The Function and Catalysis of 2-Oxoglutarate-Dependent Oxygenases Involved in Plant Flavonoid Biosynthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
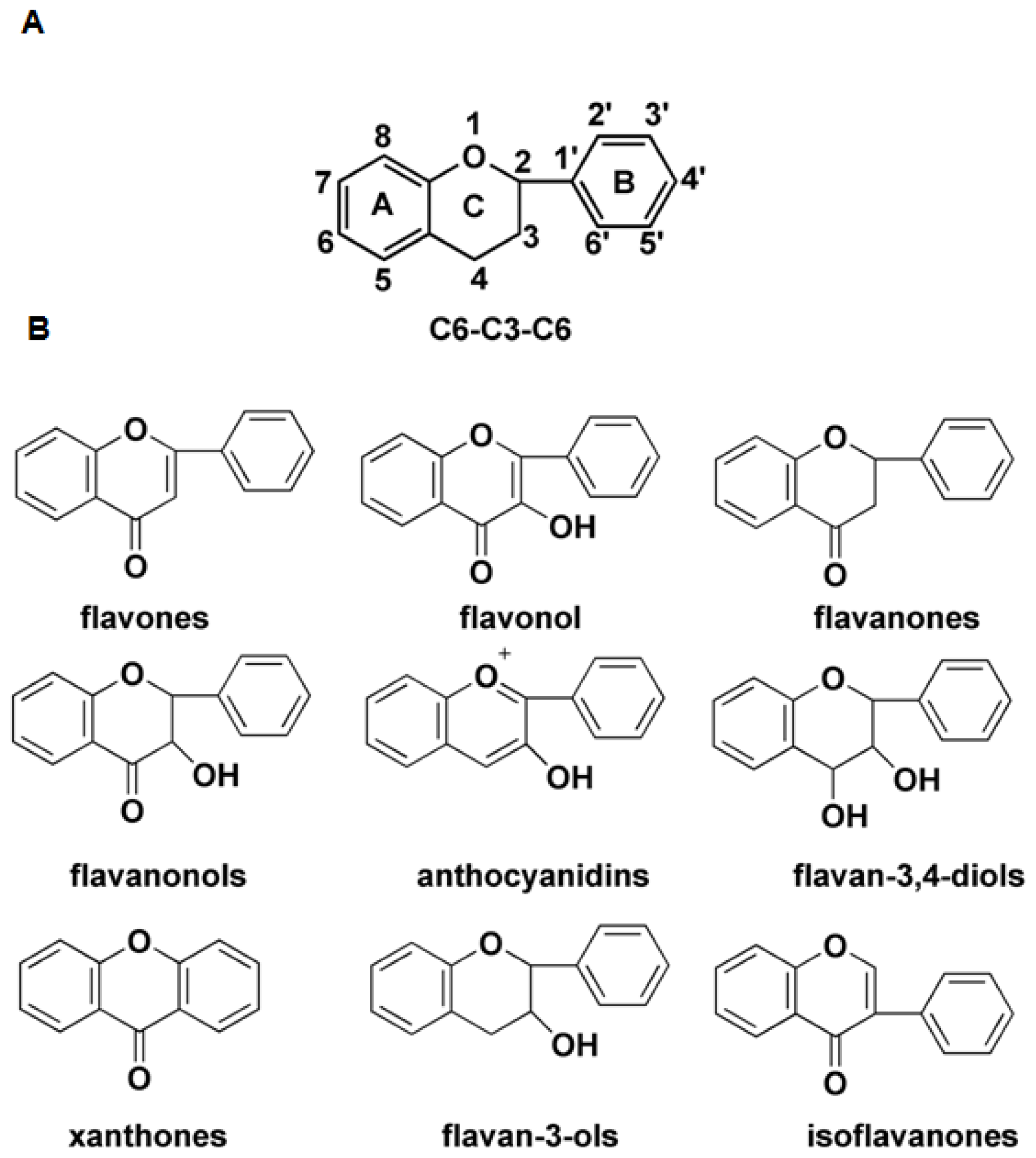
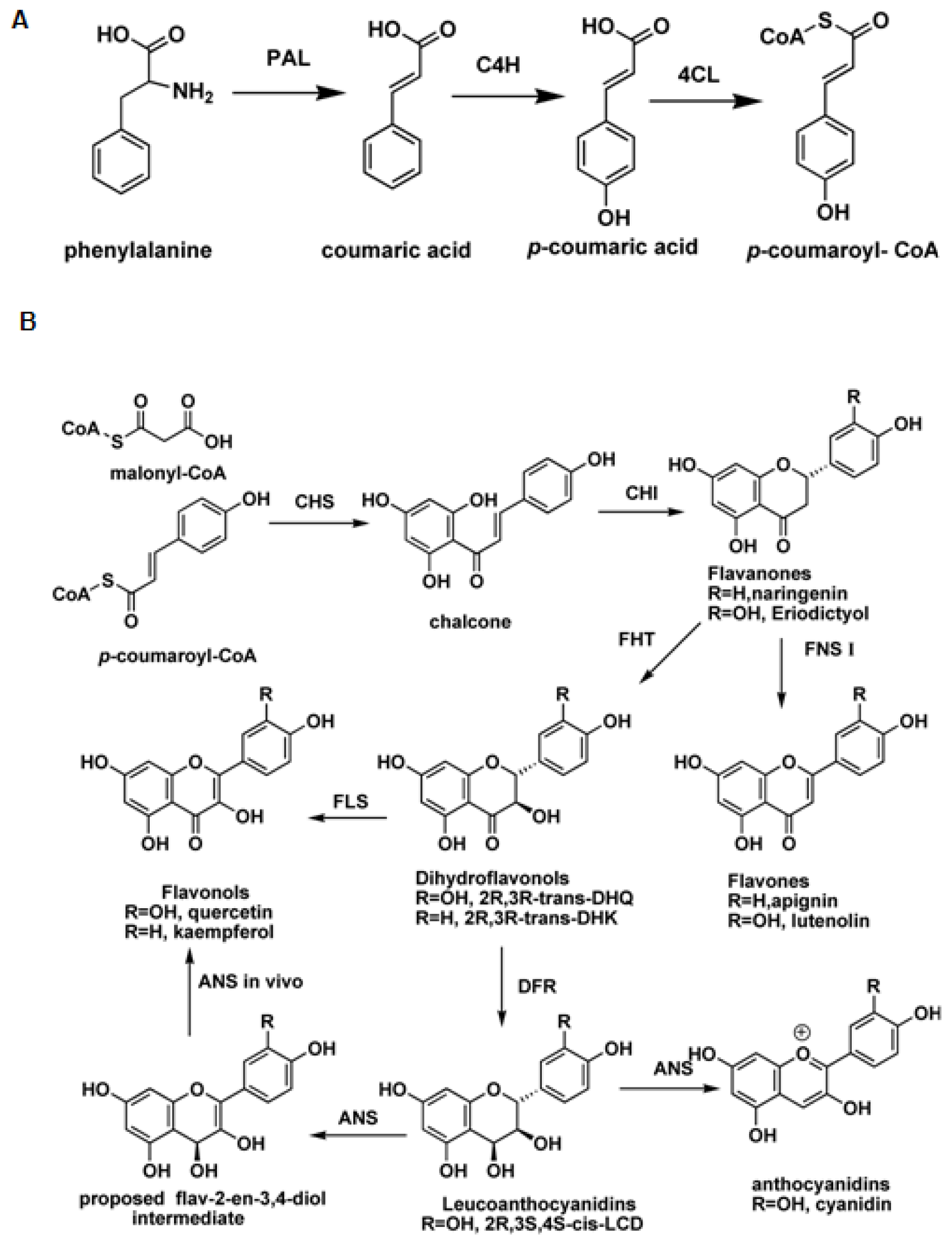
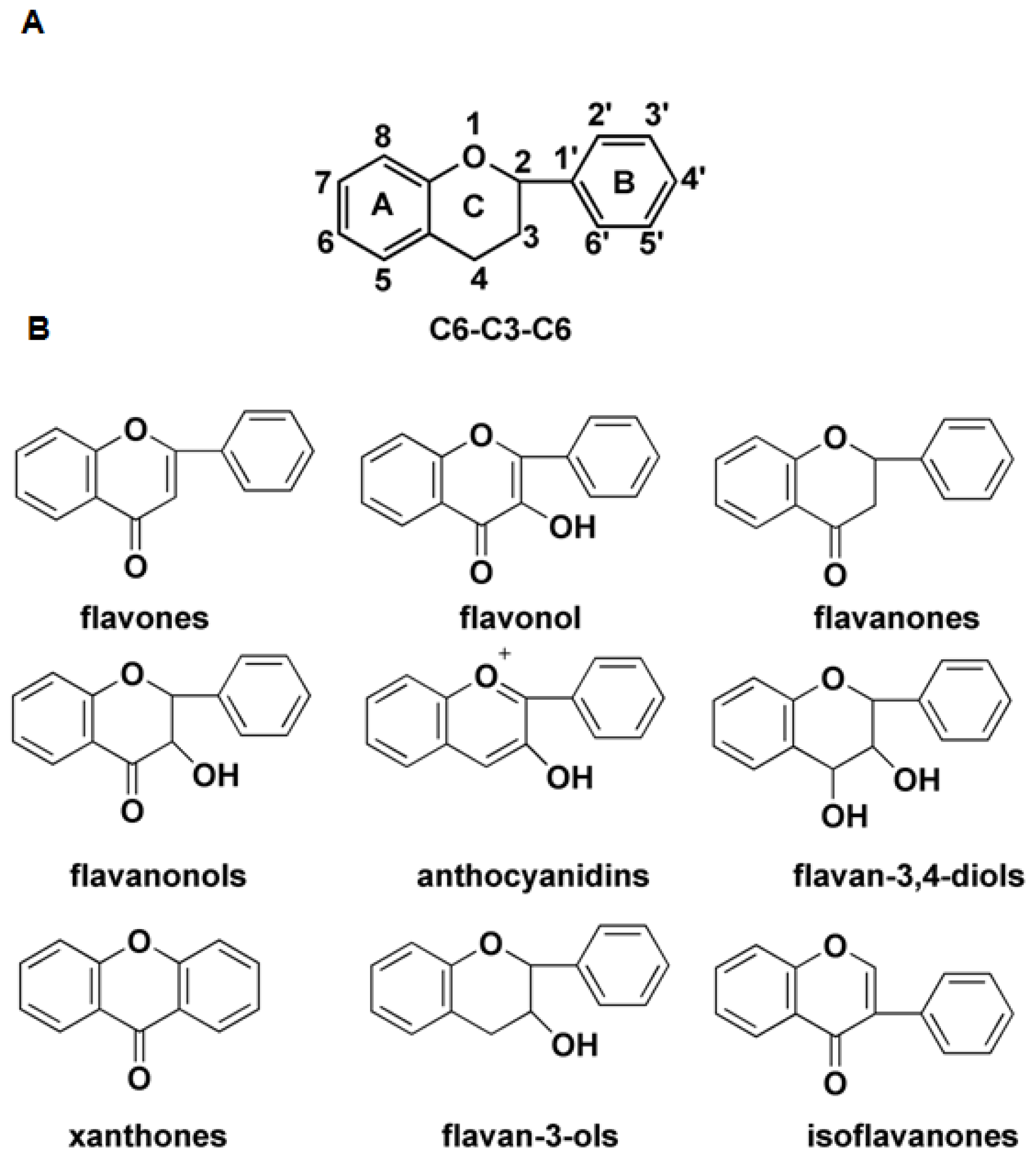
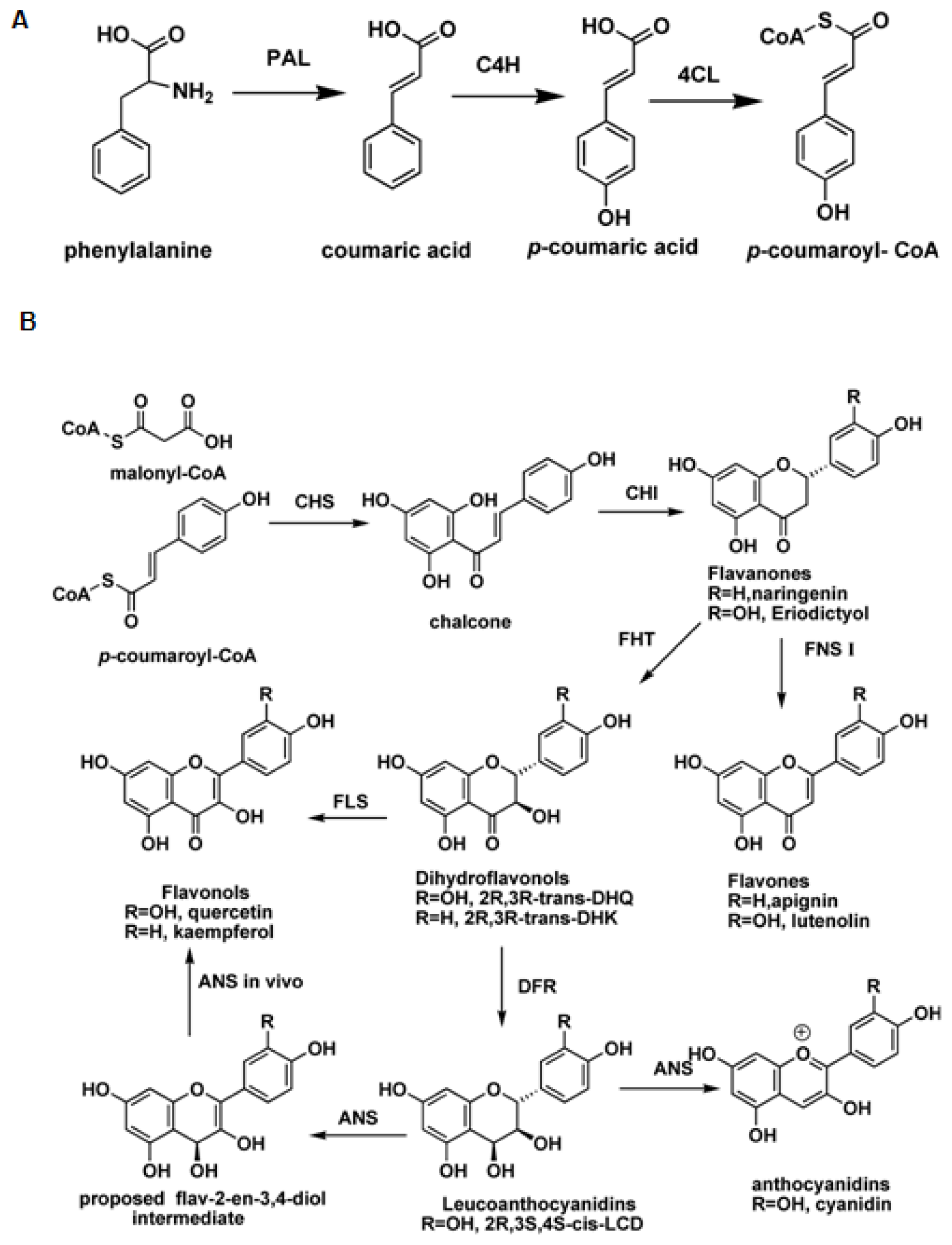
:1. Introduction
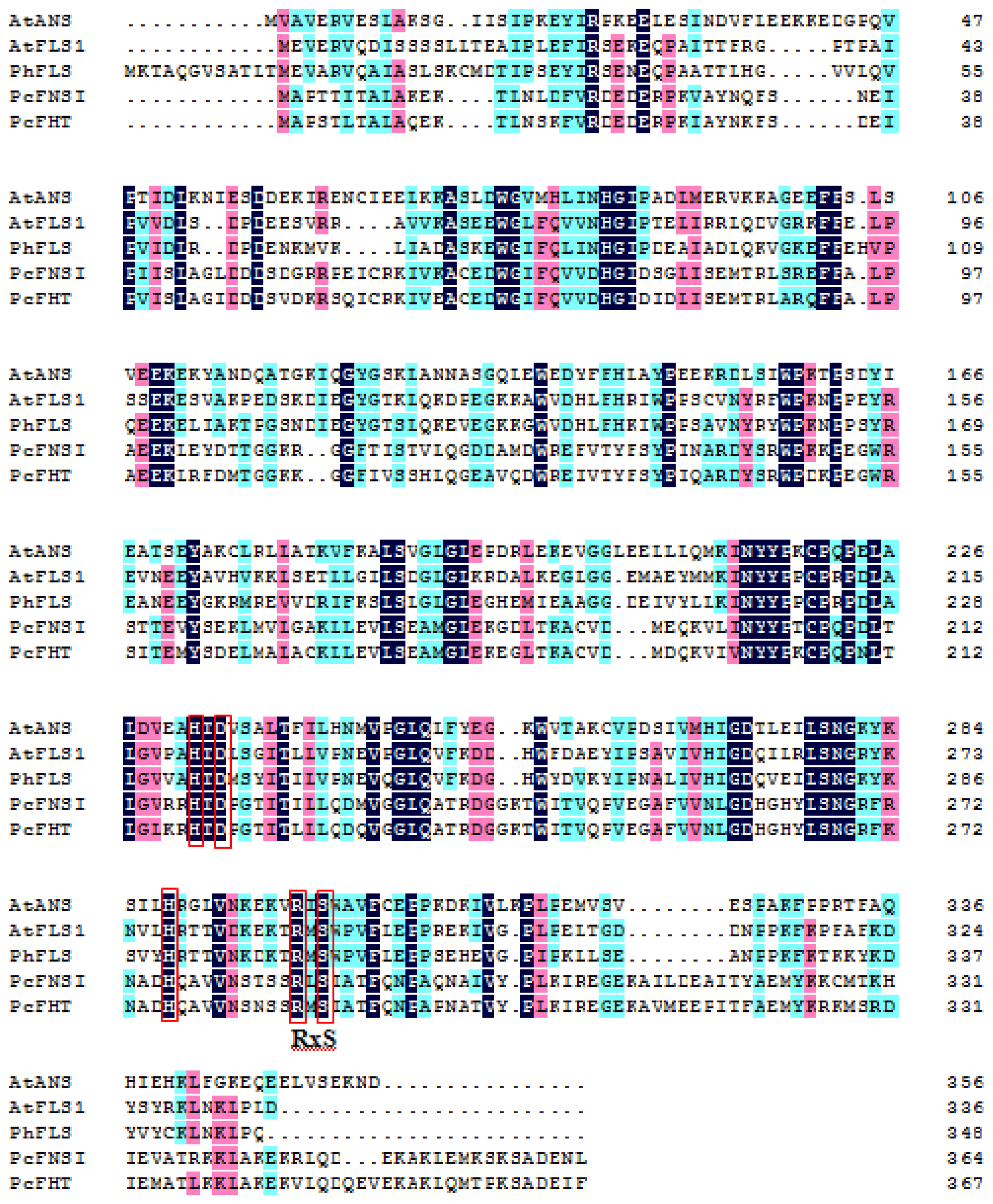
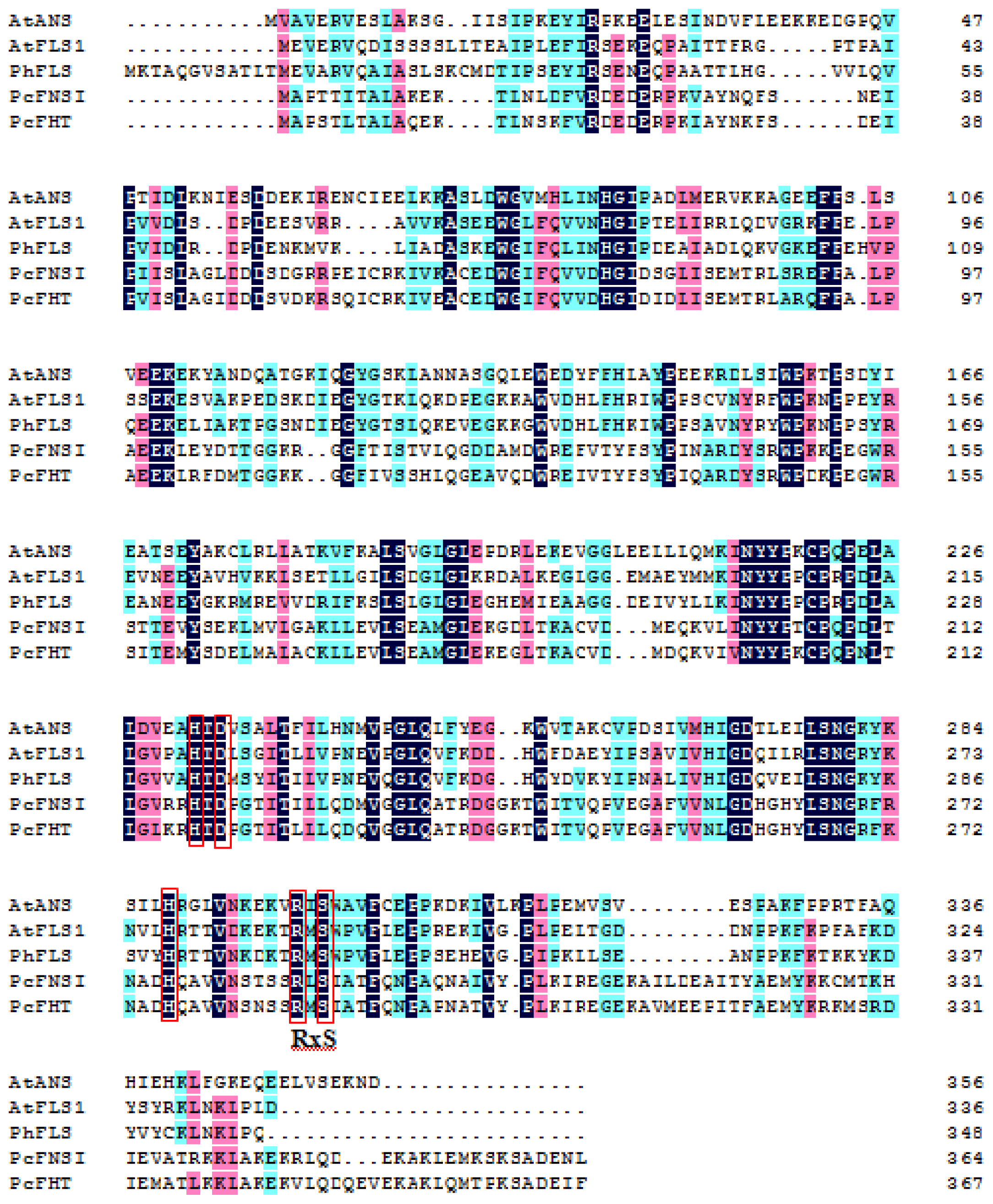
2. Flavonol Synthase and Anthocyanidin Synthase
2.1. Flavonol Synthase
2.2. Anthocyanidin Synthase
3. Flavone Synthase I (FNS I) and Flavanone 3β-Hydroxylase (FHT)
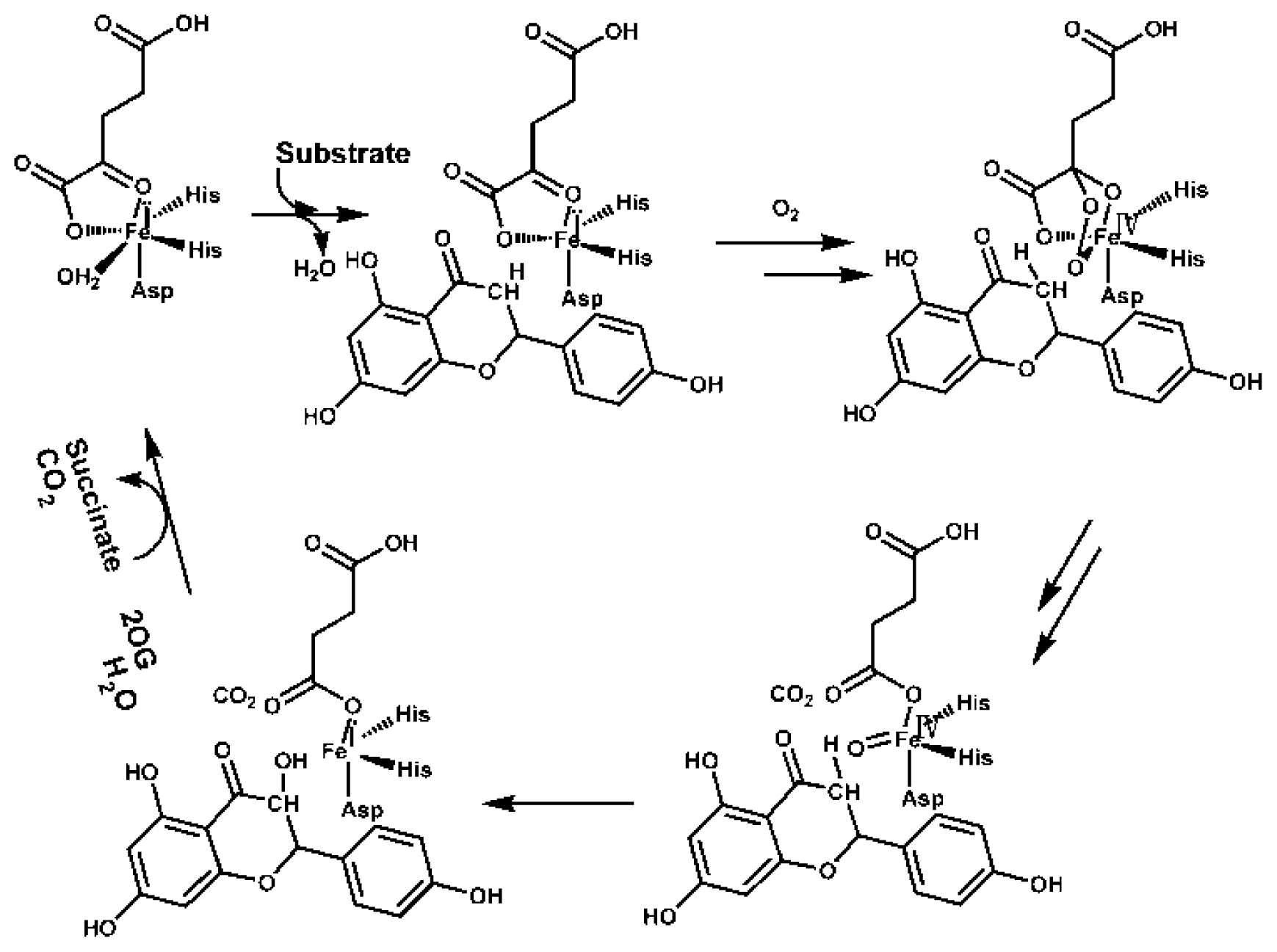
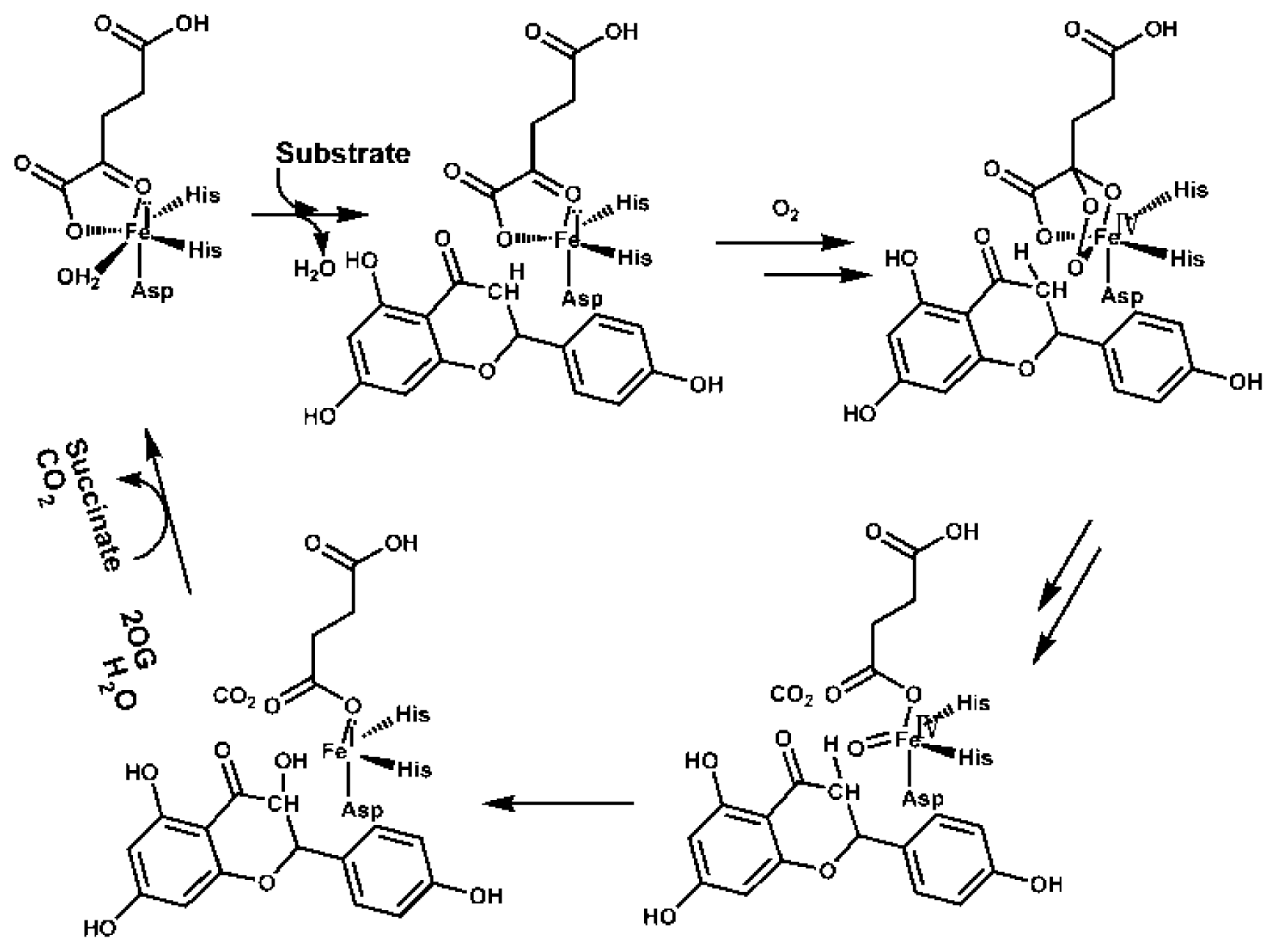
4. Varied Catalytic Mechanisms
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Takahashi, R.; Githiri, S.M.; Hatayama, K.; Dubouzet, E.G.; Shimada, N.; Aoki, T.; Ayabe, S.-I.; Iwashina, T.; Toda, K.; Matsumura, H. A single-base deletion in soybean flavonol synthase gene is associated with magenta flower color. Plant Mol. Biol 2007, 63, 125–135. [Google Scholar]
- Ziegler, J.; Facchini, P.J. Alkaloid biosynthesis: Metabolism and trafficking. Annu. Rev. Plant Biol 2008, 59, 735–769. [Google Scholar]
- Degenhardt, J.; Köllner, T.G.; Gershenzon, J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [Google Scholar]
- Williams, C.A.; Grayer, R.J. Anthocyanins and other flavonoids. Nat. Prod. Rep 2004, 21, 539–573. [Google Scholar]
- Turnbull, J.J.; Nakajima, J.-I.; Welford, R.W.; Yamazaki, M.; Saito, K.; Schofield, C.J. Mechanistic studies on three 2-oxoglutarate-dependent oxygenases of flavonoid biosynthesis anthocyanidin synthase, flavonol synthase, and flavanone 3β-hydroxylase. J. Biol. Chem 2004, 279, 1206–1216. [Google Scholar]
- Schijlen, E.G.; Ric de Vos, C.; van Tunen, A.J.; Bovy, A.G. Modification of flavonoid biosynthesis in crop plants. Phytochemistry 2004, 65, 2631–2648. [Google Scholar]
- Harborne, J.B.; Williams, C.A. Advances in flavonoid research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar]
