Chloride Channelopathies of ClC-2

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
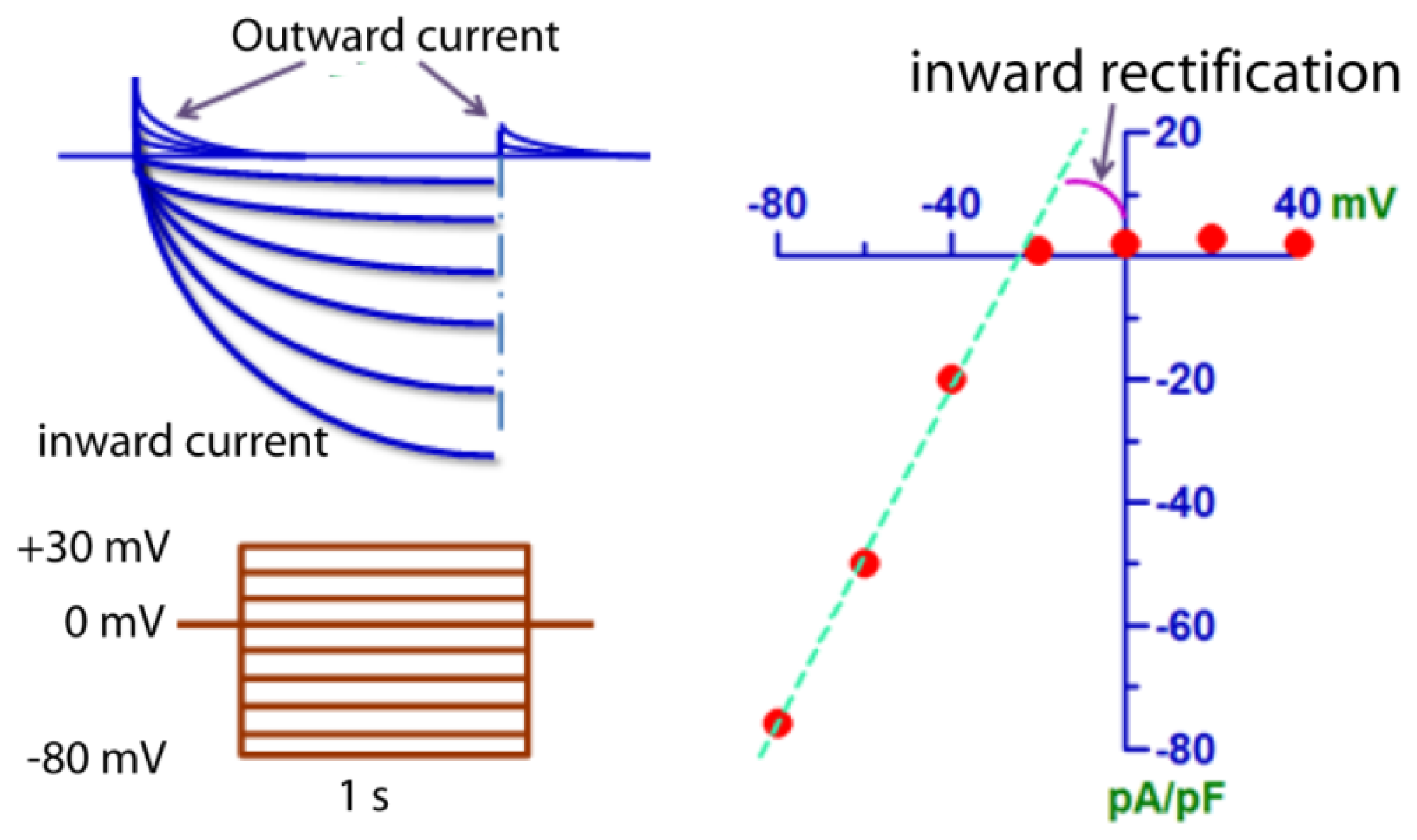
2. Biophysical Properties of ClC-2
3. Pharmacological Properties of ClLC-2
3.1. ClC-2 Inhibitors
3.2. ClC-2 Activators
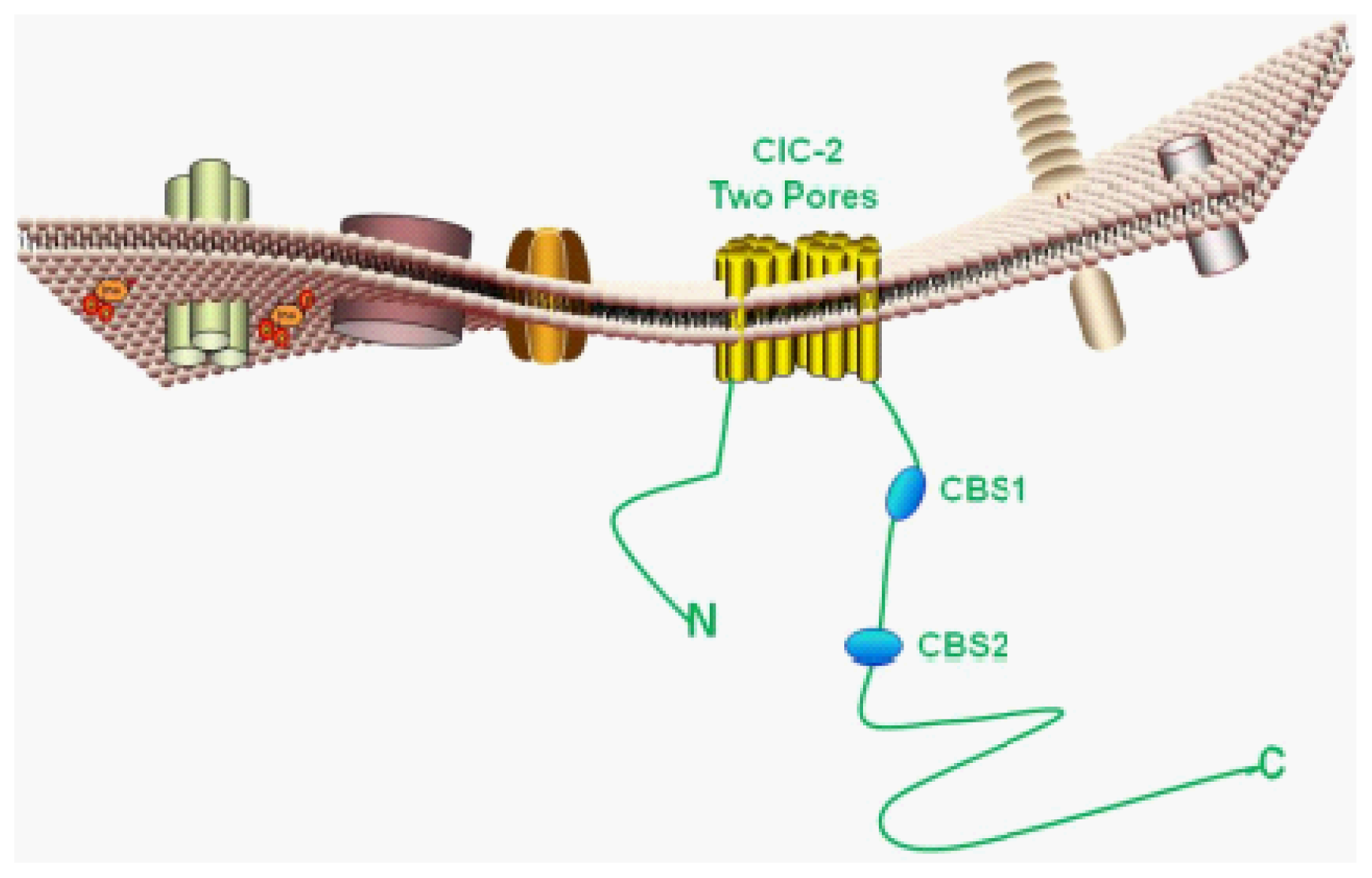
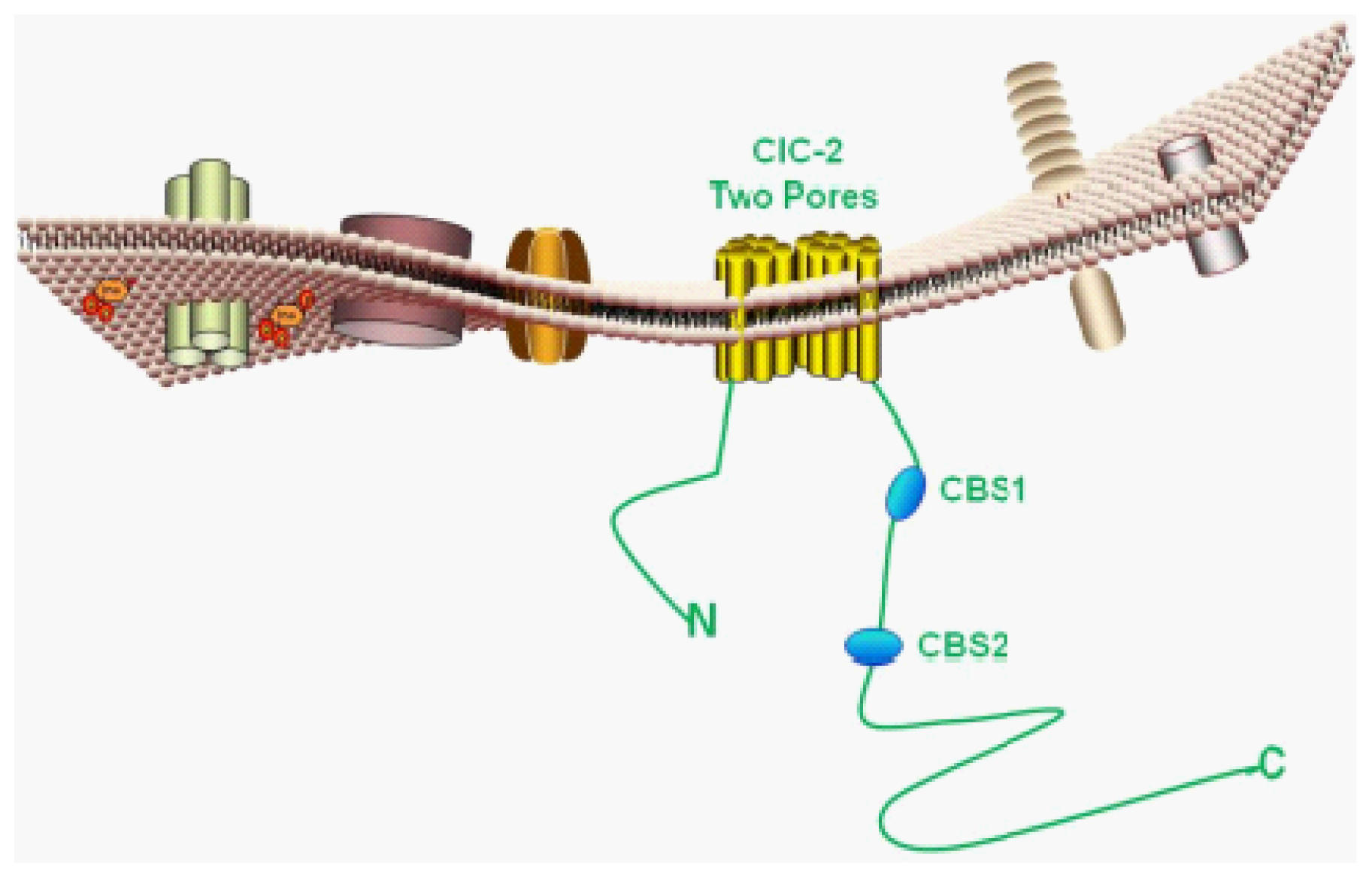
4. Molecular Features of ClC-2
4.1. Basic Protein Structure
4.2. Structure-Function Relationships
4.3. Promoter Region
5. Cellular Function of ClC-2
5.1. Regulation of Cell Volume
5.2. Regulation of Transepithelial Transport
5.3. Regulation of Tight Junction Function
5.4. Regulation of Pacemaker Activity
5.5. Regulation of Vascular Smooth Muscle Cell (VSMC) Proliferation
5.6. Regulation of Glioma Cell Proliferation
5.7. Regulation of Neuronal Excitability
5.8. Important Notes to Consider
6. Regulation of ClC-2 Expression and Function
6.1. Expression Regulation
6.1.1. Transcriptional Regulation
6.1.2. Transcript Stabilization
6.1.3. Ubiquitination
6.1.4. Trafficking Regulation
6.1.5. Regulation by Hormones
6.1.5.1. Thyroid Hormone
6.1.5.2. Aldosterone
6.1.5.3. Estrogen
6.1.6. Alpha1-Adrenoceptors
6.1.7. Dynein Motor Complex
6.2. Functional Regulation
6.2.1. Acidification
6.2.2. Protein Kinase A (PKA)
6.2.3. Epidermal Growth Factor Receptor (EGFR)
6.2.4. Arachidonic Acid and Amidation
6.2.5. Heat-Shock Proteins
6.2.6. Actin Cytoskeleton
6.2.7. Intracellular ATP
6.2.8. Membrane Cholesterol Content
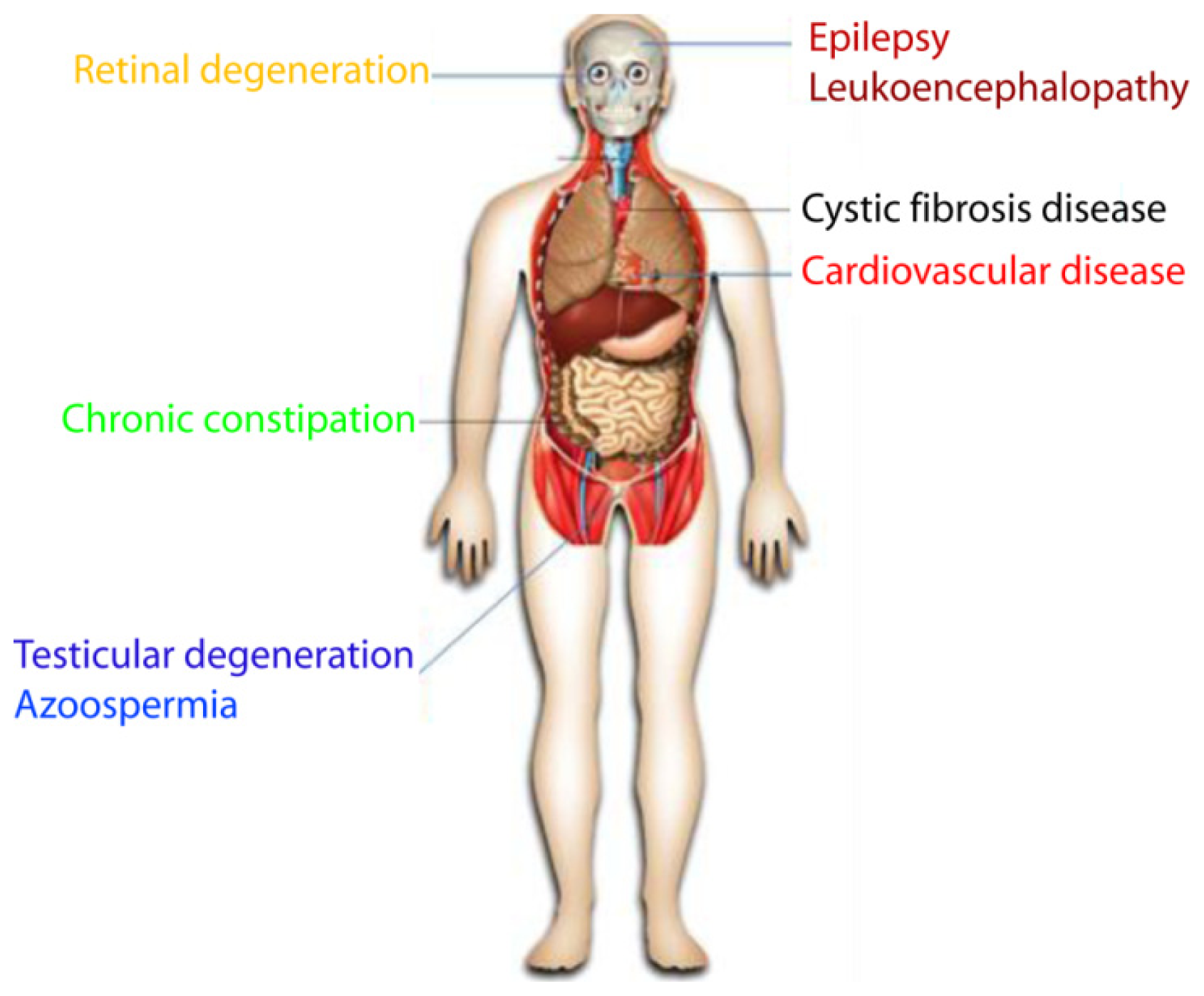
7. ClC-2 Channelopathies
7.1. Eye Disease
7.1.1. Retinal Degeneration
7.1.2. Sjögren’s Syndrome
7.2. Male Infertility
7.2.1. Testicular Degeneration
7.2.2. Azoospermia
7.3. Chronic Constipation and Irritable Bowel Syndrome
7.4. Neuronal Disease
7.4.1. Epilepsy
7.4.2. Leukoencephalopathy
7.5. Cystic Fibrosis Disease
7.6. Cardiovascular Disease
8. Conclusions and Perspectives
- Expression levels of ClC-2 in certain tissues are not sufficiently high to give rise to phenotypes. For example, six distinct types of sarcolemmal Cl− currents coexist in cardiac myocytes, and ClC-2 is one of them [16,142–145]. The expression level of ClC-2 in the heart is significantly less than that of ClC-3 [146]. Furthermore, the functional activity elicited by the molecular counterpart (ClC-2 current) exits only in a small population of cardiac myocytes, which is no match with the expression level of ClC-2 [15].
- It may be that ClC-2 plays a pathophysiological role in many tissues, but overt phenotypes are restricted to only a limited number of tissues where the loss of its effects cannot be compensated by other proteins. Again, taking the heart as an example, ClC-2 and ClC-3 were found to be co-localized in sarcolemmal membranes of cardiac cells [15], and impairment of ClC-2 may well be compensated by ClC-3 for some of the cellular functions that they share.
- The third possibility is that there are species differences between rodents and humans in terms of ClC-2 pathophysiology, and the results acquired from mouse models may not be extrapolated to humans.
Conflicts of Interest
References
- Dutzler, R. The ClC family of chloride channels and transporters. Curr. Opin. Struct. Biol 2006, 16, 439–446. [Google Scholar]
- Jentsch, T.J. CLC chloride channels and transporters: From genes to protein structure, pathology and physiology. Crit. Rev. Biochem. Mol. Biol 2008, 43, 3–36. [Google Scholar]
- Zifarelli, G.; Pusch, M. CLC Chloride channels and transporters: A biophysical and physiological perspective. Rev. Physiol. Biochem. Pharmacol 2007, 158, 23–76. [Google Scholar]
- Duran, C.; Thompson, C.H.; Xiao, Q.; Hartzell, C. Chloride channels: Often enigmatic, rarely predictable. Annu. Rev. Physiol 2010, 72, 95–121. [Google Scholar]
- Gründer, S.; Thiemann, A.; Pusch, M.; Jentsch, T.J. Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature 1992, 360, 759–762. [Google Scholar]
- Furukawa, T.; Ogura, T.; Katayama, Y.; Hiraoka, M. Characteristics of rabbit ClC-2 current expressed in Xenopus oocytes and its contribution to volume regulation. Am. J. Physiol.-Cell Physiol 1998, 274, C500–C512. [Google Scholar]
- Middleton, R.E.; Pheasant, D.J.; Miller, C. Homodimeric architecture of a ClC-type chloride ion channel. Nature 1996, 383, 337–340. [Google Scholar]
