Redox Signaling Is an Early Event in the Pathogenesis of Renovascular Hypertension

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
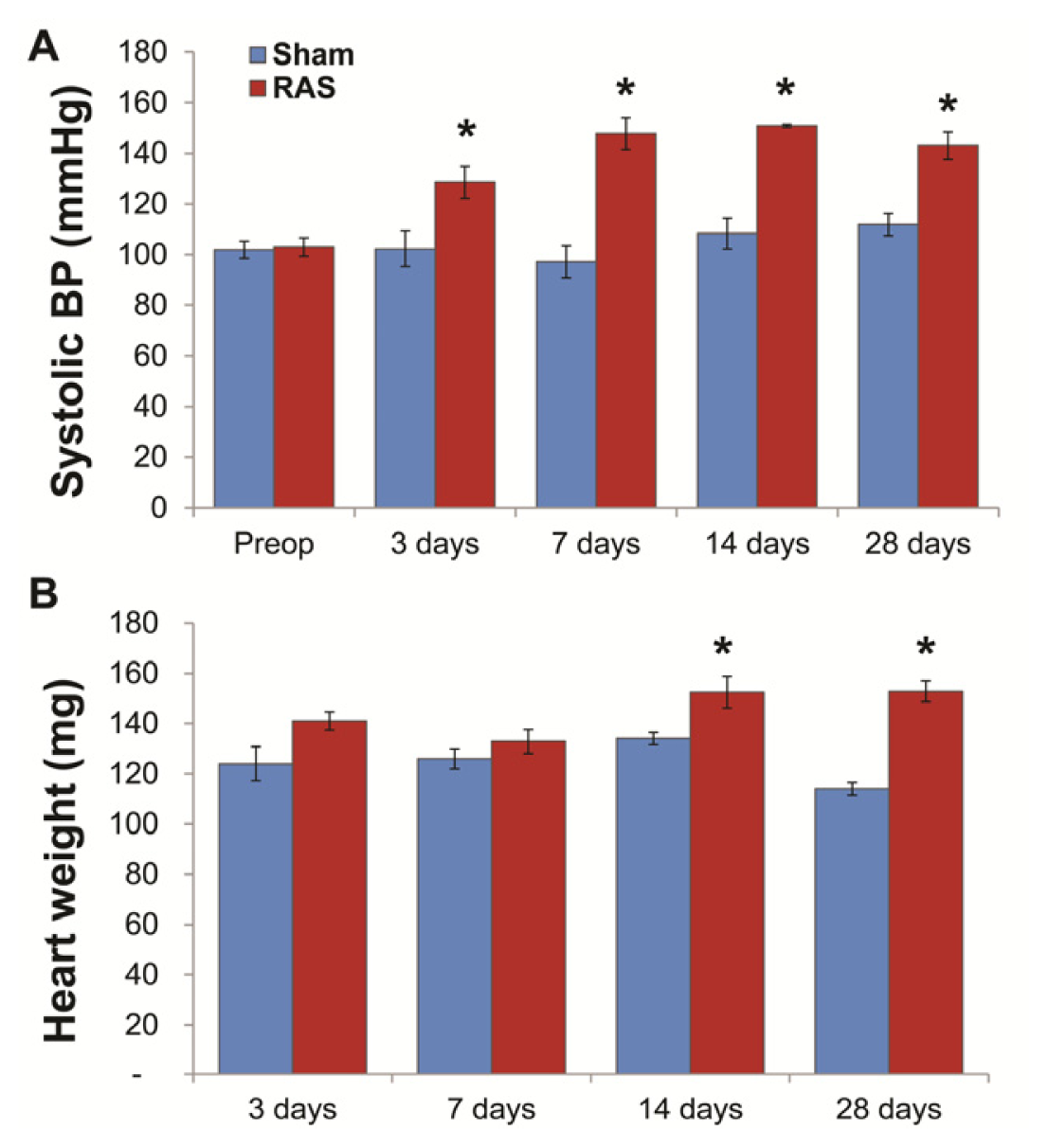
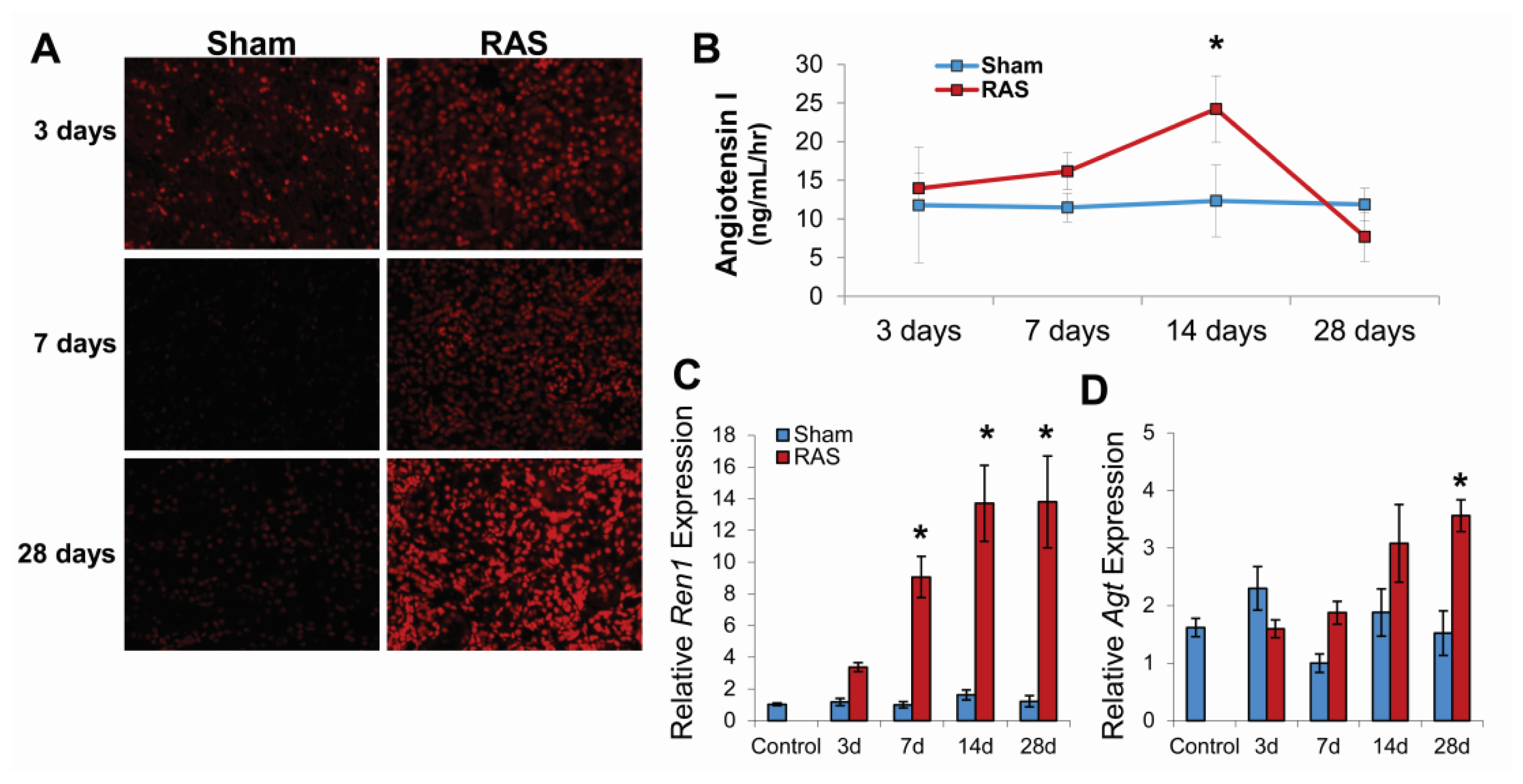
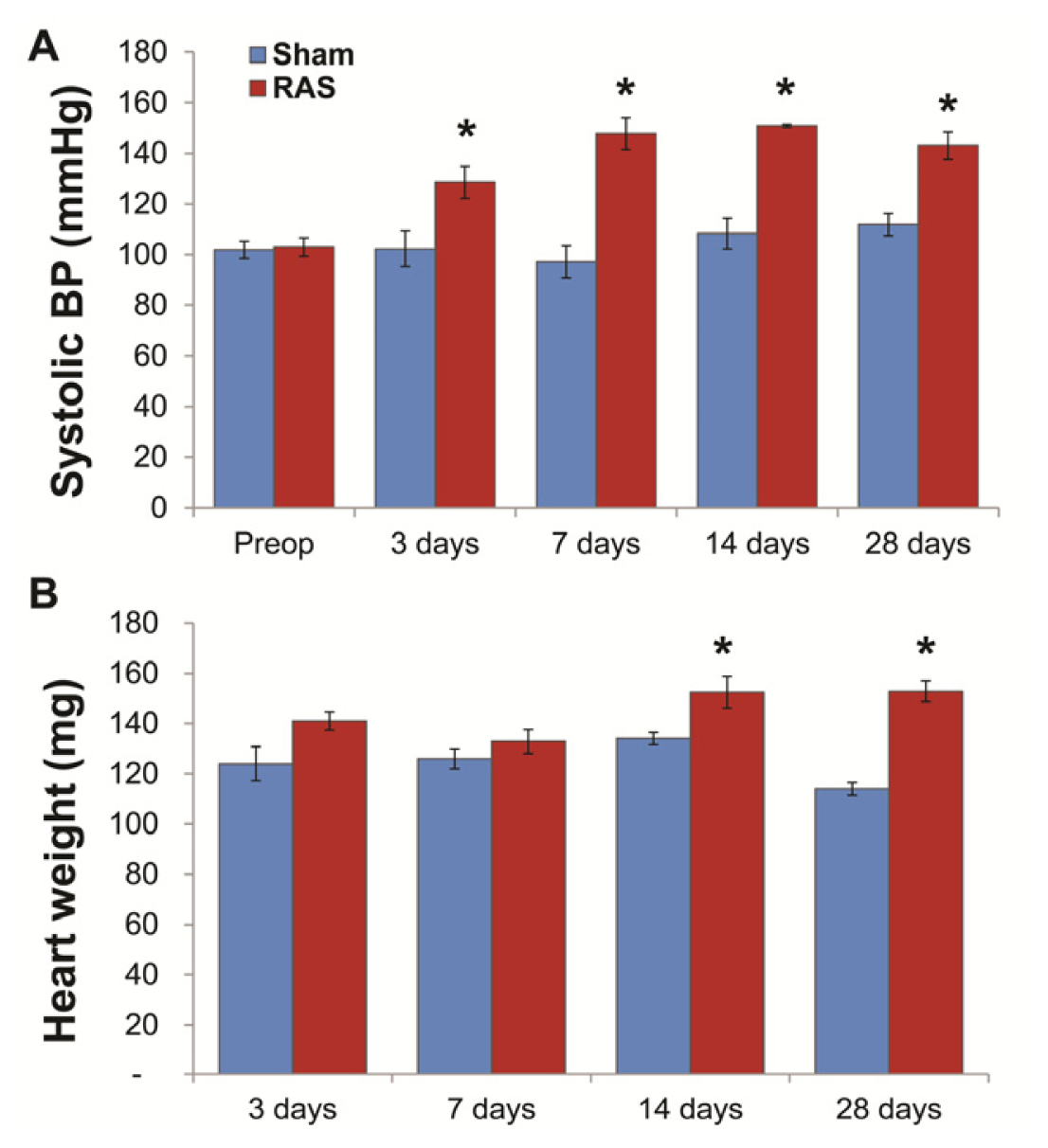
2.1. RAS Mice Rapidly Develop Hypertension and Cardiac Hypertrophy
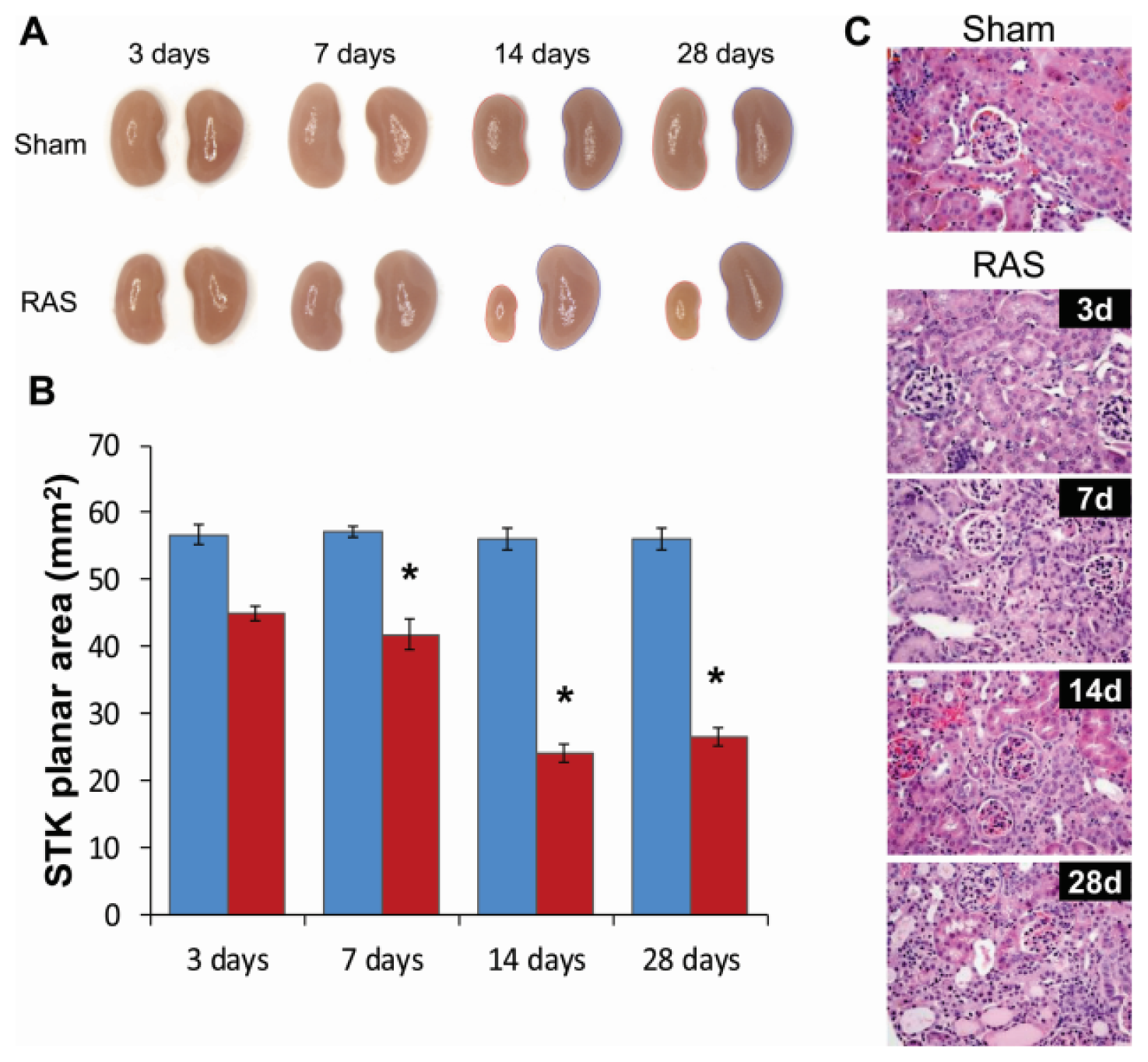
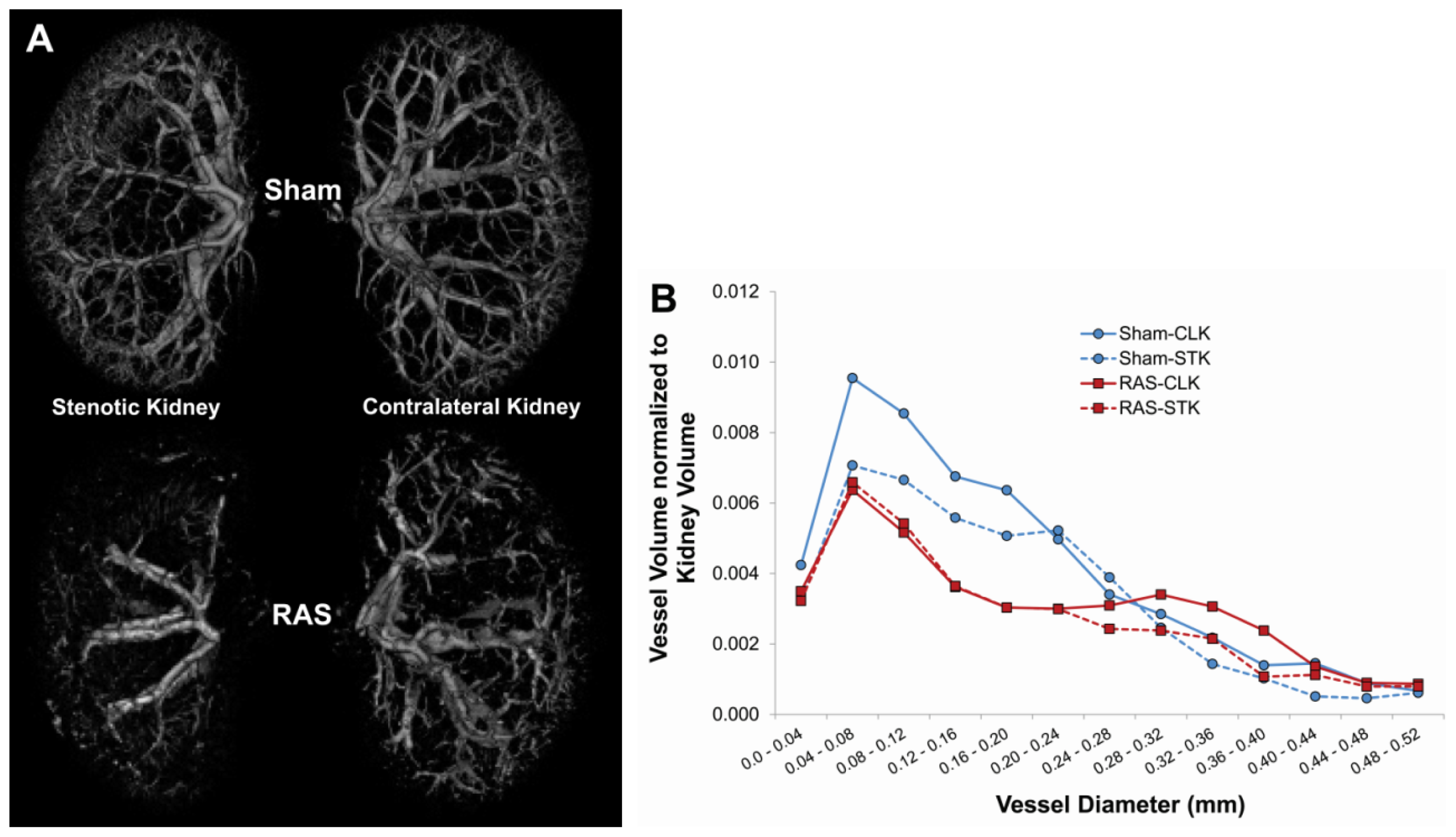
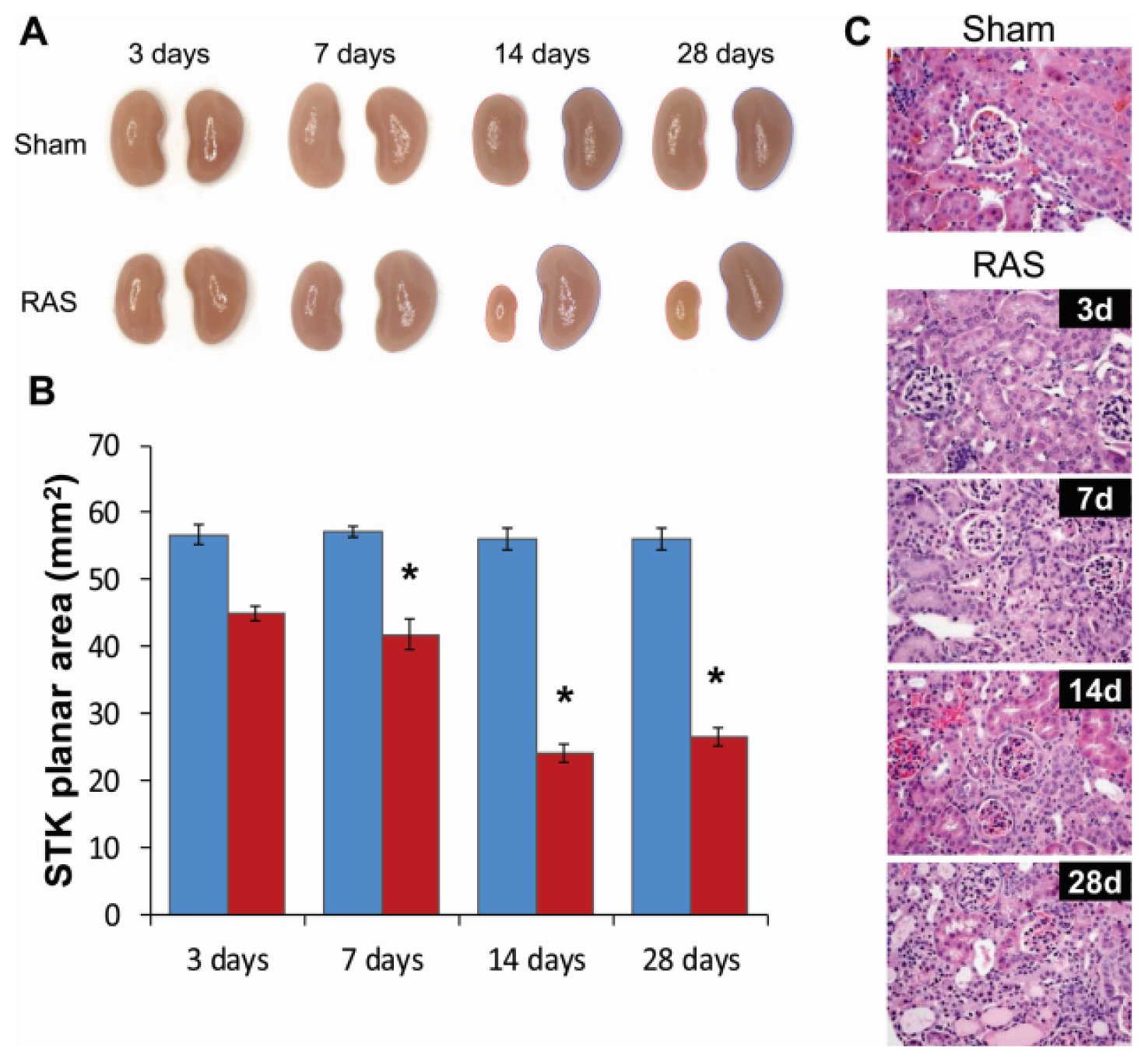
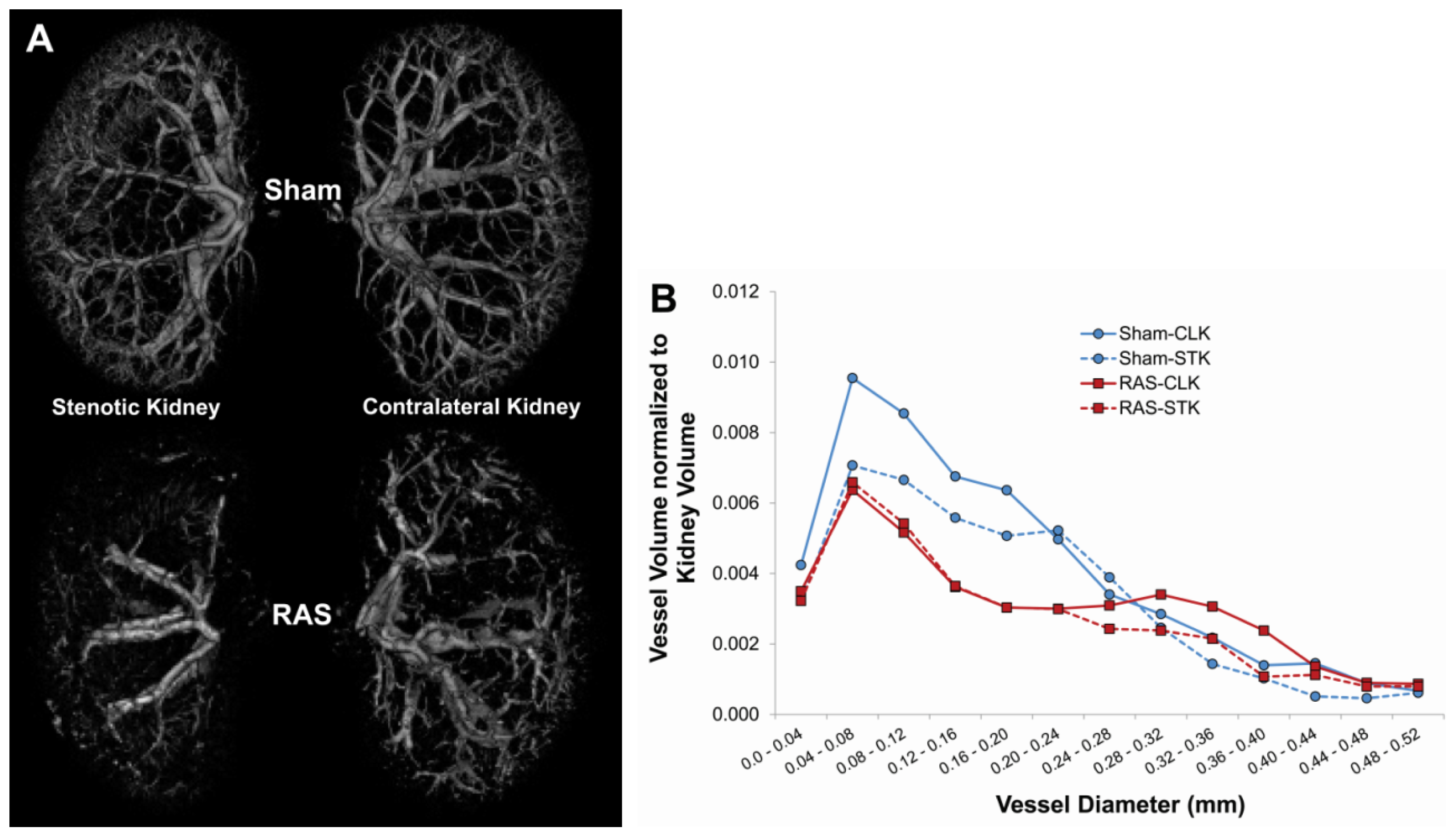
2.2. Renal Atrophy Begins to Develop at Three Days Following RAS Surgery
2.3. RAS Is Associated with Early Production of Superoxide Anion
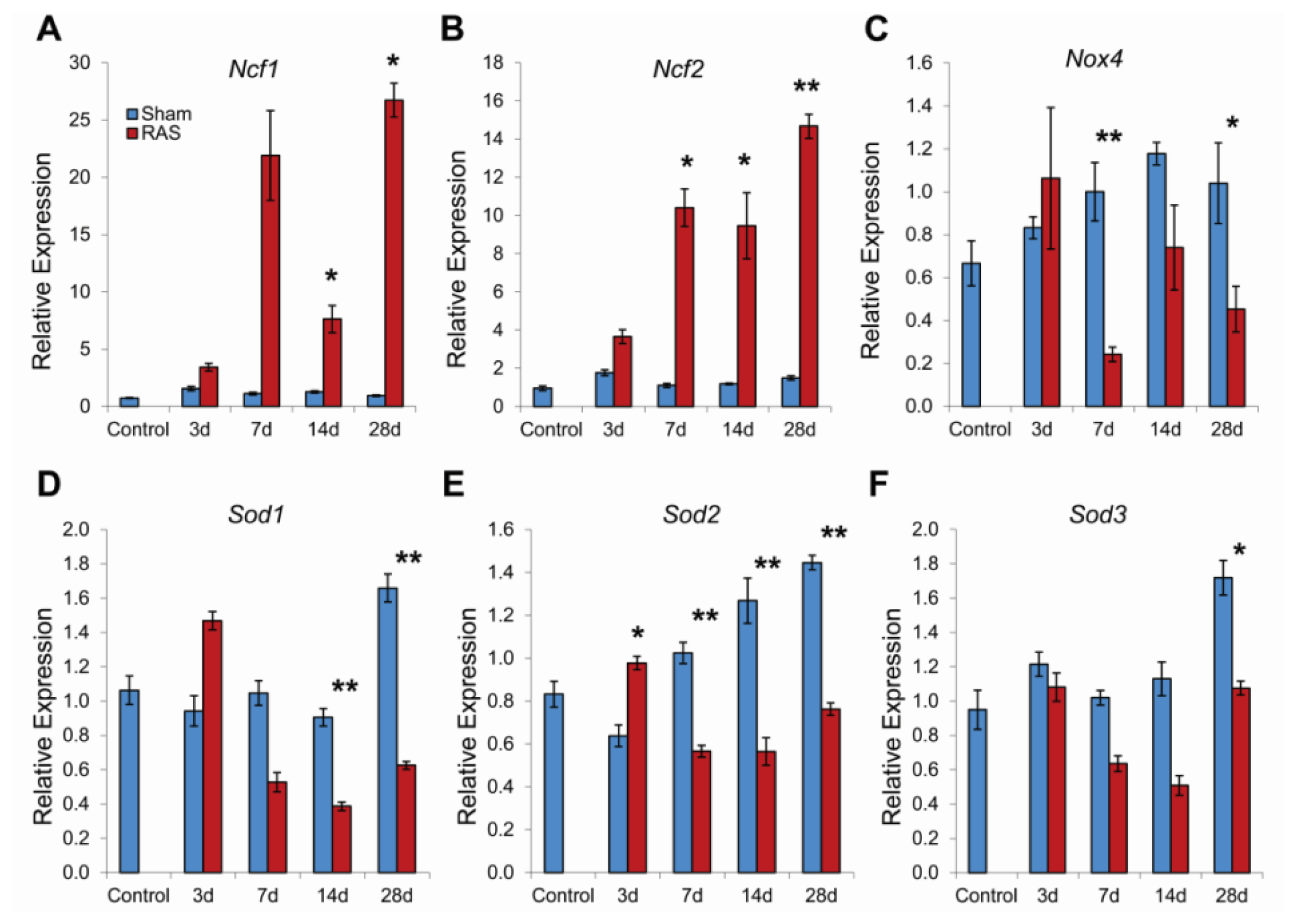
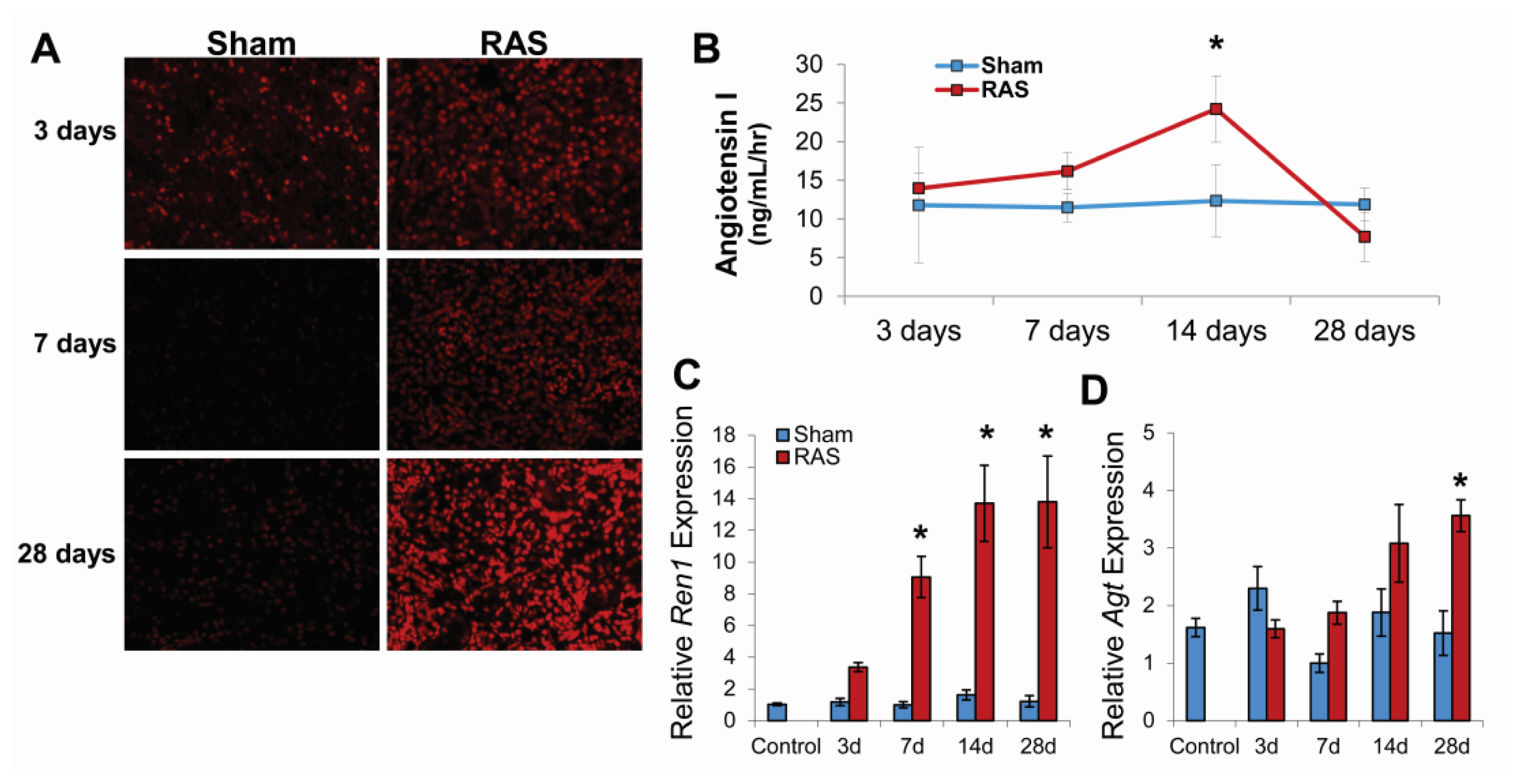
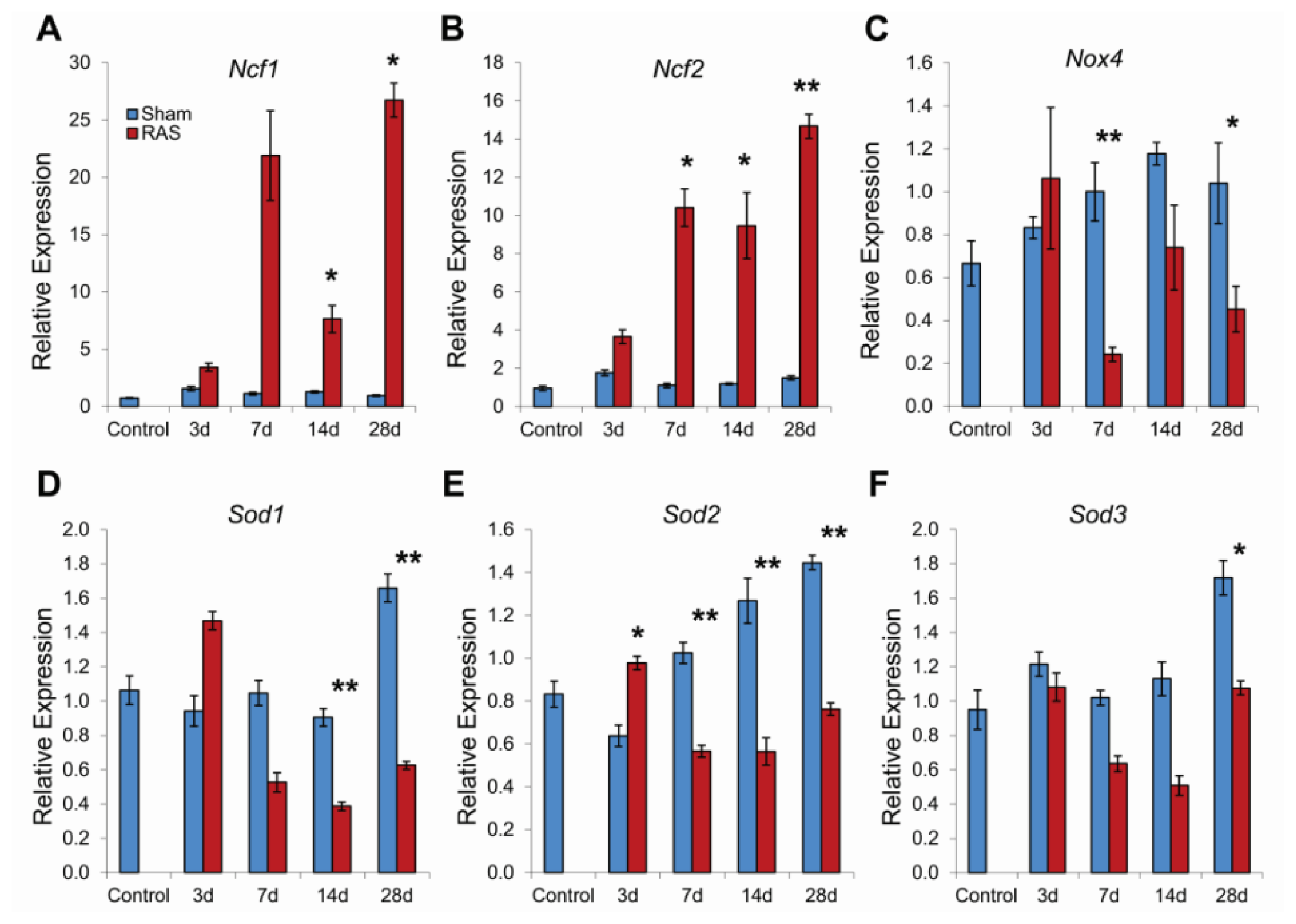
2.4. RAS Is Associated with Early Expression of Oxidant-Related Genes
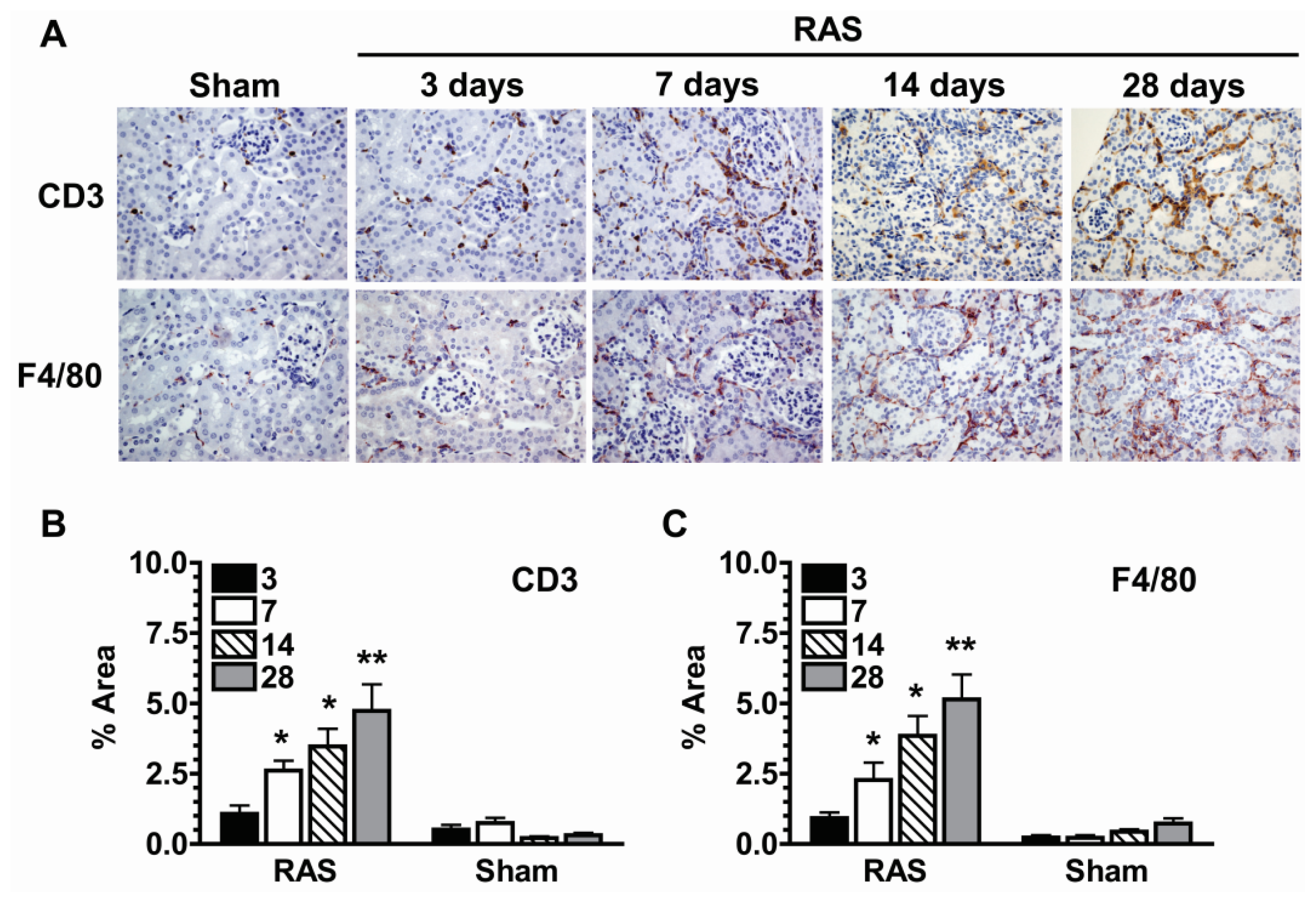
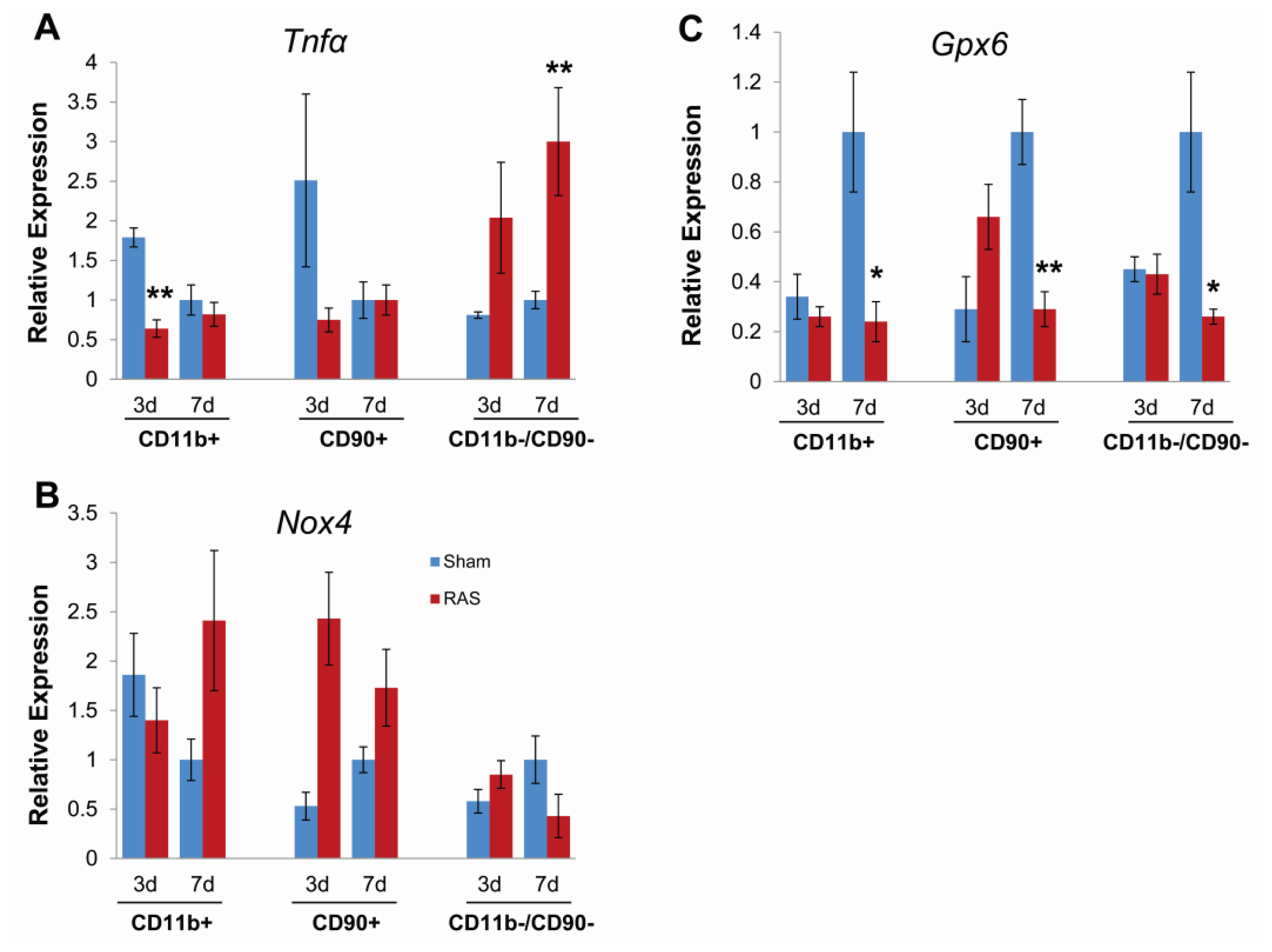
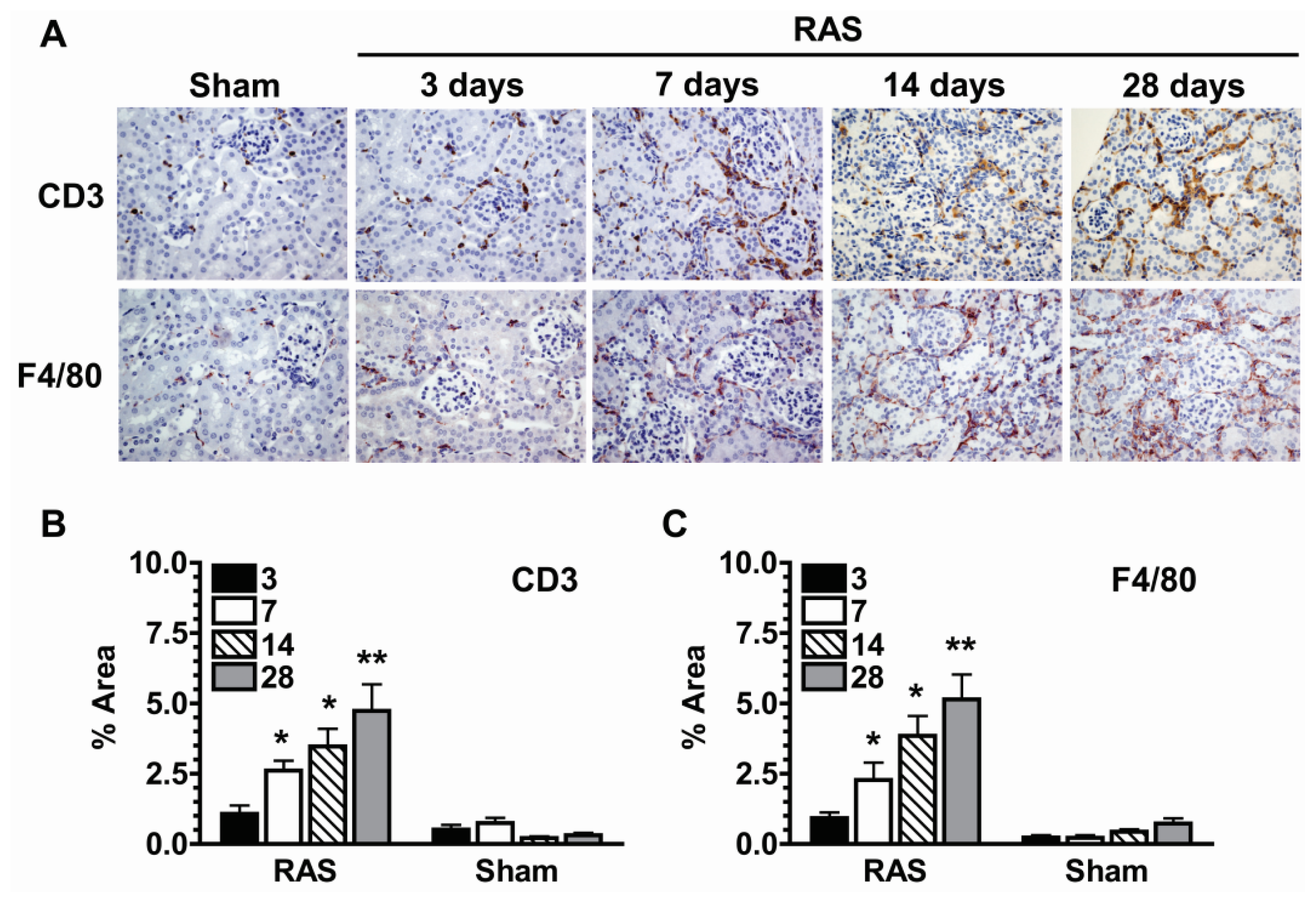
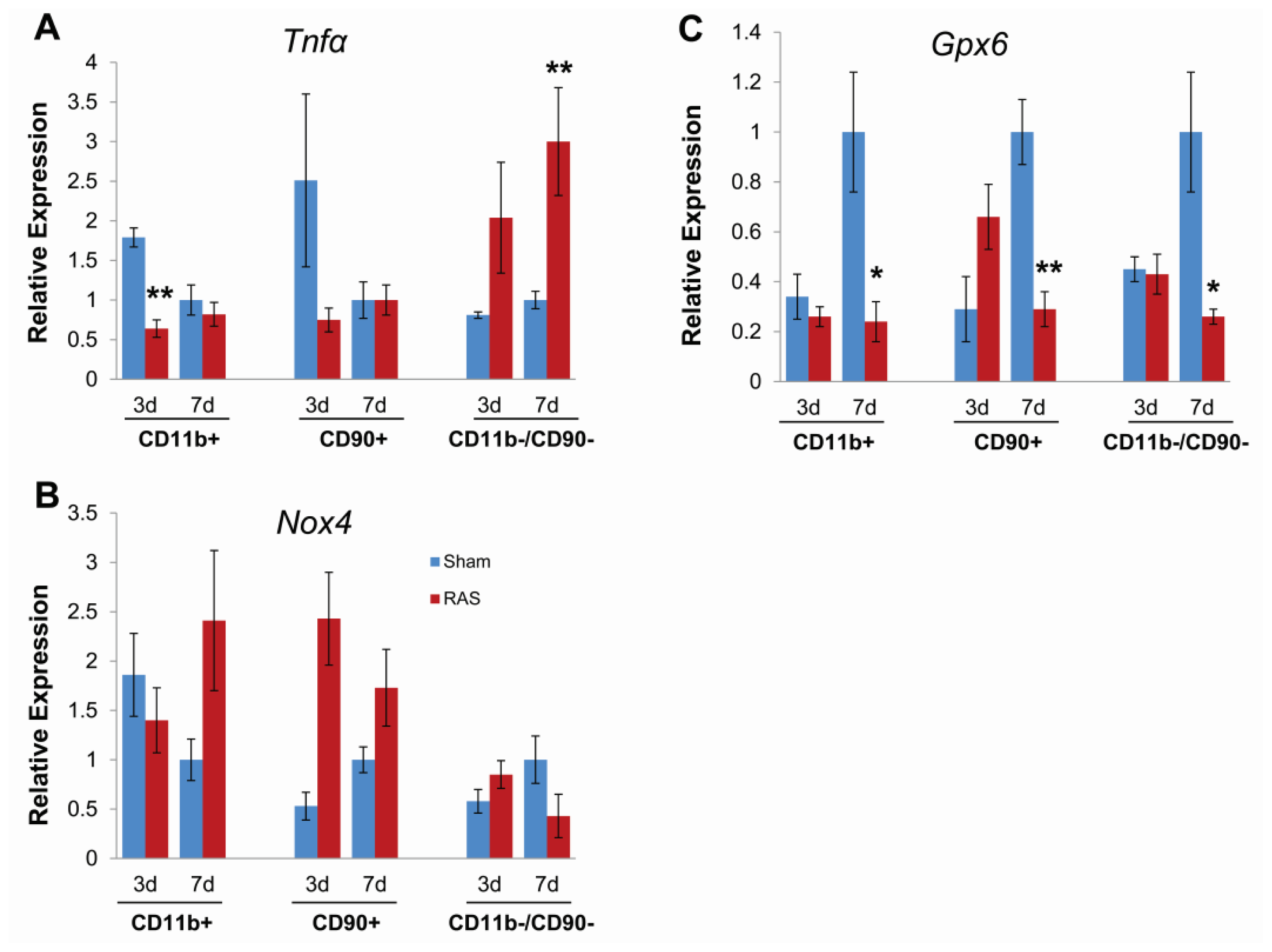
2.5. Parenchymal Cells Initiate Early Oxidative Stress Signaling
3. Discussion
4. Experimental Section
4.1. Animal Models
4.2. Fractionation of Renal Parenchymal Cells
4.3. Biochemical Analysis
4.4. Micro CT Imaging
4.5. Histology and Immunohistochemistry
4.6. PCR Array
4.7. mRNA Analysis
