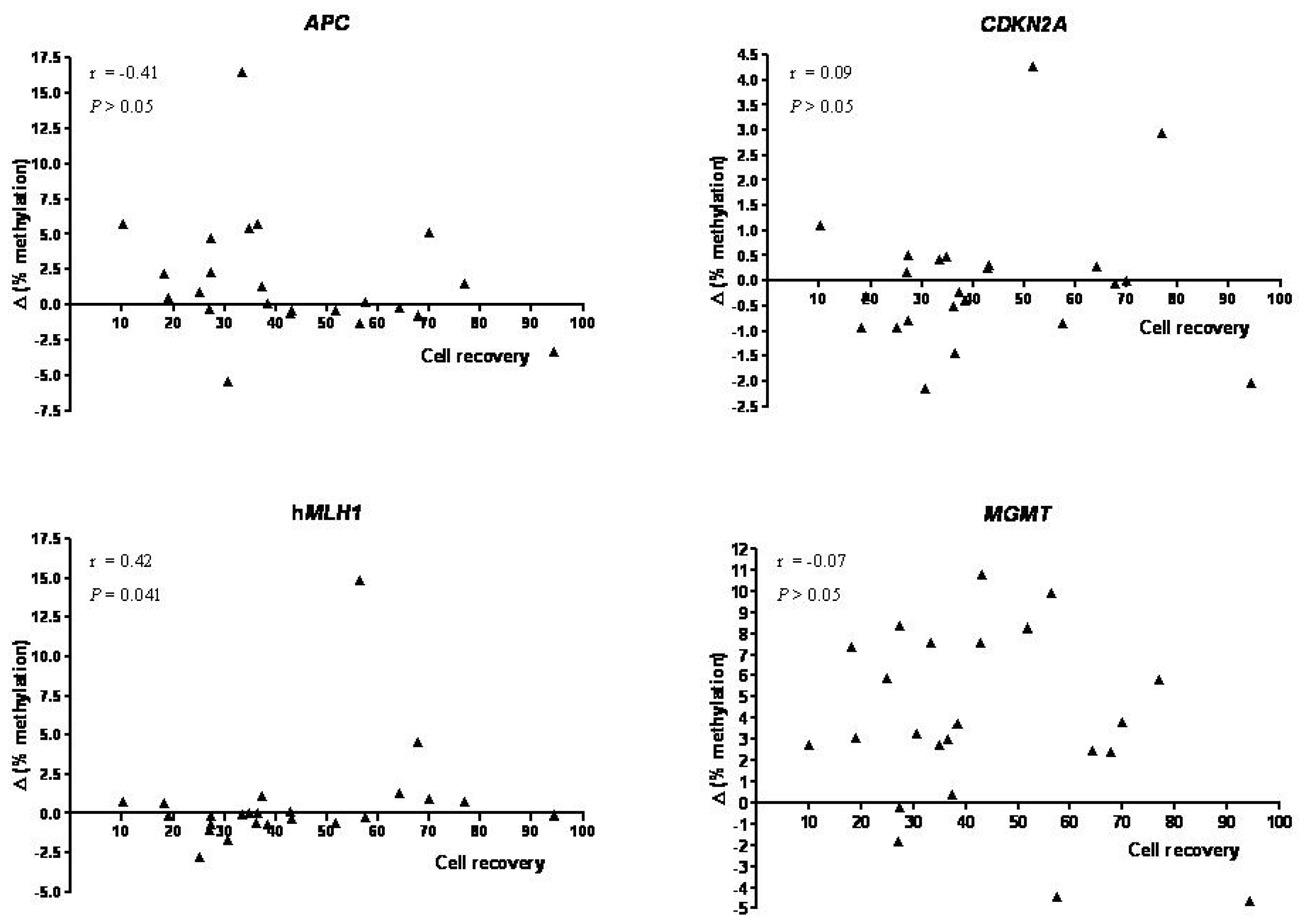
2. Results and Discussion
2.1. Results
2.1.1. Patient Characteristics
2.1.2. Recovery of EpCAM+ Cells
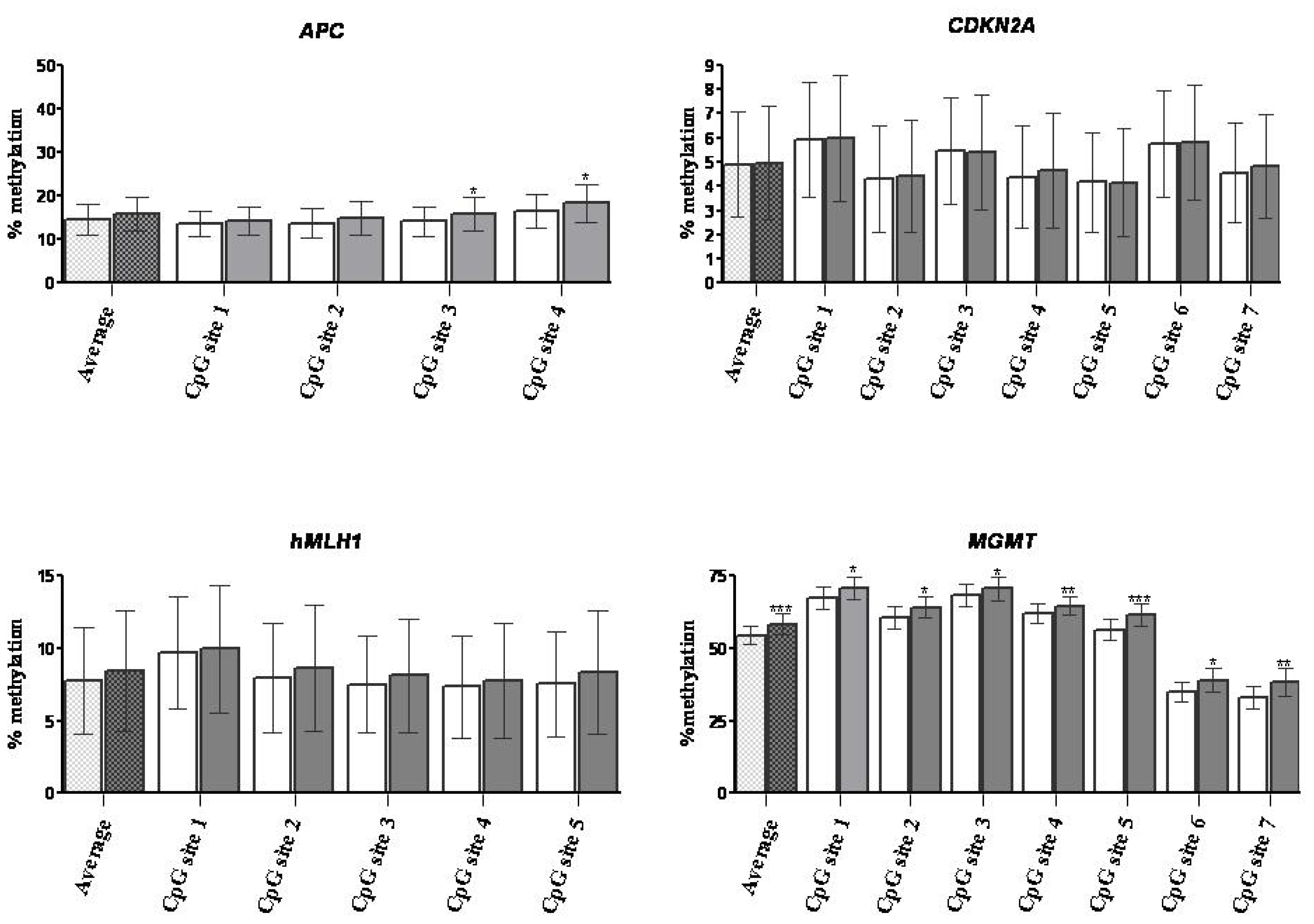
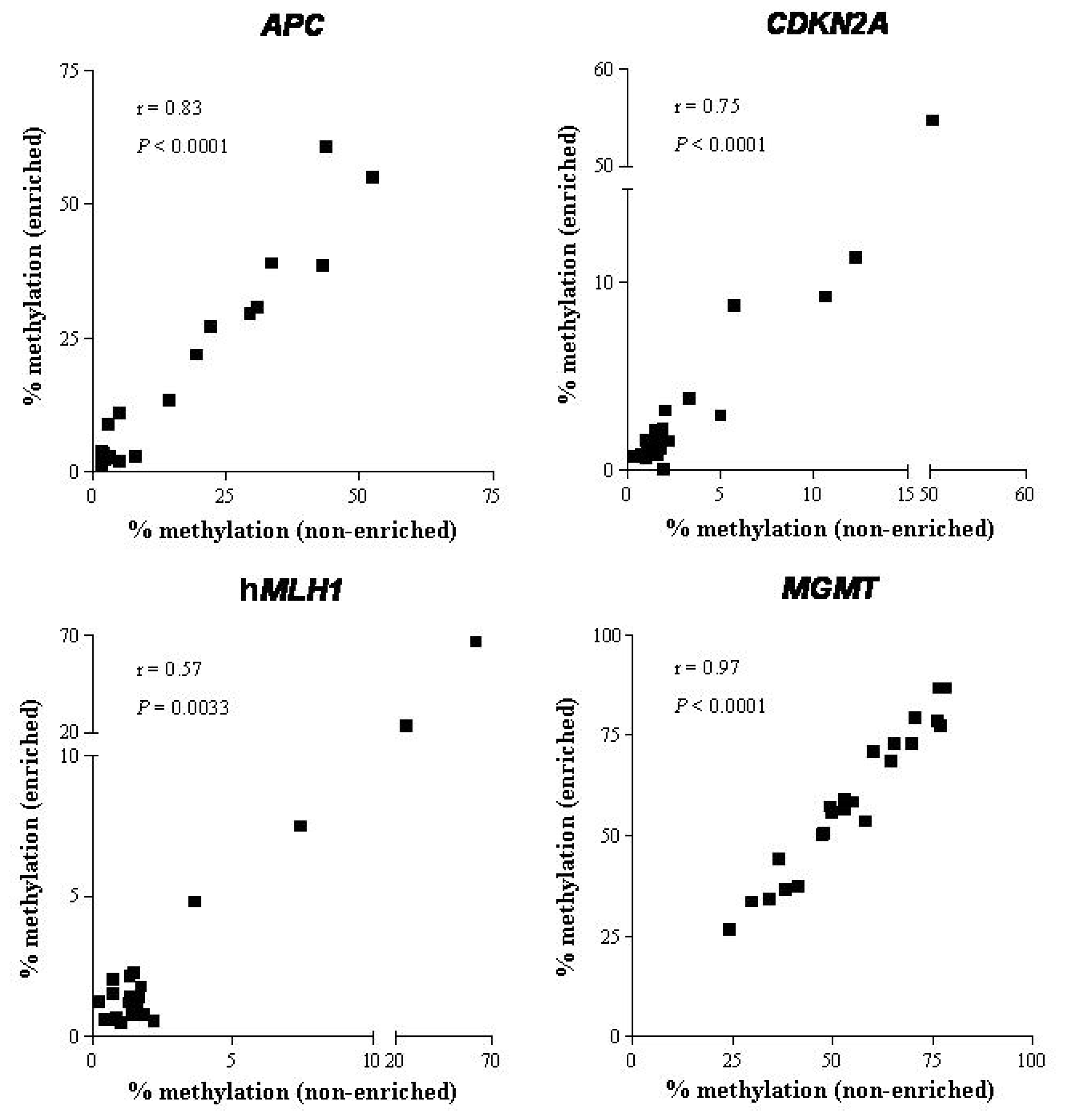
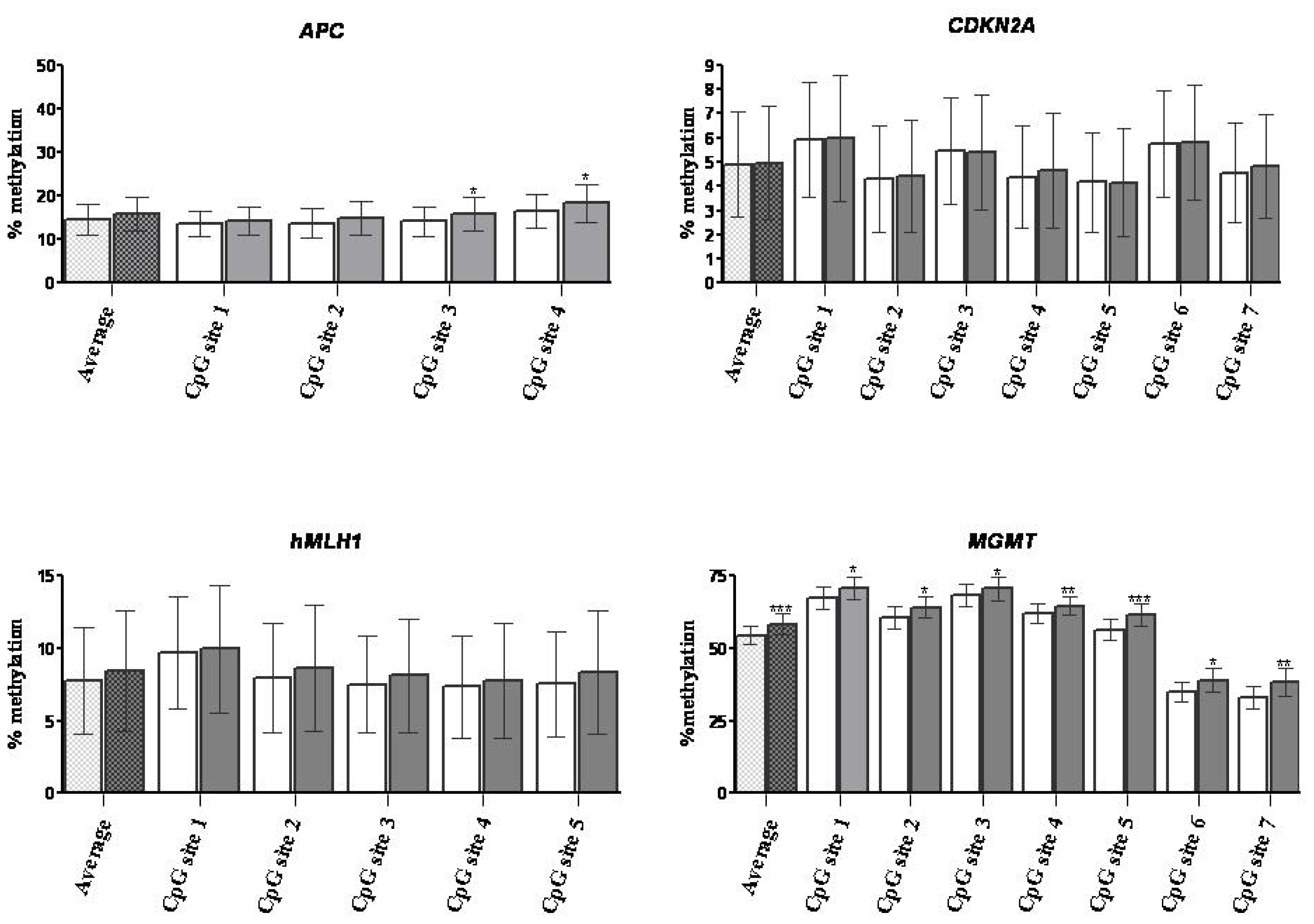
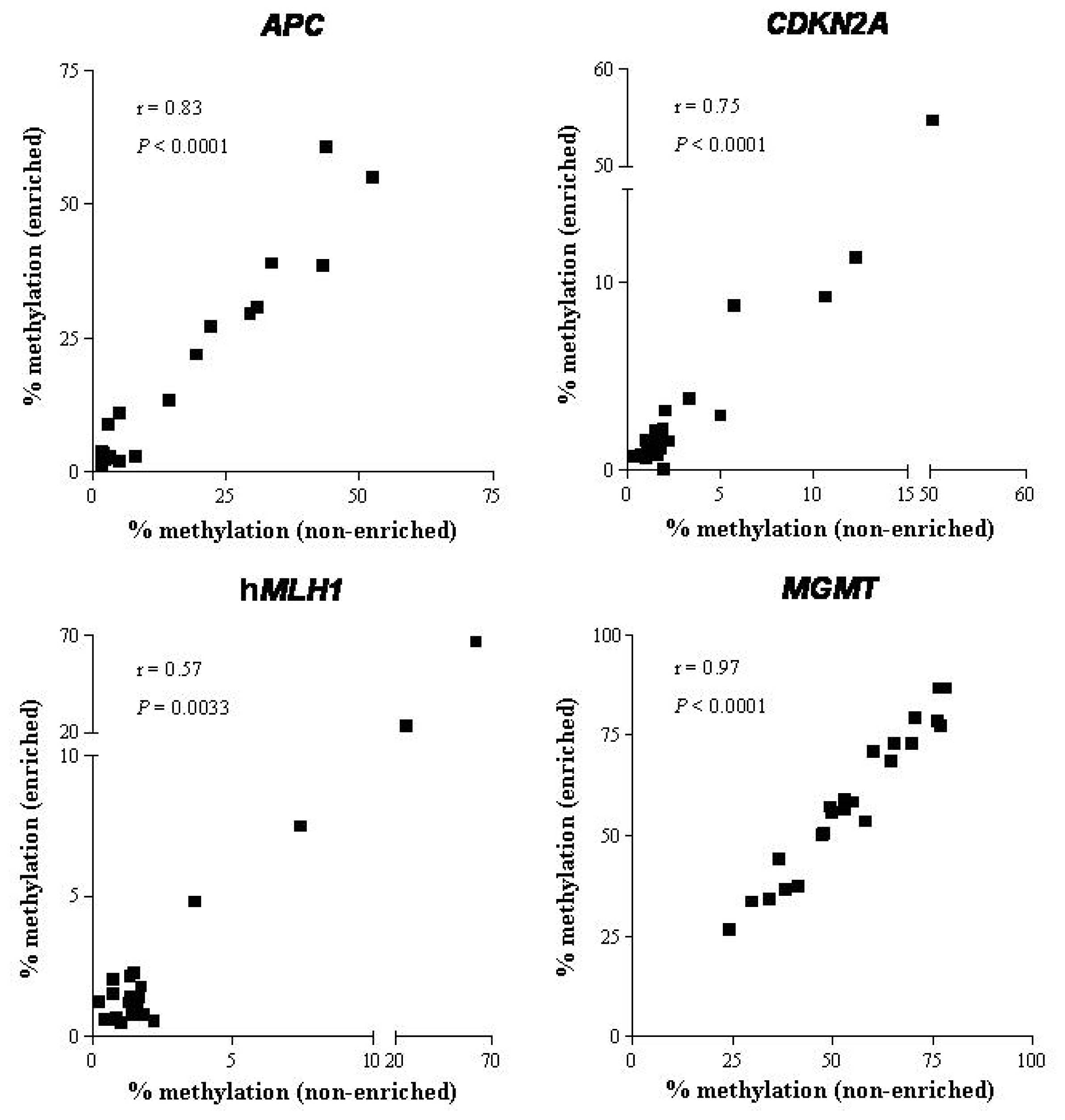
2.1.3. DNA Methylation
2.2. Discussion
3. Experimental Section
3.1. Tissue Samples
3.2. Generation of a Single Cell Suspension from Colorectal Cancer Tissues
3.3. Tumor Cell Enrichment with Anti-EpCAM Antibodies
3.4. Extraction of Genomic DNA
3.5. Bisulfite Modification
3.6. Pyrosequencing
3.7. Statistics
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar]
- Toyota, M.; Suzuki, H. Epigenetic drivers of genetic alterations. Adv. Genet 2010, 70, 309–323. [Google Scholar]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar]
- Harrison, S.; Benziger, H. The molecular biology of colorectal carcinoma and its implications: A review. Surgeon 2011, 9, 200–210. [Google Scholar]
- Migliore, L.; Migheli, F.; Spisni, R.; Coppedè, F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J. Biomed. Biotechnol 2011, 2011, 792362. [Google Scholar]