- Hsiao, S.H.; Parrish, A.R.; Nahm, S.S.; Abbott, L.C.; McCool, B.A.; Frye, G.D. Effects of early postnatal ethanol intubation on GABAergic synaptic proteins. Dev. Brain Res 2002, 138, 177–185. [Google Scholar]
- Zuniga, L.; Niemeyer, M.I.; Varela, D.; Catalan, M.; Cid, L.P.; Sepulveda, F.V. The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J. Physiol 2004, 555, 671–682. [Google Scholar]
- Cid, L.P.; Montrose-Rafizadeh, C.; Smith, D.I.; Guggino, W.B.; Cutting, G.R. Cloning of a putative human voltage-gated chloride channel (CIC-2) cDNA widely expressed in human tissues. Hum. Mol. Genet 1995, 4, 407–413. [Google Scholar]
- Planells-Cases, R.; Jentsch, T.J. Chloride channelopathies. Biochim. Biophys. Acta 2009, 1792, 173–189. [Google Scholar]
- Hartzell, C.; Qu, Z.; Putzier, I.; Artinian, L.; Chien, L.-T.; Cui, Y. Looking chloride channels straight in the eye: Bestrophins, lipofuscinosis, and retinal degeneration. Physiology 2005, 20, 292–302. [Google Scholar]
- Duan, D. Phenomics of cardiac chloride channels: The systematic study of chloride channel function in the heart. J. Physiol 2009, 587, 2163–2177. [Google Scholar]
- Strange, K. Of mice and worms: Novel insights into ClC-2 anion channel physiology. Physiology 2002, 17, 11–16. [Google Scholar]
- Duan, D.; Ye, L.; Britton, F.; Horowitz, B.; Hume, J.R. A novel anionic inward rectifier in native cardiac myocytes. Circ. Res 2000, 86, e63–e71. [Google Scholar]
- Hume, J.R.; Duan, D.; Collier, M.L.; Yamazaki, J.; Horowitz, B. Anion transport in heart. Physiol. Rev 2000, 80, 31–81. [Google Scholar]
- Niemeyer, M.I.; Cid, L.P.; Yusef, Y.R.; Briones, R.; Sepúlveda, F.V. Voltage-dependent and-independent titration of specific residues accounts for complex gating of a ClC chloride channel by extracellular protons. J. Phys 2009, 587, 1387–1400. [Google Scholar]
- Weinreich, F.; Jentsch, T.J. Pores formed by single subunits in mixed dimers of different CLC chloride channels. J. Biol. Chem 2001, 276, 2347–2353. [Google Scholar]
- Stölting, G.; Teodorescu, G.; Begemann, B.; Schubert, J.; Nabbout, R.; Toliat, M.R.; Sander, T.; Nürnberg, P.; Lerche, H.; Fahlke, C. Regulation of ClC-2 gating by intracellular ATP. Pflügers Arch 2013, 465, 1423–1437. [Google Scholar]
- Britton, F.C.; Wang, G.L.; Huang, Z.M.; Ye, L.; Horowitz, B.; Hume, J.R.; Duan, D. Functional characterization of novel alternatively spliced ClC-2 chloride channel variants in the heart. J. Biol. Chem 2005, 280, 25871–25880. [Google Scholar]
- Pusch, M.; Jordt, S.-E.; Stein, V.; Jentsch, T.J. Chloride dependence of hyperpolarization-activated chloride channel gates. J. Physiol 1999, 515, 341–353. [Google Scholar]
- Thiemann, A.; Gründer, S.; Pusch, M.; Jentsch, T.J. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature 1992, 356, 57–60. [Google Scholar]
- Thompson, C.H.; Olivetti, P.R.; Fuller, M.D.; Freeman, C.S.; McMaster, D.; French, R.J.; Pohl, J.; Kubanek, J.; McCarty, N.A. Isolation and characterization of a high affinity peptide inhibitor of ClC-2 chloride channels. J. Biol. Chem 2009, 284, 26051–26062. [Google Scholar]
- Cuppoletti, J.; Tewari, K.P.; Sherry, A.M.; Kupert, E.Y.; Malinowska, D.H. ClC-2 Cl− channels in human lung epithelia: Activation by arachidonic acid, amidation, and acid-activated omeprazole. Am. J. Physiol.-Cell Physiol 2001, 281, C46–C54. [Google Scholar]
- Besancon, M.; Simon, A.; Sachs, G.; Shin, J.M. Sites of reaction of the gastric H, K-ATPase with extracytoplasmic thiol reagents. J. Biol. Chem 1997, 272, 22438–22446. [Google Scholar]
- Keeling, D.J.; Fallowfield, C.; Milliner, K.J.; Tingley, S.K.; Ife, R.J.; Underwood, A.H. Studies on the mechanism of action of omeprazole. Biochem. Pharmacol 1985, 34, 2967–2973. [Google Scholar]
- Crowell, M.D.; Harris, L.A.; DiBaise, J.K.; Olden, K.W. Activation of type-2 chloride channels: A novel therapeutic target for the treatment of chronic constipation. Curr. Opin. Invest. Drugs 2007, 8, 66–70. [Google Scholar]
- Lacy, B.E.; Chey, W.D. Lubiprostone: Chronic constipation and irritable bowel syndrome with constipation. 2009, 10, 143–152. [Google Scholar]
- Rivkin, A.; Chagan, L. Lubiprostone: Chloride channel activator for chronic constipation. Clin. Ther 2006, 28, 2008–2021. [Google Scholar]
- Ludewig, U.; Pusch, M.; Jentsch, T.J. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature 1996, 383, 340–343. [Google Scholar]