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hansen, K.J.; Edwards, M.S.; Craven, T.E.; Cherr, G.S.; Jackson, S.A.; Appel, R.G.; Burke, G.L.; Dean, R.H. Prevalence of renovascular disease in the elderly: A population-based study. J. Vasc. Surg 2002, 36, 443–451. [Google Scholar]
- Fava, C.; Minuz, P.; Patrignani, P.; Morganti, A. Renal artery stenosis and accelerated atherosclerosis: Which comes first? J. Hypertens 2006, 24, 1687–1696. [Google Scholar]
- Kalra, P.A.; Guo, H.; Kausz, A.T.; Gilbertson, D.T.; Liu, J.; Chen, S.C.; Ishani, A.; Collins, A.J.; Foley, R.N. Atherosclerotic renovascular disease in United States patients aged 67 years or older: Risk factors, revascularization, and prognosis. Kidney Int 2005, 68, 293–301. [Google Scholar]
- Conlon, P.J.; Athirakul, K.; Kovalik, E.; Schwab, S.J.; Crowley, J.; Stack, R.; McCants, C.B., Jr; Mark, D.B.; Bashore, T.M.; Albers, F. Survival in renal vascular disease. J. Am. Soc. Nephrol. 1998, 9, 252–256. [Google Scholar]
- Conlon, P.J.; Little, M.A.; Pieper, K.; Mark, D.B. Severity of renal vascular disease predicts mortality in patients undergoing coronary angiography. Kidney Int 2001, 60, 1490–1497. [Google Scholar]
- Textor, S.C. Managing renal arterial disease and hypertension. Curr. Opin. Cardiol 2003, 18, 260–267. [Google Scholar]
- Textor, S.C.; Wilcox, C.S. Renal artery stenosis: A common, treatable cause of renal failure? Annu. Rev. Med 2001, 52, 421–442. [Google Scholar]
- Textor, S.C. Ischemic nephropathy: Where are we now? J. Am. Soc. Nephrol 2004, 15, 1974–1982. [Google Scholar]