- Lee, B.H.; Jeong, S.; Jung, S.; Lee, J.; Kim, J.; Yoon, I.; Choi, S.H.; Lee, S.M.; Chang, C.G.; Kim, H.C. Quercetin inhibits the 5-hydroxytryptamine type 3 receptor-mediated ion current by interacting with pre-transmembrane domain I. Mol. Cells 2005, 20, 69–73. [Google Scholar]
- Kim, J.D.; Liu, L.; Guo, W.; Meydani, M. Chemical structure of flavonols in relation to modulation of angiogenesis and immune-endothelial cell adhesion. J. Nutr. Biochem 2006, 17, 165–176. [Google Scholar]
- Kim, Y.H.; Lee, Y.J. TRAIL apoptosis is enhanced by quercetin through Akt dephosphorylation. J. Cell Biochem 2007, 100, 998–1009. [Google Scholar]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol 2001, 126, 485–493. [Google Scholar]
- Wellmann, F.; Griesser, M.; Schwab, W.; Martens, S.; Eisenreich, W.; Matern, U.; Lukačin, R. Anthocyanidin synthase from Gerbera hybrida catalyzes the conversion of (+)-catechin to cyanidin and a novel procyanidin. FEBS Lett 2006, 580, 1642–1648. [Google Scholar]
- Davies, K.M.; Schwinn, K.E.; Deroles, S.C.; Manson, D.G.; Lewis, D.H.; Bloor, S.J.; Bradley, J.M. Enhancing anthocyanin production by altering competition for substrate between flavonol synthase and dihydroflavonol 4-reductase. Euphytica 2003, 131, 259–268. [Google Scholar]
- Chua, C.S.; Biermann, D.; Goo, K.S.; Sim, T.S. Elucidation of active site residues of Arabidopsis thaliana flavonol synthase provides a molecular platform for engineering flavonols. Phytochemistry 2008, 69, 66–75. [Google Scholar]
- Owens, D.K.; Alerding, A.B.; Crosby, K.C.; Bandara, A.B.; Westwood, J.H.; Winkel, B.S. Functional analysis of a predicted flavonol synthase gene family in arabidopsis. Plant Physiol 2008, 147, 1046–1061. [Google Scholar]
- Martens, S.; Forkmann, G.; Matern, U.; Lukačin, R. Cloning of parsley flavone synthase I. Phytochemistry 2001, 58, 43–46. [Google Scholar]