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 2002, 415, 287–294. [Google Scholar]
- Niemeyer, M.I.; Cid, L.P.; Zúñiga, L.; Catalán, M.; Sepúlveda, F.V. A conserved pore-lining glutamate as a voltage-and chloride-dependent gate in the ClC-2 chloride channel. J. Physiol 2003, 553, 873–879. [Google Scholar]
- Jordt, S.-E.; Jentsch, T.J. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J 1997, 16, 1582–1592. [Google Scholar]
- Varela, D.; Niemeyer, M.I.; Cid, L.P.; Sepúlveda, F.V. Effect of an N-terminus deletion on voltage-dependent gating of the ClC-2 chloride channel. J. Physiol 2002, 544, 363–372. [Google Scholar]
- Haug, K.; Warnstedt, M.; Alekov, A.K.; Sander, T.; Ramírez, A.; Poser, B.; Maljevic, S.; Hebeisen, S.; Kubisch, C.; Rebstock, J. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat. Genet 2003, 33, 527–532. [Google Scholar]
- Garcia-Olivares, J.; Alekov, A.; Boroumand, M.R.; Begemann, B.; Hidalgo, P.; Fahlke, C. Gating of human ClC-2 chloride channels and regulation by carboxy-terminal domains. J. Physiol 2008, 586, 5325–5336. [Google Scholar]
- Stroffekova, K.; Kupert, E.Y.; Malinowska, D.H.; Cuppoletti, J. Identification of the pH sensor and activation by chemical modification of the ClC-2G Cl− channel. Am. J. Physiol.-Cell Physiol 1998, 275, C1113–C1123. [Google Scholar]
- Chu, S.; Blaisdell, C.J.; Liu, M.-Z.M.; Zeitlin, P.L. Perinatal regulation of the ClC-2 chloride channel in lung is mediated by Sp1 and Sp3. Am. J. Physiol.-Lung Cell. Mol. Physiol 1999, 276, L614–L624. [Google Scholar]
- Bösl, M.R.; Stein, V.; Hübner, C.; Zdebik, A.A.; Jordt, S.-E.; Mukhopadhyay, A.K.; Davidoff, M.S.; Holstein, A.-F.; Jentsch, T.J. Male germ cells and photoreceptors, both dependent on close cell–cell interactions, degenerate upon ClC-2 Cl− channel disruption. EMBO J 2001, 20, 1289–1299. [Google Scholar]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar]
- Duan, D.; Hume, J.R.; Nattel, S. Evidence that outwardly rectifying Cl− channels underlie volume-regulated Cl− currents in heart. Circ. Res 1997, 80, 103–113. [Google Scholar]
- Nehrke, K.; Arreola, J.; Nguyen, H.-V.; Pilato, J.; Richardson, L.; Okunade, G.; Baggs, R.; Shull, G.E.; Melvin, J.E. Loss of hyperpolarization-activated Cl− current in salivary acinar cells from Clcn2 knockout mice. J. Biol. Chem 2002, 277, 23604–23611. [Google Scholar]
- Moeser, A.J.; Haskell, M.M.; Shifflett, D.E.; Little, D.; Schultz, B.D.; Blikslager, A.T. ClC-2 chloride secretion mediates prostaglandin-induced recovery of barrier function in ischemia-injured porcine ileum. Gastroenterology 2004, 127, 802–815. [Google Scholar]
- Moeser, A.J.; Nighot, P.K.; Engelke, K.J.; Ueno, R.; Blikslager, A.T. Recovery of mucosal barrier function in ischemic porcine ileum and colon is stimulated by a novel agonist of the ClC-2 chloride channel, lubiprostone. Am. J. Physiol.-Gastrointest. Liver Physiol 2007, 292, G647–G656. [Google Scholar]
- Nighot, P.K.; Moeser, A.J.; Ryan, K.A.; Ghashghaei, T.; Blikslager, A.T. ClC-2 is required for rapid restoration of epithelial tight junctions in ischemic-injured murine jejunum. Exp. Cell Res 2009, 315, 110–118. [Google Scholar]
- Turner, J.R. Molecular basis of epithelial barrier regulation: From basic mechanisms to clinical application. Am. J. Pathol 2006, 169, 1901–1909. [Google Scholar]
- Nighot, P.K.; Blikslager, A.T. ClC-2 regulates mucosal barrier function associated with structural changes to the villus and epithelial tight junction. Am. J. Physiol.-Gastrointest. Liver Physiol 2010, 299, G449–G456. [Google Scholar]
- Nighot, P.K.; Blikslager, A.T. Chloride channel ClC-2 modulates tight junction barrier function via intracellular trafficking of occludin. Am. J. Physiol.-Cell Physiol 2012, 302, C178–C187. [Google Scholar]
- Boyett, M.R.; Honjo, H.; Kodama, I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res 2000, 47, 658–687. [Google Scholar]
- Dobrev, D. Ion channel portrait of the human sinus node: Useful for a better understanding of sinus node function and dysfunction in humans? Circulation 2009, 119, 1556–1558. [Google Scholar]
- Chandler, N.J.; Greener, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; Difrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular architecture of the human sinus node: Insights into the function of the cardiac pacemaker. Circulation 2009, 119, 1562–1575. [Google Scholar]