- Wheatley, K.; Ives, N.; Gray, R.; Kalra, P.A.; Moss, J.G.; Baigent, C.; Carr, S.; Chalmers, N.; Eadington, D.; Hamilton, G.; et al. Revascularization versus medical therapy for renal-artery stenosis. N. Engl. J. Med 2009, 361, 1953–1962. [Google Scholar]
- Warner, G.M.; Cheng, J.; Knudsen, B.E.; Gray, C.E.; Deibel, A.; Juskewitch, J.E.; Lerman, L.O.; Textor, S.C.; Nath, K.A.; Grande, J.P. Genetic deficiency of Smad3 protects the kidneys from atrophy and interstitial fibrosis in 2K1C hypertension. Am. J. Physiol. Ren. Physiol 2012, 302, F1455–F1464. [Google Scholar]
- Cheng, J.; Zhou, W.; Warner, G.M.; Knudsen, B.E.; Garovic, V.D.; Gray, C.E.; Lerman, L.O.; Platt, J.L.; Romero, J.C.; Textor, S.C.; et al. Temporal analysis of signaling pathways activated in a murine model of 2-kidney, 1-clip hypertension. Am. J. Physiol. Ren. Physiol 2009, 297, F1055–F1068. [Google Scholar]
- Wang, D.; Warner, G.M.; Yin, P.; Knudsen, B.E.; Cheng, J.; Butters, K.A.; Lien, K.R.; Gray, C.E.; Garovic, V.D.; Lerman, L.O.; et al. Grande, Inhibition of p38 MAPK attenuates renal atrophy and fibrosis in a murine renal artery stenosis model. Am. J. Physiol. Ren. Physiol 2013, 304, F938–F947. [Google Scholar]
- Edwards, M.; Craven, T.; Burke, G.; Dean, R.; Hansen, K. Renovascular disease and the risk of adverse coronary events in the elderly: A prospective, population-based study. Arch. Intern. Med 2005, 165, 207–213. [Google Scholar]
