Structural Variations in Articular Cartilage Matrix Are Associated with Early-Onset Osteoarthritis in the Spondyloepiphyseal Dysplasia Congenita (Sedc) Mouse

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
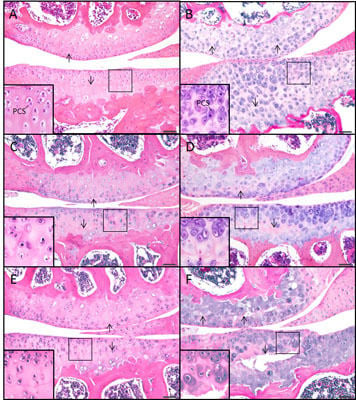
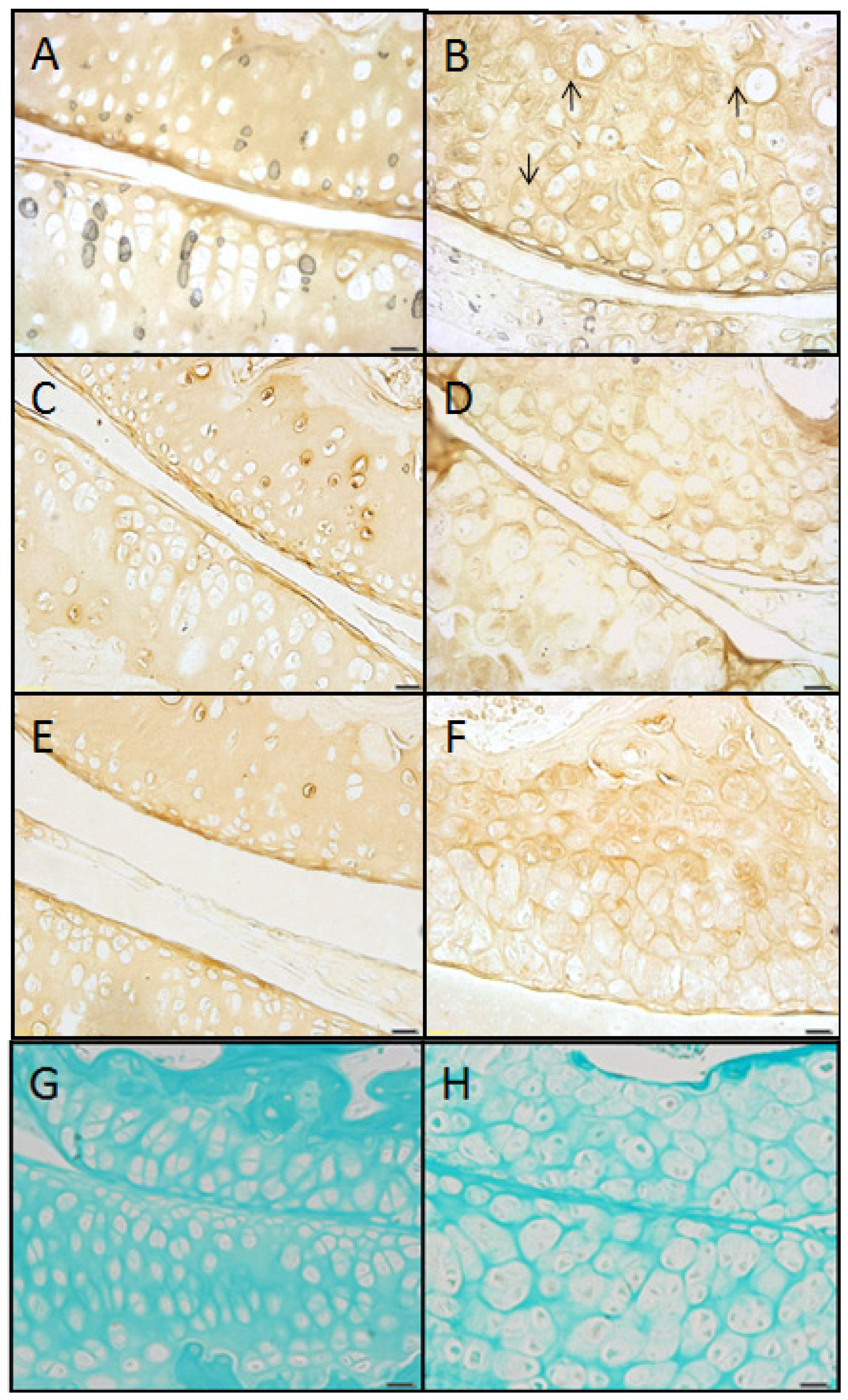
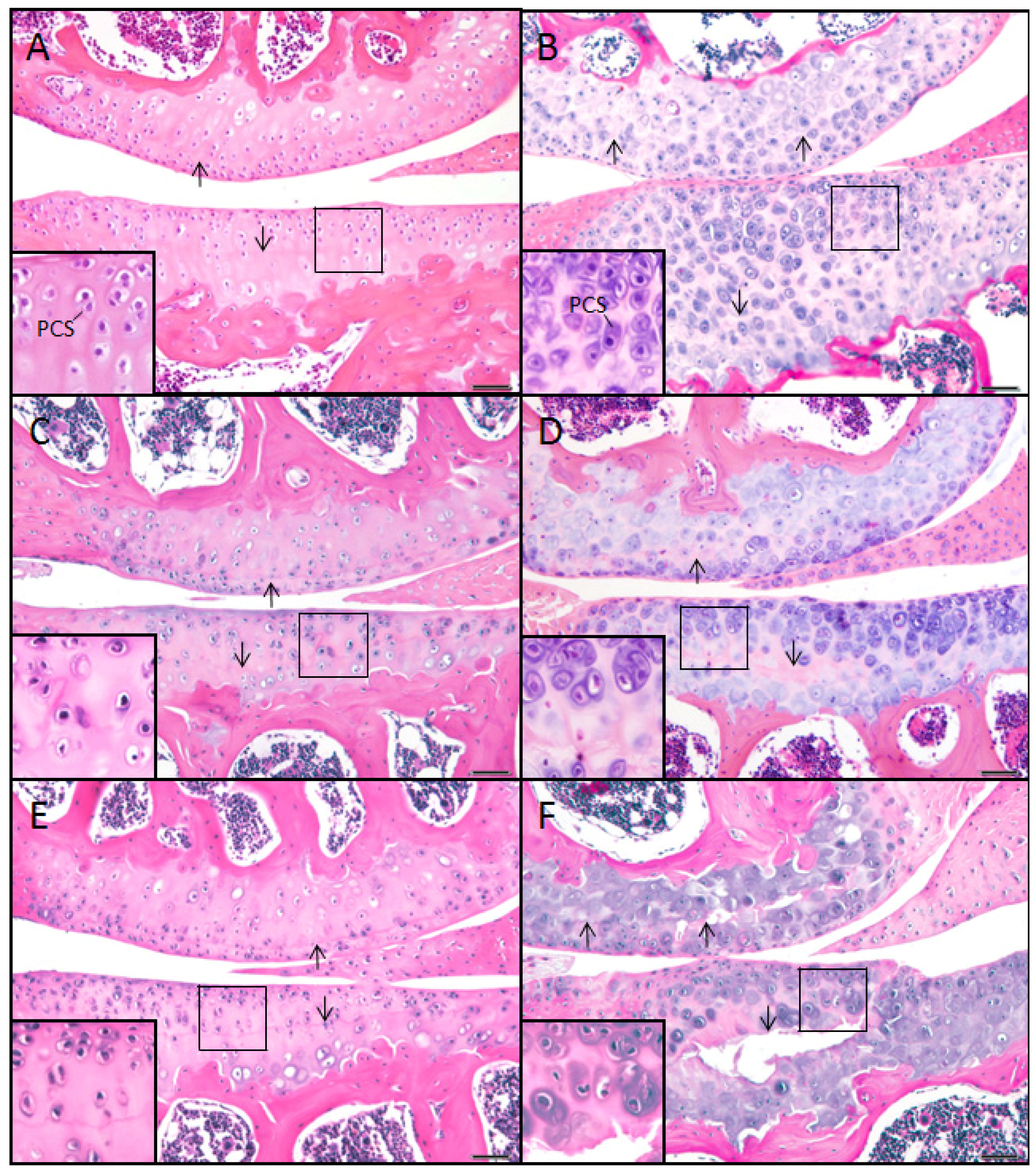
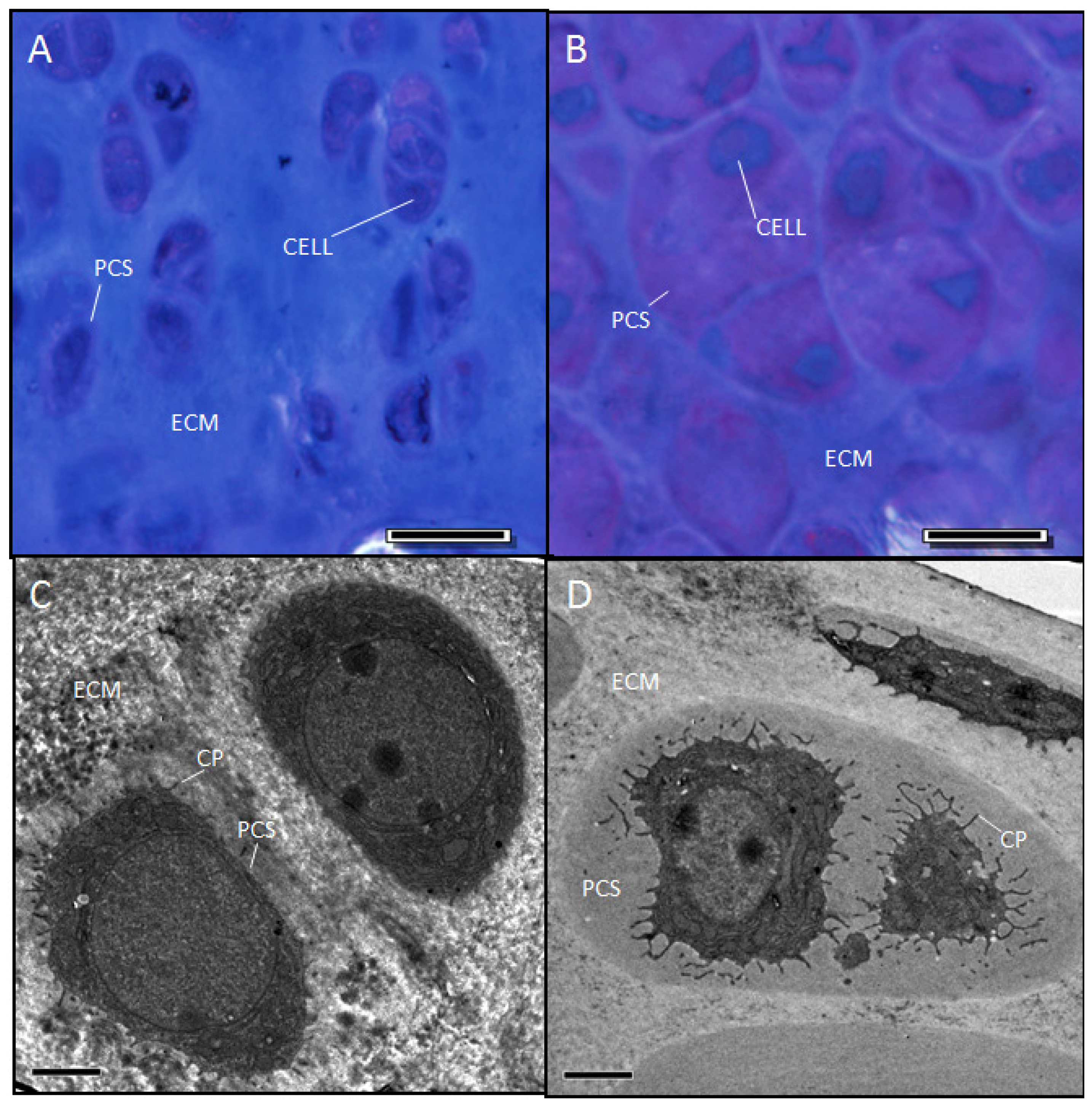