- Verkerk, A.O.; van Ginneken, A.C.; Wilders, R. Pacemaker activity of the human sinoatrial node: Role of the hyperpolarization-activated current, I(f). Int. J. Cardiol 2009, 132, 318–336. [Google Scholar]
- Monfredi, O.; Dobrzynski, H.; Mondal, T.; Boyett, M.R.; Morris, G.M. The anatomy and physiology of the sinoatrial node--a contemporary review. Pacing Clin. Electrophysiol 2010, 33, 1392–1406. [Google Scholar]
- Huang, Z.M.; Prasad, C.; Britton, F.C.; Ye, L.L.; Hatton, W.J.; Duan, D. Functional role of CLC-2 chloride inward rectifier channels in cardiac sinoatrial nodal pacemaker cells. J. Mol. Cell. Cardiol 2009, 47, 121–132. [Google Scholar]
- Enz, R.; Ross, B.J.; Cutting, G.R. Expression of the voltage-gated chloride channel ClC-2 in rod bipolar cells of the rat retina. J. Neurosci 1999, 19, 9841–9847. [Google Scholar]
- Bayes-Genis, A.; Conover, C.A.; Schwartz, R.S. The insulin-like growth factor axis a review of atherosclerosis and restenosis. Circ. Res 2000, 86, 125–130. [Google Scholar]
- Delafontaine, P. Insulin-like growth factor I and its binding proteins in the cardiovascular system. Cardiovasc. Res 1995, 30, 825–834. [Google Scholar]
- Cheng, G.; Kim, M.-J.; Jia, G.; Agrawal, D.K. Involvement of chloride channels in IGF-I-induced proliferation of porcine arterial smooth muscle cells. Cardiovasc. Res 2007, 73, 198–207. [Google Scholar]
- Yang, X.; Lai, X.; Zhang, Y.; Pei, J.; Yang, A.; Zhou, S. siRNA-mediated silencing of ClC-2 gene inhibits proliferation of human U-87 glioma cells. Ai Zheng 2006, 25, 805–810. (in Chinese). [Google Scholar]
- Staley, K. The role of an inwardly rectifying chloride conductance in postsynaptic inhibition. J. Neurophysiol 1994, 72, 273–284. [Google Scholar]
- Smith, R.; Clayton, G.; Wilcox, C.; Escudero, K.; Staley, K. Differential expression of an inwardly rectifying chloride conductance in rat brain neurons: A potential mechanism for cell-specific modulation of postsynaptic inhibition. J. Neurosci 1995, 15, 4057–4067. [Google Scholar]
- Staley, K.; Smith, R.; Schaack, J.; Wilcox, C.; Jentsch, T.J. Alteration of GABAA receptor function following gene transfer of the CLC-2 chloride channel. Neuron 1996, 17, 543–551. [Google Scholar]
- Rinke, I.; Artmann, J.; Stein, V. ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J. Neurosci 2010, 30, 4776–4786. [Google Scholar]
- Foldy, C.; Lee, S.H.; Morgan, R.J.; Soltesz, I. Regulation of fast-spiking basket cell synapses by the chloride channel ClC-2. Nat. Neurosci 2010, 13, 1047–1049. [Google Scholar]
- Ratté, S.; Prescott, S.A. ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J. Neurosci 2011, 31, 15838–15843. [Google Scholar]
- Murray, C.B.; Morales, M.M.; Flotte, T.R.; McGrath-Morrow, S.A.; Guggino, W.B.; Zeitlin, P.L. CIC-2: A developmentally dependent chloride channel expressed in the fetal lung and downregulated after birth. Am. J. Respir. Cell Mol. Biol 1995, 12, 597–604. [Google Scholar]
- Schwiebert, E.M.; Cid-Soto, L.P.; Stafford, D.; Carter, M.; Blaisdell, C.J.; Zeitlin, P.L.; Guggino, W.B.; Cutting, G.R. Analysis of ClC-2 channels as an alternative pathway for chloride conduction in cystic fibrosis airway cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3879–3884. [Google Scholar]
- Huber, S.; Schröppel, B.; Kretzler, M.; Schlöndorff, D.; Horster, M. Single cell RT-PCR analysis of ClC-2 mRNA expression in ureteric bud tip. Am. J. Physiol.-Renal Physiol 1998, 274, F951–F957. [Google Scholar]
- Malinowska, D.H.; Kupert, E.Y.; Bahinski, A.; Sherry, A.; Cuppoletti, J. Cloning, functional expression, and characterization of a PKA-activated gastric Cl-channel. Am. J. Physiol.-Cell Physiol 1995, 268, C191–C200. [Google Scholar]
- Sander, T.; Schulz, H.; Saar, K.; Gennaro, E.; Riggio, M.C.; Bianchi, A.; Zara, F.; Luna, D.; Bulteau, C.; Kaminska, A. Genome search for susceptibility loci of common idiopathic generalised epilepsies. Hum. Mol. Genet 2000, 9, 1465–1472. [Google Scholar]
- Protopopov, A.; Gizatullin, R.; Vorobieva, N.; Protopopova, M.; Kiss, C.; Kashuba, V.; Klein, G.; Kisselev, L.; Graphodatsky, A.; Zabarovsky, E. Human chromosome 3: High-resolution fluorescencein situ hybridization mapping of 40 uniqueNotl linking clones homologous to genes and cDNAs. Chromosome Res 1996, 4, 443–447. [Google Scholar]