- Favreau, F.; Zhu, X.-Y.; Krier, J.D.; Lin, J.; Warner, L.; Textor, S.C.; Lerman, L.O. Revascularization of swine renal artery stenosis improves renal function but not the changes in vascular structure. Kidney Int 2010, 78, 1110–1118. [Google Scholar]
- Eirin, A.; Zhu, X.Y.; Urbieta-Caceres, V.H.; Grande, J.P.; Lerman, A.; Textor, S.C.; Lerman, L.O. Persistent kidney dysfunction in swine renal artery stenosis correlates with outer cortical microvascular remodeling. Am. J. Physiol. Ren. Physiol 2011, 300, F1394–F1401. [Google Scholar]
- Chade, A.R.; Rodriguez-Porcel, M.; Grande, J.P.; Krier, J.D.; Lerman, A.; Romero, J.C.; Napoli, C.; Lerman, L.O. Distinct renal injury in early atherosclerosis and renovascular disease. Circulation 2002, 106, 1165–1171. [Google Scholar]
- Eirin, A.; Zhu, X.Y.; Li, Z.; Ebrahimi, B.; Zhang, X.; Tang, H.; Korsmo, M.J.; Chade, A.R.; Grande, J.P.; Ward, C.J.; et al. Endothelial outgrowth cells shift macrophage phenotype and improve kidney viability in swine renal artery stenosis. Arterioscler. Thromb. Vasc. Biol 2013, 33, 1006–1013. [Google Scholar]
- Lavoie, J.L.; Sigmund, C.D. Minireview: Overview of the renin-angiotensin system—An endocrine and paracrine system. Endocrinology 2003, 144, 2179–2183. [Google Scholar]