- Gebhardt, Y.; Witte, S.; Forkmann, G.; Lukačin, R.; Matern, U.; Martens, S. Molecular evolution of flavonoid dioxygenases in the family apiaceae. Phytochemistry 2005, 66, 1273–1284. [Google Scholar]
- Gebhardt, Y.H.; Witte, S.; Steuber, H.; Matern, U.; Martens, S. Evolution of flavone synthase I from parsley flavanone 3β-hydroxylase by site-directed mutagenesis. Plant Physiol 2007, 144, 1442–1454. [Google Scholar]
- Wellmann, F.; Lukačin, R.; Moriguchi, T.; Britsch, L.; Schiltz, E.; Matern, U. Functional expression and mutational analysis of flavonol synthase from Citrus unshiu. Eur. J. Biochem 2002, 269, 4134–4142. [Google Scholar]
- Prescott, A.G.; Lloyd, M.D. The iron (II) and 2-oxoacid-dependent dioxygenases and their role in metabolism. Nat. Prod. Rep 2000, 17, 367–383. [Google Scholar]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J. Targeting of HIF-α to the von hippel-lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar]
- Trewick, S.C.; Henshaw, T.F.; Hausinger, R.P.; Lindahl, T.; Sedgwick, B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 2002, 419, 174–178. [Google Scholar]
- Falnes, P.Ø.; Johansen, R.F.; Seeberg, E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 2002, 419, 178–182. [Google Scholar]
- Bleijlevens, B.; Shivarattan, T.; Flashman, E.; Yang, Y.; Simpson, P.J.; Koivisto, P.; Sedgwick, B.; Schofield, C.J.; Matthews, S.J. Dynamic states of the DNA repair enzyme AlkB regulate product release. EMBO Rep 2008, 9, 872–877. [Google Scholar]
- Stubbs, C.J.; Loenarz, C.; Mecinović, J.; Yeoh, K.K.; Hindley, N.; Liénard, B.T.M.; Sobott, F.; Schofield, C.J.; Flashman, E. Application of a proteolysis/mass spectrometry method for investigating the effects of inhibitors on hydroxylase structure. J. Med. Chem 2009, 52, 2799–2805. [Google Scholar]
- Hausinger, R.P. Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol 2004, 39, 21–68. [Google Scholar]
- Schofield, C.J.; Zhang, Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr. Opin. Struct. Biol 1999, 9, 722–731. [Google Scholar]