- Holmes, K.W.; Hales, R.; Chu, S.; Maxwell, M.J.; Mogayzel, P.J., Jr.; Zeitlin, P.L. Modulation of Sp1 and Sp3 in lung epithelial cells regulates ClC-2 chloride channel expression. Am. J. Respirat. Cell Mol. Biol 2003, 29, 499–505. [Google Scholar]
- Vij, N.; Zeitlin, P.L. Regulation of the ClC-2 lung epithelial chloride channel by glycosylation of SP1. Am. J. Respirat. Cell Mol. Biol 2006, 34, 754. [Google Scholar]
- Palmada, M.; Dieter, M.; Boehmer, C.; Waldegger, S.; Lang, F. Serum and glucocorticoid inducible kinases functionally regulate ClC-2 channels. Biochem. Biophys. Res. Commun 2004, 321, 1001–1006. [Google Scholar]
- Blaisdell, C.J.; Pellettieri, J.P.; Loughlin, C.E.; Chu, S.; Zeitlin, P.L. Keratinocyte growth factor stimulates CLC-2 expression in primary fetal rat distal lung epithelial cells. Am. J. Respirat. Cell Mol. Biol 1999, 20, 842–847. [Google Scholar]
- Chu, S.; Blaisdell, C.J.; Bamford, P.; Ferro, T.J. Interferon-γ regulates ClC-2 chloride channel in lung epithelial cells. Biochem. Biophys. Res. Commun 2004, 324, 31–39. [Google Scholar]
- Hicke, L. A new ticket for entry into budding vesicles—ubiquitin. Cell 2001, 106, 527–530. [Google Scholar]
- Shen, M.-R.; Droogmans, G.; Eggermont, J.; Voets, T.; Ellory, J.C.; Nilius, B. Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J. Physiol 2000, 529, 385–394. [Google Scholar]
- Voets, T.; Szücs, G.; Droogmans, G.; Nilius, B. Blockers of volume-activated Cl− currents inhibit endothelial cell proliferation. Pflügers Arch 1995, 431, 132–134. [Google Scholar]
- Furukawa, T.; Ogura, T.; Zheng, Y.-J.; Tsuchiya, H.; Nakaya, H.; Katayama, Y.; Inagaki, N. Phosphorylation and functional regulation of CIC-2 chloride channels expressed in Xenopus oocytes by M cyclin-dependent protein kinase. J. Physiol 2002, 540, 883–893. [Google Scholar]
- Dhani, S.U.; Kim Chiaw, P.; Huan, L.-J.; Bear, C.E. ATP depletion inhibits the endocytosis of ClC-2. J. Cell. Physiol 2008, 214, 273–280. [Google Scholar]
- Cornejo, I.; Niemeyer, M.I.; Zúñiga, L.; Yusef, Y.R.; Sepúlveda, F.V.; Cid, L.P. Rapid recycling of ClC-2 chloride channels between plasma membrane and endosomes: Role of a tyrosine endocytosis motif in surface retrieval. J. Cell. Physiol 2009, 221, 650–657. [Google Scholar]
- Hosseinzadeh, Z.; Bhavsar, S.K.; Lang, F. Downregulation of ClC-2 by JAK2. Cell. Physiol. Biochem 2012, 29, 737–742. [Google Scholar]
- Ornellas, D.S.; Grozovsky, R.; Goldenberg, R.; Carvalho, D.; Fong, P.; Guggino, W.; Morales, M. Thyroid hormone modulates ClC-2 chloride channel gene expression in rat renal proximal tubules. J. Endocrinol 2003, 178, 503–511. [Google Scholar]
- Ornellas, D.S.; Nascimento, D.S.; Christoph, D.H.; Guggino, W.B.; Morales, M.M. Aldosterone and high-NaCl diet modulate ClC-2 chloride channel gene expression in rat kidney. Pflügers Arch 2002, 444, 193–201. [Google Scholar]
- Nascimento, D.S.; Reis, C.U.; Goldenberg, R.C.; Ortiga-Carvalho, T.M.; Pazos-Moura, C.C.; Guggino, S.E.; Guggino, W.B.; Morales, M.M. Estrogen modulates ClC-2 chloride channel gene expression in rat kidney. Pflügers Arch 2003, 446, 593–599. [Google Scholar]
- Baglole, C.J.; Sigalet, D.L.; Meddings, J.B. α1-adrenoceptors down-regulate ClC-2 chloride channels in epithelial cells from the acutely denervated jejunum. Eur. J. Pharmacol 2007, 565, 202–206. [Google Scholar]
- Dhani, S.U.; Mohammad-Panah, R.; Ahmed, N.; Ackerley, C.; Ramjeesingh, M.; Bear, C.E. Evidence for a functional interaction between the ClC-2 chloride channel and the retrograde motor dynein complex. J. Biol. Chem 2003, 278, 16262–16270. [Google Scholar]
- Cuppoletti, J.; Baker, A.; Malinowska, D.H. Cl-channels of the gastric parietal cell that are active at low pH. Am. J. Physiol.-Cell Physiol 1993, 264, C1609–C1618. [Google Scholar]
- Sherry, A.; Stroffekova, K.; Knapp, L.M.; Kupert, E.Y.; Cuppoletti, J.; Malinowska, D. Characterization of the human pH-and PKA-activated ClC-2G (2 alpha) Cl-channel. Am. J. Physiol.-Cell Physiol 1997, 273, C384–C393. [Google Scholar]
- Tewari, K.P.; Malinowska, D.H.; Sherry, A.M.; Cuppoletti, J. PKA and arachidonic acid activation of human recombinant ClC-2 chloride channels. Am. J. Physiol.-Cell Physiol 2000, 279, C40–C50. [Google Scholar]
- Klaus, F.; Laufer, J.; Czarkowski, K.; Strutz-Seebohm, N.; Seebohm, G.; Lang, F. PIKfyve-dependent regulation of the Cl− channel ClC-2. Biochem. Biophys. Res. Commun 2009, 381, 407–411. [Google Scholar]