- Shen, L.; Toyota, M.; Kondo, Y.; Zhang, E.L.L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; Hamilton, S.R.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.M.; van den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012, 22, 271–282. [Google Scholar]
- Coppedè, F. Epigenetic biomarkers of colorectal cancer: Focus on DNA methylation. Cancer Lett 2014, 342, 238–247. [Google Scholar]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet 2012, 13, 679–692. [Google Scholar]
- Marsh, S. Pyrosequencing applications. Methods Mol. Biol 2007, 373, 15–24. [Google Scholar]
- Novak, P.; Stampfer, M.R.; Munoz-Rodriguez, J.L.; Garbe, J.C.; Ehrich, M.; Futscher, B.W.; Jensen, T.J. Cell-type specific DNA methylation patterns define human breast cellular identity. PLoS One 2012, 7, e52299. [Google Scholar]
- Van der Gun, B.T.F.; de Groote, M.L.; Kazemier, H.G.; Arendzen, A.J.; Terpstra, P.; Ruiters, M.H.; McLaughlin, P.M.; Rots, M.G. Transcription factors and molecular epigenetic marks underlying EpCAM overexpression in ovarian cancer. Br. J. Cancer 2011, 105, 312–319. [Google Scholar]
- Königsberg, R.; Obermayr, E.; Bises, G.; Pfeiler, G.; Gneist, M.; Wrba, F.; de Santis, M.; Zeillinger, R.; Hudec, M.; Dittrich, C. Detection of EpCAM positive and negative circulating tumor cells in metastatic breast cancer patients. Acta Oncol 2011, 50, 700–710. [Google Scholar]