- Ryle, M.J.; Hausinger, R.P. Non-heme iron oxygenases. Curr. Opin. Chem. Biol 2002, 6, 193–201. [Google Scholar]
- Zhang, Z.; Ren, J.; Stammers, D.K.; Baldwin, J.E.; Harlos, K.; Schofield, C.J. Structural origins of the selectivity of the trifunctional oxygenase clavaminic acid synthase. Nat. Struct. Mol. Biol 2000, 7, 127–133. [Google Scholar]
- Zhou, J.; Kelly, W.L.; Bachmann, B.O.; Gunsior, M.; Townsend, C.A.; Solomon, E.I. Spectroscopic studies of substrate interactions with clavaminate synthase 2, a multifunctional α-KG-dependent non-heme iron enzyme: Correlation with mechanisms and reactivities. J. Am. Chem. Soc 2001, 123, 7388–7398. [Google Scholar]
- Rohde, J.-U.; In, J.-H.; Lim, M.H.; Brennessel, W.W.; Bukowski, M.R.; Stubna, A.; Münck, E.; Nam, W.; Que, L., Jr. Crystallographic and spectroscopic characterization of a nonheme Fe(IV)=O complex. Science 2003, 299, 1037–1039. [Google Scholar]
- Clifton, I.J.; McDonough, M.A.; Ehrismann, D.; Kershaw, N.J.; Granatino, N.; Schofield, C.J. Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J. Inorg. Biochem 2006, 100, 644–669. [Google Scholar]
- Koehntop, K.D.; Emerson, J.P.; Que, L., Jr. The 2-His-1-carboxylate facial triad: A versatile platform for dioxygen activation by mononuclear non-heme iron (II) enzymes. J. Biol. Inorg. Chem 2005, 10, 87–93. [Google Scholar]
- Lee, Y.J.; Kim, J.H.; Kim, B.G.; Lim, Y.; Ahn, J.H. Characterization of flavone synthase I from rice. BMB Rep 2008, 41, 68–71. [Google Scholar]
- Anzellotti, D.; Ibrahim, R.K. Molecular characterization and functional expression of flavonol 6-hydroxylase. BMC Plant Biol 2004, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Latunde-Dada, A.O.; Cabello-Hurtado, F.; Czittrich, N.; Didierjean, L.; Schopfer, C.; Hertkorn, N.; Werck-Reichhart, D.; Ebel, J. Flavonoid 6-hydroxylase from soybean (Glycine maxl.), a novel plant p-450 monooxygenase. J. Biol. Chem 2001, 276, 1688–1695. [Google Scholar]