- Duan, D.; Ye, L.; Britton, F.; Miller, L.J.; Yamazaki, J.; Horowitz, B.; Hume, J.R. Purinoceptor-coupled Cl− channels in mouse heart: A novel, alternative pathway for CFTR regulation. J. Physiol 1999, 521, 43–56. [Google Scholar]
- Nagasaki, M.; Ye, L.; Duan, D.; Horowitz, B.; Hume, J.R. Intracellular cyclic AMP inhibits native and recombinant volume-regulated chloride channels from mammalian heart. J. Physiol 2000, 523, 705–717. [Google Scholar]
- Kajita, H.; Omori, K.; Matsuda, H. The chloride channel ClC-2 contributes to the inwardly rectifying Cl− conductance in cultured porcine choroid plexus epithelial cells. J. Physiol 2000, 523, 313–324. [Google Scholar]
- Cuppoletti, J.; Tewari, K.P.; Sherry, A.M.; Ferrante, C.J.; Malinowska, D.H. Sites of protein kinase A activation of the human ClC-2 Cl–channel. J. Biol. Chem 2004, 279, 21849–21856. [Google Scholar]
- Charles-Schoeman, C.; Watanabe, J.; Lee, Y.Y.; Furst, D.E.; Amjadi, S.; Elashoff, D.; Park, G.; McMahon, M.; Paulus, H.E.; Fogelman, A.M. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheum 2009, 60, 2870–2879. [Google Scholar]
- Sorota, S. Tyrosine protein kinase inhibitors prevent activation of cardiac swelling-induced chloride current. Pflügers Arch 1995, 431, 178–185. [Google Scholar]
- Tilly, B.; van den Berghe, N.; Tertoolen, L.; Edixhoven, M.J.; de Jonge, H.R. Protein tyrosine phosphorylation is involved in osmoregulation of ionic conductances. J. Biol. Chem 1993, 268, 19919–19922. [Google Scholar]
- Voets, T.; Manolopoulos, V.; Eggermont, J.; Ellory, C.; Droogmans, G.; Nilius, B. Regulation of a swelling-activated chloride current in bovine endothelium by protein tyrosine phosphorylation and G proteins. J. Physiol 1998, 506, 341–352. [Google Scholar]
- Bali, M.; Lipecka, J.; Edelman, A.; Fritsch, J. Regulation of ClC-2 chloride channels in T84 cells by TGF-α. Am. J. Physiol.-Cell Physiol 2001, 280, C1588–C1598. [Google Scholar]
- Smith, D.F.; Whitesell, L.; Katsanis, E. Molecular chaperones: Biology and prospects for pharmacological intervention. Pharmacol. Rev 1998, 50, 493–514. [Google Scholar]
- Pratt, W.B. The role of Thehsp90-based chaperone system in signal transduction by nuclear receptors and receptors signaling via map kinase. Annu. Rev. Pharmacol. Toxicol 1997, 37, 297–326. [Google Scholar]
- Hinzpeter, A.; Lipecka, J.; Brouillard, F.; Baudoin-Legros, M.; Dadlez, M.; Edelman, A.; Fritsch, J. Association between Hsp90 and the ClC-2 chloride channel upregulates channel function. Am. J. Physiol.-Cell Physiol 2006, 290, C45–C56. [Google Scholar]
- Ahmed, N.; Ramjeesingh, M.; Wong, S.; Varga, A.; Garami, E.; Bear, C. Chloride channel activity of ClC-2 is modified by the actin cytoskeleton. Biochem. J 2000, 352, 789–794. [Google Scholar]
- Hinzpeter, A.; Fritsch, J.; Borot, F.; Trudel, S.; Vieu, D.-L.; Brouillard, F.; Baudouin-Legros, M.; Clain, J.; Edelman, A.; Ollero, M. Membrane cholesterol content modulates ClC-2 gating and sensitivity to oxidative stress. J. Biol. Chem 2007, 282, 2423–2432. [Google Scholar]
- Zeuthen, T.; Hamann, S.; La Cour, M. Cotransport of H+, lactate and H2O by membrane proteins in retinal pigment epithelium of bullfrog. J. Physiol 1996, 497, 3–17. [Google Scholar]
- Philp, N.J.; Yoon, H.; Grollman, E.F. Monocarboxylate transporter MCT1 is located in the apical membrane and MCT3 in the basal membrane of rat RPE. Am. J. Physiol.-Regul. Integr. Comp. Physiol 1998, 274, R1824–R1828. [Google Scholar]
- Edwards, M.M.; de Evsikova, C.M.; Collin, G.B.; Gifford, E.; Wu, J.; Hicks, W.L.; Whiting, C.; Varvel, N.H.; Maphis, N.; Lamb, B.T. Photoreceptor degeneration, azoospermia, leukoencephalopathy, and abnormal RPE cell function in mice expressing an early stop mutation in CLCN2. Invest. Ophthalmol. Visual Sci 2010, 51, 3264–3272. [Google Scholar]
- Pflugfelder, S.C.; Tseng, S.C.; Sanabria, O.; Kell, H.; Garcia, C.G.; Felix, C.; Feuer, W.; Reis, B.L. Evaluation of subjective assessments and objective diagnostic tests for diagnosing tear-film disorders known to cause ocular irritation. Cornea 1998, 17, 38. [Google Scholar]
- Nandoskar, P.; Wang, Y.; Wei, R.; Liu, Y.; Zhao, P.; Lu, M.; Huang, J.; Thomas, P.; Trousdale, M.D.; Ding, C. Changes of chloride channels in the lacrimal glands of a rabbit model of Sjögren’s Syndrome. Cornea 2012, 31, 273. [Google Scholar]