- Bader, M.; Peters, J.; Baltatu, O.; Müller, D.; Luft, F.; Ganten, D. Tissue renin-angiotensin systems: New insights from experimental animal models in hypertension research. J. Mol. Med. (Berl. Ger. ) 2001, 79, 76–102. [Google Scholar]
- Rohrwasser, A.; Morgan, T.; Dillon, H.; Zhao, L.; Callaway, C.; Hillas, E.; Zhang, S.; Cheng, T.; Inagami, T.; Ward, K.; et al. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension 1999, 34, 1265–1274. [Google Scholar]
- Chade, A.R.; Rodriguez-Porcel, M.; Grande, J.P.; Zhu, X.Y.; Sica, V.; Napoli, C.; Sawamura, T.; Textor, S.C.; Lerman, A.; Lerman, L.O. Mechanisms of renal structural alterations in combined hypercholesterolemia and renal artery stenosis. Arterioscler. Thromb. Vasc. Biol 2003, 23, 1295–1301. [Google Scholar]
- Chade, A.R.; Rodriguez-Porcel, M.; Herrmann, J.; Krier, J.D.; Zhu, X.; Lerman, A.; Lerman, L.O. Beneficial effects of antioxidant vitamins on the stenotic kidney. Hypertension 2003, 42, 605–612. [Google Scholar]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Investig 1996, 97, 1916–1923. [Google Scholar]
- Chabrashvili, T.; Tojo, A.; Onozato, M.L.; Kitiyakara, C.; Quinn, M.T.; Fujita, T.; Welch, W.J.; Wilcox, C.S. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 2002, 39, 269–274. [Google Scholar]
- Tojo, A.; Onozato, M.L.; Kobayashi, N.; Goto, A.; Matsuoka, H.; Fujita, T. Angiotensin II and oxidative stress in Dahl Salt-sensitive rat with heart failure. Hypertension 2002, 40, 834–839. [Google Scholar]