- Went, P.; Vasei, M.; Bubendorf, L.; Terracciano, L.; Tornillo, L.; Riede, U.; Kononen, J.; Simon, R.; Sauter, G.; Baeuerle, P.A. Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Br. J. Cancer 2006, 94, 128–135. [Google Scholar]
- Spizzo, G.; Went, P.; Dirnhofer, S.; Obrist, P.; Moch, H.; Baeuerle, P.A.; Mueller-Holzner, E.; Marth, C.; Gastl, G.; Zeimet, A.G. Overexpression of epithelial cell adhesion molecule (Ep-CAM) is an independent prognostic marker for reduced survival of patients with epithelial ovarian cancer. Gynecol. Oncol 2006, 103, 483–488. [Google Scholar]
- Maetzel, D.; Denzel, S.; Mack, B.; Canis, M.; Went, P.; Benk, M.; Kieu, C.; Papior, P.; Baeuerle, P.A.; Munz, M.; et al. Nuclear signalling by tumorassociated antigen EpCAM. Nat. Cell Biol 2009, 11, 162–171. [Google Scholar]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumors: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treat. Rev 2012, 38, 68–75. [Google Scholar]
- Migheli, F.; Stoccoro, A.; Coppedè, F.; Omar, W.A.W.; Failli, A.; Consolini, R.; Seccia, M.; Spisni, R.; Miccoli, P.; Mathers, J.C.; Migliore, L. comparison study of ms-hrm and pyrosequencing techniques for quantification of APC and CDKN2A gene methylation. PLoS One 2013, 8, e52501. [Google Scholar]