- Gyömörey, K.; Yeger, H.; Ackerley, C.; Garami, E.; Bear, C.E. Expression of the chloride channel ClC-2 in the murine small intestine epithelium. Am. J. Physiol.-Cell Physiol 2000, 279, C1787–C1794. [Google Scholar]
- Murek, M.; Kopic, S.; Geibel, J. Evidence for intestinal chloride secretion. Exp. Physiol 2010, 95, 471–478. [Google Scholar]
- Catalán, M.; Cornejo, I.; Figueroa, C.D.; Niemeyer, M.I.; Sepúlveda, F.V.; Cid, L.P. ClC-2 in guinea pig colon: mRNA, immunolabeling, and functional evidence for surface epithelium localization. Am. J. Physiol.-Gastrointest. Liver Physiol 2002, 283, G1004–G1013. [Google Scholar]
- Lipecka, J.; Bali, M.; Thomas, A.; Fanen, P.; Edelman, A.; Fritsch, J. Distribution of ClC-2 chloride channel in rat and human epithelial tissues. Am. J. Physiol.-Cell Physiol 2002, 282, C805–C816. [Google Scholar]
- Catalán, M.; Niemeyer, M.I.; Cid, L.P.; Sepúlveda, F.V. Basolateral ClC-2 chloride channels in surface colon epithelium: Regulation by a direct effect of intracellular chloride. Gastroenterology 2004, 126, 1104–1114. [Google Scholar]
- Catalán, M.A.; Flores, C.A.; González–Begne, M.; Zhang, Y.; Sepúlveda, F.V.; Melvin, J.E. Severe defects in absorptive ion transport in distal colons of mice that lack ClC-2 channels. Gastroenterology 2012, 142, 346–354. [Google Scholar]
- Inagaki, A.; Yamaguchi, S.; Takahashi-Iwanaga, H.; Iwanaga, T.; Ishikawa, T. Functional characterization of a ClC-2-like Cl− conductance in surface epithelial cells of rat rectal colon. J. Membr. Biol 2010, 235, 27–41. [Google Scholar]
- Ben-Ari, Y. Excitatory actions of GABA during development: The nature of the nurture. Nat. Rev. Neurosci 2002, 3, 728–739. [Google Scholar]
- Madison, D.V.; Malenka, R.C.; Nicoll, R.A. Phorbol esters block a voltage-sensitive chloride current in hippocampal pyramidal cells. Nature 1986, 321, 695–697. [Google Scholar]
- Cossette, P.; Liu, L.; Brisebois, K.; Dong, H.; Lortie, A.; Vanasse, M.; Saint-Hilaire, J.-M.; Carmant, L.; Verner, A.; Lu, W.-Y. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet 2002, 31, 184–189. [Google Scholar]
- Ge, Y.-X.; Liu, Y.; Tang, H.-Y.; Liu, X.-G.; Wang, X. ClC-2 contributes to tonic inhibition mediated by α5 subunit-containing GABAA receptor in experimental temporal lobe epilepsy. Neuroscience 2011, 186, 120–127. [Google Scholar]
- D’Agostino, D.; Bertelli, M.; Gallo, S.; Cecchin, S.; Albiero, E.; Garofalo, P.; Gambardella, A.; Hilaire, J.-M.S.; Kwiecinski, H.; Andermann, E. Mutations and polymorphisms of the CLCN2 gene in idiopathic epilepsy. Neurology 2004, 63, 1500–1502. [Google Scholar]
- Everett, K.; Chioza, B.; Aicardi, J.; Aschauer, H.; Brouwer, O.; Callenbach, P.; Covanis, A.; Dooley, J.; Dulac, O.; Durner, M. Linkage and mutational analysis of CLCN2 in childhood absence epilepsy. Epilepsy Res 2007, 75, 145–153. [Google Scholar]
- Klassen, T.; Davis, C.; Goldman, A.; Burgess, D.; Chen, T.; Wheeler, D.; McPherson, J.; Bourquin, T.; Lewis, L.; Villasana, D.; et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 2011, 145, 1036–1048. [Google Scholar]
- Stogmann, E.; Lichtner, P.; Baumgartner, C.; Schmied, M.; Hotzy, C.; Asmus, F.; Leutmezer, F.; Bonelli, S.; Assem-Hilger, E.; Vass, K. Mutations in the CLCN2 gene are a rare cause of idiopathic generalized epilepsy syndromes. Neurogenetics 2006, 7, 265–268. [Google Scholar]
- Combi, R.; Grioni, D.; Contri, M.; Redaelli, S.; Redaelli, F.; Bassi, M.T.; Barisani, D.; Lavitrano, M.L.; Tredici, G.; Tenchini, M.L. Clinical and genetic familial study of a large cohort of Italian children with idiopathic epilepsy. Brain Res. Bull 2009, 79, 89–96. [Google Scholar]
- Saint-Martin, C.; Gauvain, G.; Teodorescu, G.; Gourfinkel-An, I.; Fedirko, E.; Weber, Y.G.; Maljevic, S.; Ernst, J.P.; Garcia-Olivares, J.; Fahlke, C. Two novel CLCN2 mutations accelerating chloride channel deactivation are associated with idiopathic generalized epilepsy. Hum. Mutat 2009, 30, 397–405. [Google Scholar]
- Niemeyer, M.I.; Yusef, Y.R.; Cornejo, I.; Flores, C.A.; Sepúlveda, F.V.; Cid, L.P. Functional evaluation of human ClC-2 chloride channel mutations associated with idiopathic generalized epilepsies. Physiol. Genomics 2004, 19, 74–83. [Google Scholar]