- Liu, G.; Fang, F.; Zhou, J.; Koulajian, K.; Yang, S.; Lam, L.; Reich, H.; John, R.; Herzenberg, A.; Giacca, A.; et al. Deletion of p47phox attenuates the progression of diabetic nephropathy and reduces the severity of diabetes in the Akita mouse. Diabetologia 2012, 55, 2522–2532. [Google Scholar]
- Geiszt, M.; Kopp, J.B.; Varnai, P.; Leto, T.L. Identification of Renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar]
- Chabrashvili, T.; Kitiyakara, C.; Blau, J.; Karber, A.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am. J. Physiol. Regul. Integr. Comp. Physiol 2003, 285, R117–R124. [Google Scholar]
- Lassegue, B.; Sorescu, D.; Szocs, K.; Yin, Q.Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91(phox) homologues in vascular smooth muscle cells - Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res 2001, 88, 888–894. [Google Scholar]
- Nlandu Khodo, S.; Dizin, E.; Sossauer, G.; Szanto, I.; Martin, P.-Y.; Feraille, E.; Krause, K.H.; de Seigneux, S. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J. Am. Soc. Nephrol 2012, 23, 1967–1976. [Google Scholar]
- Babelova, A.; Avaniadi, D.; Jung, O.; Fork, C.; Beckmann, J.; Kosowski, J.; Weissmann, N.; Anilkumar, N.; Shah, A.M.; Schaefer, L.; et al. Role of Nox4 in murine models of kidney disease. Free Radic. Biol. Med 2012, 53, 842–853. [Google Scholar]