- Prescott, A.G.; Stamford, N.P.; Wheeler, G.; Firmin, J.L. In vitro properties of a recombinant flavonol synthase from Arabidopsis thaliana. Phytochemistry 2002, 60, 589–593. [Google Scholar]
- Britsch, L.; Heller, W.; Grisebach, H. Conversion of flavanone to flavone, dihydroflavonol and flavonol with an enzyme system from cell culture of parsley. Z. Naturforsch C 1981, 36, 742–750. [Google Scholar]
- Spribille, R.; Forkmann, G. Conversion of dihydroflavonols to flavonols with enzyme extracts from flower buds of Matthiola incana R. Z Naturforsch C 1984, 39, 714–719. [Google Scholar]
- Forkmann, G.; de Vlaming, P.; Spribille, R.; Wiering, H.; Schram, A.W. Genetic and biochemical studies on the conversion of dihydroflavonols to flavonols in flowers of Petunia hybrida. Z Naturforsch C 1986, 41, 179–186. [Google Scholar]
- Forkmann, G. Flavonoids as flower pigments: The formation of the natural spectrum and its extension by genetic engineering. Plant Breed 1991, 106, 1–26. [Google Scholar]
- Holton, T.A.; Brugliera, F.; Tanaka, Y. Cloning and expression of flavonol synthase from Petunia hybrida. Plant J 1993, 4, 1003–1010. [Google Scholar]
- Pelletier, M.K.; Murrell, J.R.; Shirley, B.W. Characterization of flavonol synthase and leucoanthocyanidin dioxygenase genes in Arabidopsis (further evidence for differential regulation of “early” and “late” genes). Plant Physiol 1997, 113, 1437–1445. [Google Scholar]
- Stracke, R.; de Vos, R.C.; Bartelniewoehner, L.; Ishihara, H.; Sagasser, M.; Martens, S.; Weisshaar, B. Metabolomic and genetic analyses of flavonol synthesis in Arabidopsis thaliana support the in vivo involvement of leucoanthocyanidin dioxygenase. Planta 2009, 229, 427–445. [Google Scholar]
- Preuß, A.; Stracke, R.; Weisshaar, B.; Hillebrecht, A.; Matern, U.; Martens, S. Arabidopsis thaliana expresses a second functional flavonol synthase. FEBS Lett 2009, 583, 1981–1986. [Google Scholar]