- Haug, K.; Warnstedt, M.; Alekov, A.K.; Sander, T.; Ramírez, A.; Poser, B.; Maljevic, S.; Hebeisen, S.; Kubisch, C.; Rebstock, J. Retraction: Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat. Genet 2009, 41, 1043. [Google Scholar]
- Blanz, J.; Schweizer, M.; Auberson, M.; Maier, H.; Muenscher, A.; Hübner, C.A.; Jentsch, T.J. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J. Neurosci 2007, 27, 6581–6589. [Google Scholar]
- Cortez, M.; Li, C.; Whitehead, S.; Dhani, S.; D’Antonio, C.; Huan, L.; Bennett, S.; Snead, O.; Bear, C. Disruption of ClC-2 expression is associated with progressive neurodegeneration in aging mice. Neuroscience 2010, 167, 154–162. [Google Scholar]
- Van der Knaap, M.; Barth, P.; Stroink, H.; van Nieuwenhuizen, O.; Arts, W.; Hoogenraad, F.; Valk, J. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann. Neurol 1995, 37, 324–334. [Google Scholar]
- Duarri, A.; Lopez de Heredia, M.; Capdevila-Nortes, X.; Ridder, M.C.; Montolio, M.; López-Hernández, T.; Boor, I.; Lien, C.-F.; Hagemann, T.; Messing, A. Knockdown of MLC1 in primary astrocytes causes cell vacuolation: A MLC disease cell model. Neurobiol. Dis 2011, 43, 228–238. [Google Scholar]
- Brignone, M.S.; Lanciotti, A.; Macioce, P.; Macchia, G.; Gaetani, M.; Aloisi, F.; Petrucci, T.C.; Ambrosini, E. The β1 subunit of the Na, K-ATPase pump interacts with megalencephalic leucoencephalopathy with subcortical cysts protein 1 (MLC1) in brain astrocytes: New insights into MLC pathogenesis. Hum. Mol. Genet 2011, 20, 90–103. [Google Scholar]
- Jeworutzki, E.; López-Hernández, T.; Capdevila-Nortes, X.; Sirisi, S.; Bengtsson, L.; Montolio, M.; Zifarelli, G.; Arnedo, T.; Müller, C.S.; Schulte, U. GlialCAM, a Protein Defective in a Leukodystrophy, Serves as a ClC-2 Cl− Channel Auxiliary Subunit. Neuron 2012, 73, 951–961. [Google Scholar]
- Scheper, G.C.; van Berkel, C.G.; Leisle, L.; de Groot, K.E.; Errami, A.; Jentsch, T.J.; van der Knaap, M.S. Analysis of CLCN2 as candidate gene for megalencephalic leukoencephalopathy with subcortical cysts. Genet. Test. Mol. Biomark 2010, 14, 255–257. [Google Scholar]
- Yankaskas, J.R.; Marshall, B.C.; Sufian, B.; Simon, R.H.; Rodman, D. Cystic fibrosis adult careconsensus conference report. CHEST J 2004, 125, 1S–39S. [Google Scholar]
- Cuppoletti, J.; Malinowska, D.H. Ca2+-activated Cl− channels Focus on “Molecular cloning and transmembrane structure of hCLCA2 from human lung, trachea, and mammary gland”. Am. J. Physiol.-Cell Physiol 1999, 276, C1259–C1260. [Google Scholar]
- Blaisdell, C.J.; Howard, T.D.; Stern, A.; Bamford, P.; Bleecker, E.R.; Stine, O.C. CLC-2 single nucleotide polymorphisms (SNPs) as potential modifiers of cystic fibrosis disease severity. BMC Med. Genet 2004, 5, 26. [Google Scholar]
- Komukai, K.; Brette, F.; Pascarel, C.; Orchard, C.H. Electrophysiological response of rat ventricular myocytes to acidosis. Am. J. Physiol.-Heart Circ. Physiol 2002, 283, H412–H422. [Google Scholar]
- Ackerman, M.J.; Clapham, D.E. Cardiac chloride channels. Trends Cardiovasc. Med 1993, 3, 23–28. [Google Scholar]
- Harvey, R.D. Cardiac chloride currents. Physiology 1996, 11, 175–181. [Google Scholar]
- Hiraoka, M.; Kawano, S.; Hirano, Y.; Furukawa, T. Role of cardiac chloride currents in changes in action potential characteristics and arrhythmias. Cardiovasc. Res 1998, 40, 23–33. [Google Scholar]
- Sorota, S. Insights into the structure, distribution and function of the cardiac chloride channels. Cardiovasc. Res 1999, 42, 361–376. [Google Scholar]
- Britton, F.C.; Hatton, W.J.; Rossow, C.F.; Duan, D.; Hume, J.R.; Horowitz, B. Molecular distribution of volume-regulated chloride channels (ClC-2 and ClC-3) in cardiac tissues. Am. J. Physiol. Heart Circ. Physiol 2000, 279, H2225–H2233. [Google Scholar]


© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bi, M.M.; Hong, S.; Zhou, H.Y.; Wang, H.W.; Wang, L.N.; Zheng, Y.J. Chloride Channelopathies of ClC-2. Int. J. Mol. Sci. 2014, 15, 218-249. https://doi.org/10.3390/ijms15010218
Bi MM, Hong S, Zhou HY, Wang HW, Wang LN, Zheng YJ. Chloride Channelopathies of ClC-2. International Journal of Molecular Sciences. 2014; 15(1):218-249. https://doi.org/10.3390/ijms15010218
Chicago/Turabian StyleBi, Miao Miao, Sen Hong, Hong Yan Zhou, Hong Wei Wang, Li Na Wang, and Ya Juan Zheng. 2014. "Chloride Channelopathies of ClC-2" International Journal of Molecular Sciences 15, no. 1: 218-249. https://doi.org/10.3390/ijms15010218