- Vaziri, N.D.; Dicus, M.; Ho, N.D.; Boroujerdi-Rad, L.; Sindhu, R.K. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int 2003, 63, 179–185. [Google Scholar]
- Lerman, L.O.; Nath, K.A.; Rodriguez-Porcel, M.; Krier, J.D.; Schwartz, R.S.; Napoli, C.; Romero, J.C. Increased oxidative stress in experimental renovascular hypertension. Hypertension 2001, 37, 541–546. [Google Scholar]
- Chade, A.R.; Rodriguez-Porcel, M.; Herrmann, J.; Zhu, X.Y.; Grande, J.P.; Napoli, C.; Lerman, A.; Lerman, L.O. Antioxidant intervention blunts renal injury in experimental renovascular disease. J. Am. Soc. Nephrol 2004, 15, 958–966. [Google Scholar]
- Adler, S.; Huang, H. Oxidant stress in kidneys of spontaneously hypertensive rats involves both oxidase overexpression and loss of extracellular superoxide dismutase. Am. J. Physiol. Ren. Physiol 2004, 287, F907–F913. [Google Scholar]
- Zhao, W.; Chen, S.S.; Chen, Y.; Ahokas, R.A.; Sun, Y. Kidney fibrosis in hypertensive rats: Role of oxidative stress. Am. J. Nephrol 2008, 28, 548–554. [Google Scholar]
- Vielhauer, V.; Anders, H.J.; Perez de Lema, G.; Luckow, B.; Schlondorff, D.; Mack, M. Phenotyping renal leukocyte subsets by four-color flow cytometry: Characterization of chemokine receptor expression. Nephron Exp. Nephrol 2003, 93, e63. [Google Scholar]