- Fujita, A.; Goto-Yamamoto, N.; Aramaki, I.; Hashizume, K. Organ-specific transcription of putative flavonol synthase genes of grapevine and effects of plant hormones and shading on flavonol biosynthesis in grape berry skins. Biosci. Biotechnol. Biochem 2006, 70, 632–638. [Google Scholar]
- Lin, G.-Z.; Lian, Y.-J.; Ryu, J.-H.; Sung, M.-K.; Park, J.-S.; Park, H.-J.; Park, B. K.; Shin, J.-S.; Lee, M.-S.; Cheon, C.-I. Expression and purification of his-tagged flavonol synthase of Camellia sinensis from Escherichia coli. Protein Expr. Purif 2007, 55, 287–292. [Google Scholar]
- Ferreyra, M.L.; Rius, S.; Emiliani, J.; Pourcel, L.; Feller, A.; Morohashi, K.; Casati, P.; Grotewold, E. Cloning and characterization of a UV-B-inducible maize flavonol synthase. Plant J 2010, 62, 77–91. [Google Scholar]
- Lukačin, R.; Britsch, L. Identification of strictly conserved histidine and arginine residues as part of the active site in Petunia hybrida flavanone 3β-hydroxylase. Eur. J. Biochem 1997, 249, 748–757. [Google Scholar]
- Stracke, R.; Ishihara, H.; Huep, G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J 2007, 50, 660–677. [Google Scholar]
- Wisman, E.; Hartmann, U.; Sagasser, M.; Baumann, E.; Palme, K.; Hahlbrock, K.; Saedler, H.; Weisshaar, B. Knock-out mutants from an en-1 mutagenized Arabidopsis thaliana population generate phenylpropanoid biosynthesis phenotypes. Proc. Natl. Acad. Sci. USA 1998, 95, 12432–12437. [Google Scholar]
- Wilmouth, R.C.; Turnbull, J.J.; Welford, R.W.; Clifton, I.J.; Prescott, A.G.; Schofield, C.J. Structure and mechanism of anthocyanidin synthase from Arabidopsis thaliana. Structure 2002, 10, 93–103. [Google Scholar]
- Welford, R.W.; Clifton, I.J.; Turnbull, J.J.; Wilson, S.C.; Schofield, C.J. Structural and mechanistic studies on anthocyanidin synthase catalysed oxidation of flavanone substrates: The effect of C-2 stereochemistry on product selectivity and mechanism. Org. Biomol. Chem 2005, 3, 3117–3126. [Google Scholar]