- Croner, R.S.; Guenther, K.; Foertsch, T.; Siebenhaar, R.; Brueckl, W.M.; Stremmel, C.; Hlubek, F.; Hohenberger, W.; Reingruber, B. Tissue preparation for gene expression profiling of colorectal carcinoma: Three alternatives to laser microdissection with preamplification. J. Lab. Clin. Med 2004, 143, 344–351. [Google Scholar]
- McKay, J.A.; Xie, L.; Harris, S.; Wong, Y.K.; Mathers, J.C. Blood as a surrogate marker for tissue-specific DNA methylation and changes due to folate depletion in post-partum female mice. Mol. Nutr. Food Res 2011, 55, 1026–1235. [Google Scholar]
- De Bruin, E.C.; van de Pas, S.; Lips, E.H.; van Eijk, R.; van der Zee, M.M.; Lombaerts, M.; van Wezel, T.; Marijnen, C.A.; van Krieken, J.H.; Medema, J.P.; et al. Macrodissection versus microdissection of rectal carcinoma: Minor influence of stroma cells to tumor cell gene expression profiles. BMC Genomics 2005, 14, 142. [Google Scholar]
- Tveito, S.; Andersen, K.; Kåresen, R.; Fodstad, Ø. Analysis of EpCAM positive cells isolated from sentinel lymph nodes of breast cancer patients identifies subpopulations of cells with distinct transcription profiles. Breast Cancer Res 2011, 13, R75. [Google Scholar]
- Tveito, S.; Maelandsmo, G.M.; Hoifodt, H.K.; Rasmussen, H.; Fodstad, O. Specific isolation of disseminated cancer cells: A new method permitting sensitive detection of target molecules of diagnostic and therapeutic value. Clin. Exp. Metastasis 2007, 24, 317–332. [Google Scholar]