- Zhang, X.; Urbieta-Caceres, V.H.; Eirin, A.; Bell, C.C.; Crane, J.A.; Tang, H.; Jordan, K.L.; Oh, Y.K.; Zhu, X.Y.; Korsmo, M.J.; et al. Humanin prevents intra-renal microvascular remodeling and inflammation in hypercholesterolemic ApoE deficient mice. Life Sci 2012, 91, 199–206. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hartono, S.P.; Knudsen, B.E.; Zubair, A.S.; Nath, K.A.; Textor, S.J.; Lerman, L.O.; Grande, J.P. Redox Signaling Is an Early Event in the Pathogenesis of Renovascular Hypertension. Int. J. Mol. Sci. 2013, 14, 18640-18656. https://doi.org/10.3390/ijms140918640
Hartono SP, Knudsen BE, Zubair AS, Nath KA, Textor SJ, Lerman LO, Grande JP. Redox Signaling Is an Early Event in the Pathogenesis of Renovascular Hypertension. International Journal of Molecular Sciences. 2013; 14(9):18640-18656. https://doi.org/10.3390/ijms140918640
Chicago/Turabian StyleHartono, Stella P., Bruce E. Knudsen, Adeel S. Zubair, Karl A. Nath, Stephen J. Textor, Lilach O. Lerman, and Joseph P. Grande. 2013. "Redox Signaling Is an Early Event in the Pathogenesis of Renovascular Hypertension" International Journal of Molecular Sciences 14, no. 9: 18640-18656. https://doi.org/10.3390/ijms140918640
APA StyleHartono, S. P., Knudsen, B. E., Zubair, A. S., Nath, K. A., Textor, S. J., Lerman, L. O., & Grande, J. P. (2013). Redox Signaling Is an Early Event in the Pathogenesis of Renovascular Hypertension. International Journal of Molecular Sciences, 14(9), 18640-18656. https://doi.org/10.3390/ijms140918640