- Martens, S.; Forkmann, G.; Britsch, L.; Wellmann, F.; Matern, U.; Lukačin, R. Divergent evolution of flavonoid 2-oxoglutarate-dependent dioxygenases in parsley. FEBS Lett 2003, 544, 93–98. [Google Scholar]
- Xu, F.; Li, L.; Zhang, W.; Cheng, H.; Sun, N.; Cheng, S.; Wang, Y. Isolation, characterization, and function analysis of a flavonolsynthase gene from Ginkgo biloba. Mol. Biol. Rep 2012, 1–12. [Google Scholar]
- Menssen, A.; Höhmann, S.; Martin, W.; Schnable, P.; Peterson, P.; Saedler, H.; Gierl, A. The En/Spm transposable element of Zea mays contains splice sites at the termini generating a novel intron from a dSpm element in the A2 gene. EMBO J 1990, 9, 3051–3057. [Google Scholar]
- Saito, K.; Kobayashi, M.; Gong, Z.; Tanaka, Y.; Yamazaki, M. Direct evidence for anthocyanidin synthase as a 2-oxoglutarate-dependent oxygenase: Molecular cloning and functional expression of cDNA from a red forma of Perilla frutescens. Plant J 1999, 17, 181–189. [Google Scholar]
- Turnbull, J.J.; Sobey, W.J.; Aplin, R.T.; Hassan, A.; Firmin, J.L.; Schofield, C.J.; Prescott, A.G. Are anthocyanidins the immediate products of anthocyanidin synthase? Chem. Commun 2000, 2473–2474. [Google Scholar]
- Nakajima, J.-I.; Tanaka, Y.; Yamazaki, M.; Saito, K. Reaction mechanism from leucoanthocyanidin to anthocyanidin 3-glucoside, a key reaction for coloring in anthocyanin biosynthesis. J. Biol. Chem 2001, 276, 25797–25803. [Google Scholar]
- Welford, R.W.D.; Turnbull, J.J.; Claridge, T.M.W.; Prescott, A.G.; Schofield, C.J. Evidence for oxidation at C-3 of the flavonoid C-ring during anthocyanin biosynthesis. Chem. Commun 2001, 1828–1829. [Google Scholar]
- Lange, T.; Kegler, C.; Hedden, P.; Phillips, A.L.; Graebe, J.E. Molecular characterisation of gibberellin 20-oxidases. Structure-function studies on recombinant enzymes and chimaeric proteins. Physiol. Plant 1997, 100, 543–549. [Google Scholar]