- Chimonidou, M.; Strati, A.; Tzitzira, A.; Sotiropoulou, G.; Malamos, N.; Georgoulias, V.; Lianidou, E.S. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin. Chem 2011, 57, 1169–1177. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nature Rev. Genet 2002, 3, 415–428. [Google Scholar]
- Munz, M.; Baeuerle, P.A.; Gires, O. The emerging role of EpCAM in cancer and stem cell signalling. Cancer Res 2009, 69, 5627–5629. [Google Scholar]
- Gires, O.; Klein, C.A.; Baeuerle, P.A. On the abundance of EpCAM on cancer stem cells. Nat. Rev. Cancer 2009, 9, 143. [Google Scholar]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumor cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar]
- Sieuwerts, A.M.; Kraan, J.; Bolt, J.; van der Spoel, P.; Elstrodt, F.; Schutte, M.; Martens, J.W.; Gratama, J.W.; Sleijfer, S.; Foekens, J.A. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J. Natl. Cancer Inst 2009, 101, 61–66. [Google Scholar]
- Failli, A.; Consolini, R.; Legitimo, A.; Spisni, R.; Castagna, M.; Romanini, A.; Crimaldi, G.; Miccoli, P. The challenge of culturing human colorectal tumor cells: Establishment of a cell culture model by the comparison of different methodological approaches. Tumori 2009, 95, 343–347. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Characteristic | No. of patients |
|---|---|
| Age (range years) | |
| ≤50 (47–50) | 3 |
| 51–69 (53–65) | 5 |
| 70–79 (70–76) | 5 |
| ≥80 (81–91) | 11 |
| Gender | |
| Female | 10 |
| Male | 14 |
| Stage | |
| I | 4 |
| II | 9 |
| III | 6 |
| IV | 5 |
| Primary tumor site | |
| Colon dx | 15 |
| Colon sx | 9 |
| Tumor differentiation | |
| Well | 0 |
| Moderate | 15 |
| Poor | 9 |
| Additional pathologic characteristics | |
| Signet ring cell | 0 |
| Lymphatic invasion | 11 |
| Vascular invasion | 8 |
| Tumor budding | 12 |
| Clinical stage | Recovery | APC | CDKN2A | hMLH1 | MGMT | ||||
|---|---|---|---|---|---|---|---|---|---|
| Non-enriched | Enriched | Non-enriched | Enriched | Non-enriched | Enriched | Non-enriched | Enriched | ||
| I (n = 4) | 64.4% (27.3%–95.3%) | 13.7% (2.1%–31.0%) | 15.3% (1.8%–30.7%) | 4.1% (2.0%–50.4%) | 5.1% (0%–54.7%) | 1.2% (0.7%–1.4%) | 0.7% (0.5%–2.1%) | 64.4% (49.9%–78.3%) | 67.4% (53.4%–83.6%) |
| II (n = 9) | 36.5% (10.1%–70.0%) | 5.2% (2.1%–33.8%) | 11% (2.4%–38.9%) | 1.8% (0.8%–12.3%) | 1.8% (0.7%–11.3%) | 1.7% (0.3%–64.7%) | 1.5% (0.6%–79.6%) | 53.1% (36.6%–76.7%) | 59.0% * (36.4%–86.6%) |