- Lee, H.-J.; Lloyd, M.D.; Harlos, K.; Clifton, I.J.; Baldwin, J.E.; Schofield, C.J. Kinetic and crystallographic studies on deacetoxycephalosporin c synthase (DAOCS). J. Mol. Biol 2001, 308, 937–948. [Google Scholar]
- Wellmann, F.; Matern, U.; Lukačin, R. Significance of C-terminal sequence elements for Petunia flavanone 3β-hydroxylase activity. FEBS Lett 2004, 561, 149–154. [Google Scholar]
- Britsch, L. Purification and characterization of flavone synthase I, a 2-oxoglutarate-dependent desaturase. Arch. Biochem. Biophys 1990, 282, 152–160. [Google Scholar]
- Lukačin, R.; Wellmann, F.; Britsch, L.; Martens, S.; Matern, U. Flavonol synthase from Citrus unshiu is a bifunctional dioxygenase. Phytochemistry 2003, 62, 287–292. [Google Scholar]
- Britsch, L. Purification of flavanone 3β-hydroxylase from Petunia hybrida: Antibody preparation and characterization of a chemogenetically defined mutant. Arch. Biochem. Biophys 1990, 276, 348–354. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cheng, A.-X.; Han, X.-J.; Wu, Y.-F.; Lou, H.-X. The Function and Catalysis of 2-Oxoglutarate-Dependent Oxygenases Involved in Plant Flavonoid Biosynthesis. Int. J. Mol. Sci. 2014, 15, 1080-1095. https://doi.org/10.3390/ijms15011080
Cheng A-X, Han X-J, Wu Y-F, Lou H-X. The Function and Catalysis of 2-Oxoglutarate-Dependent Oxygenases Involved in Plant Flavonoid Biosynthesis. International Journal of Molecular Sciences. 2014; 15(1):1080-1095. https://doi.org/10.3390/ijms15011080
Chicago/Turabian StyleCheng, Ai-Xia, Xiao-Juan Han, Yi-Feng Wu, and Hong-Xiang Lou. 2014. "The Function and Catalysis of 2-Oxoglutarate-Dependent Oxygenases Involved in Plant Flavonoid Biosynthesis" International Journal of Molecular Sciences 15, no. 1: 1080-1095. https://doi.org/10.3390/ijms15011080
APA StyleCheng, A.-X., Han, X.-J., Wu, Y.-F., & Lou, H.-X. (2014). The Function and Catalysis of 2-Oxoglutarate-Dependent Oxygenases Involved in Plant Flavonoid Biosynthesis. International Journal of Molecular Sciences, 15(1), 1080-1095. https://doi.org/10.3390/ijms15011080