| III (n = 6) | 36.1% (18.9%–64.2%) | 2.5% (2.0%–14.5%) | 2.7% (2.3%–19.9%) | 1.1% (1.0%–1.9%) | 1.4% (0.7%–2.1%) | 1.6% (0.7%–3.7%) | 1.8% (0.6%–4.8%) | 50.4% (24.2%–76.9%) | 53.5% ** (26.6%–77.3%) |
| IV (n = 4) | 38.1% (27.3%–67.7%) | 23.8% (1.8%–55.6%) | 28.8% (1.2%–60.6%) | 1.8% (0.5%–3.4%) | 1.5% (0.7%–3.8%) | 1.5% (0.5%–61.8%) | 1.2% (0.6%–66.4%) | 57.4% (34.4%–76.2%) | 64.9% (34.2%–78.6%) |
| Genes | Primer pyrosequencing | Ta | RefSeqGene | CpG sites analyzed |
|---|---|---|---|---|
| APC | F-TATTAATTTTTTTGTTTGTTGGGGA | 55 °C | NG_008481.4 | C/TGGAGTGC/TGGGTC/TGGGAAGC/TGG |
| R-AACTACACCAATACAACCACATATC | AGAGAGAAGTAGTTGTGTAATTC/TGTTG | |||
| Sequencing primer: GGGGTTTTGTGTTTTATTG | GATGC/TGGATTAGGGC/TGT | |||
| CDKN2A | F-AGAGGATTTGAGGGATAG | 50 °C | NG_007485.1 | GAGGGTGGGGC/TGGATC/TGC/TGTGC/TG |
| R-AATTCCCCTACAAACTTC | TTC/TGGC/TGGTTGC/TGGAGAGGGGGAGAGT | |||
| Sequencing primer: GGGTTGGTTGGTTATTA | AGGTAGC/TGGGC/TGGC/TG | |||
| hMLH1 | F-GGTTATAAGAGTAGGGTTAA | 45 °C | NG_007109.2 | TTC/TGTATTTTTC/TGAGTTTTTAAAAAC/TGA |
| R-ATACCAATCAAATTTCTC | ATTAATAGGAAGAGC/TGGATAGC/TGATTTTT | |||
| Sequencing primer: TGTTTTTATTGGTTGGATAT | AAC/TGC/TGTAAGC/TGTA | |||
| MGMT | F-AGTTTTTTTGGTGGATATA | 47 °C | NP_002403.2 | TC/TGC/TGTTTC/TGGGTTTAGC/TGTAGTC/TGT |
| R-TACCTTTTCCTATCACAA | TTC/TGAGTAGGATC/TGGGATTTTTATTAAG | |||
| Sequencing primer: TTTAGGAGGGGAGAGAT | ||||
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Failli, A.; Legitimo, A.; Migheli, F.; Coppedè, F.; Mathers, J.C.; Spisni, R.; Miccoli, P.; Migliore, L.; Consolini, R. Efficacy and Feasibility of the Epithelial Cell Adhesion Molecule (EpCAM) Immunomagnetic Cell Sorter for Studies of DNA Methylation in Colorectal Cancer. Int. J. Mol. Sci. 2014, 15, 44-57. https://doi.org/10.3390/ijms15010044
Failli A, Legitimo A, Migheli F, Coppedè F, Mathers JC, Spisni R, Miccoli P, Migliore L, Consolini R. Efficacy and Feasibility of the Epithelial Cell Adhesion Molecule (EpCAM) Immunomagnetic Cell Sorter for Studies of DNA Methylation in Colorectal Cancer. International Journal of Molecular Sciences. 2014; 15(1):44-57. https://doi.org/10.3390/ijms15010044
Chicago/Turabian StyleFailli, Alessandra, Annalisa Legitimo, Francesca Migheli, Fabio Coppedè, John C. Mathers, Roberto Spisni, Paolo Miccoli, Lucia Migliore, and Rita Consolini. 2014. "Efficacy and Feasibility of the Epithelial Cell Adhesion Molecule (EpCAM) Immunomagnetic Cell Sorter for Studies of DNA Methylation in Colorectal Cancer" International Journal of Molecular Sciences 15, no. 1: 44-57. https://doi.org/10.3390/ijms15010044