2.1. The sedc/sedc Mouse Shows Unique Histological Variations in Articular Cartilage
Wildtype control (+/+)
Mutant (sedc/sedc)
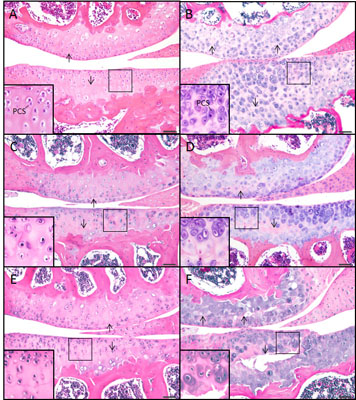
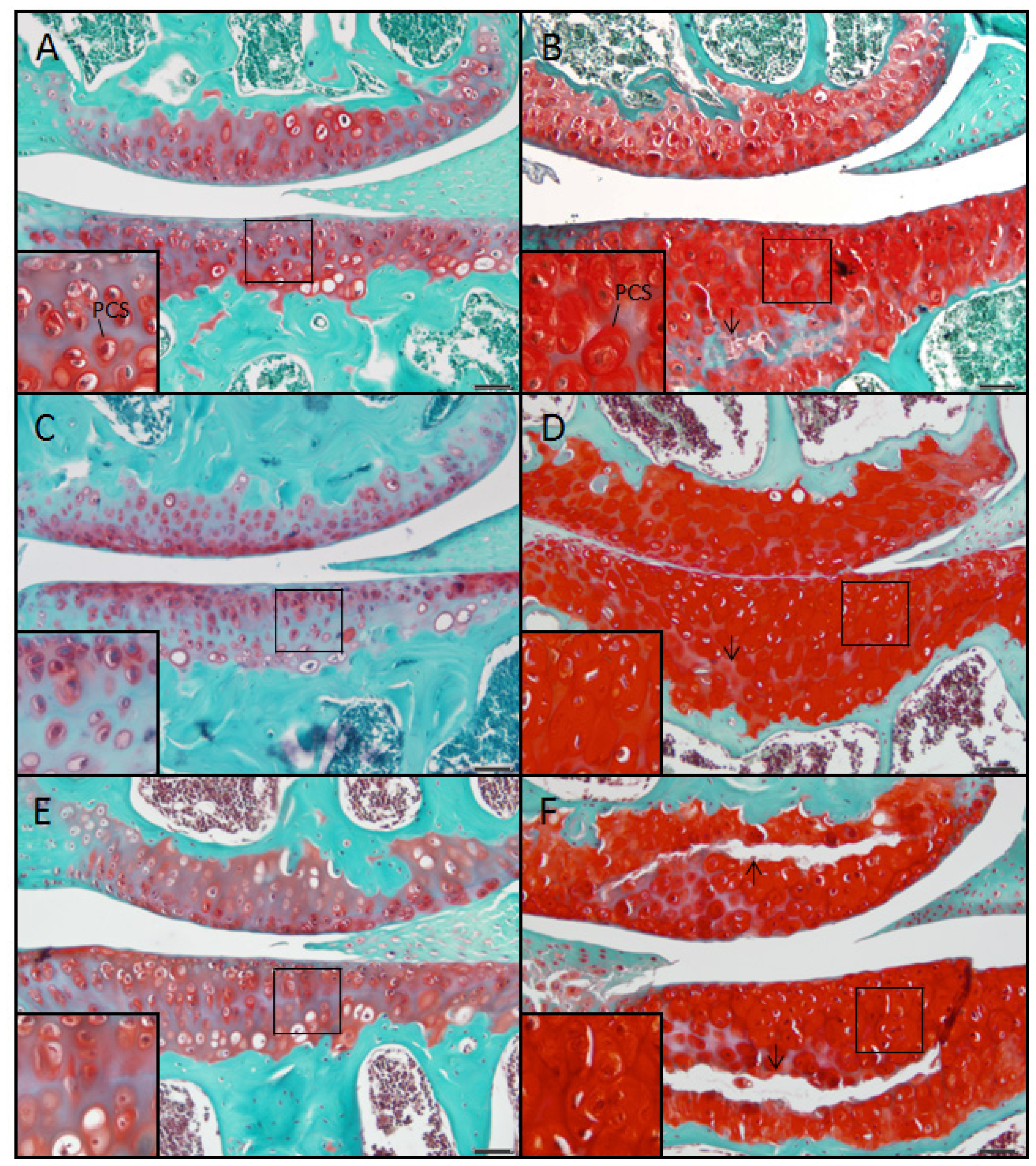
2.2. The sedc/sedc Mouse Shows Increased Safranin O Staining in the PCS Confirming Presence of Basophilic Proteoglycan
Wildtype control (+/+)
Mutant (sedc/sedc)
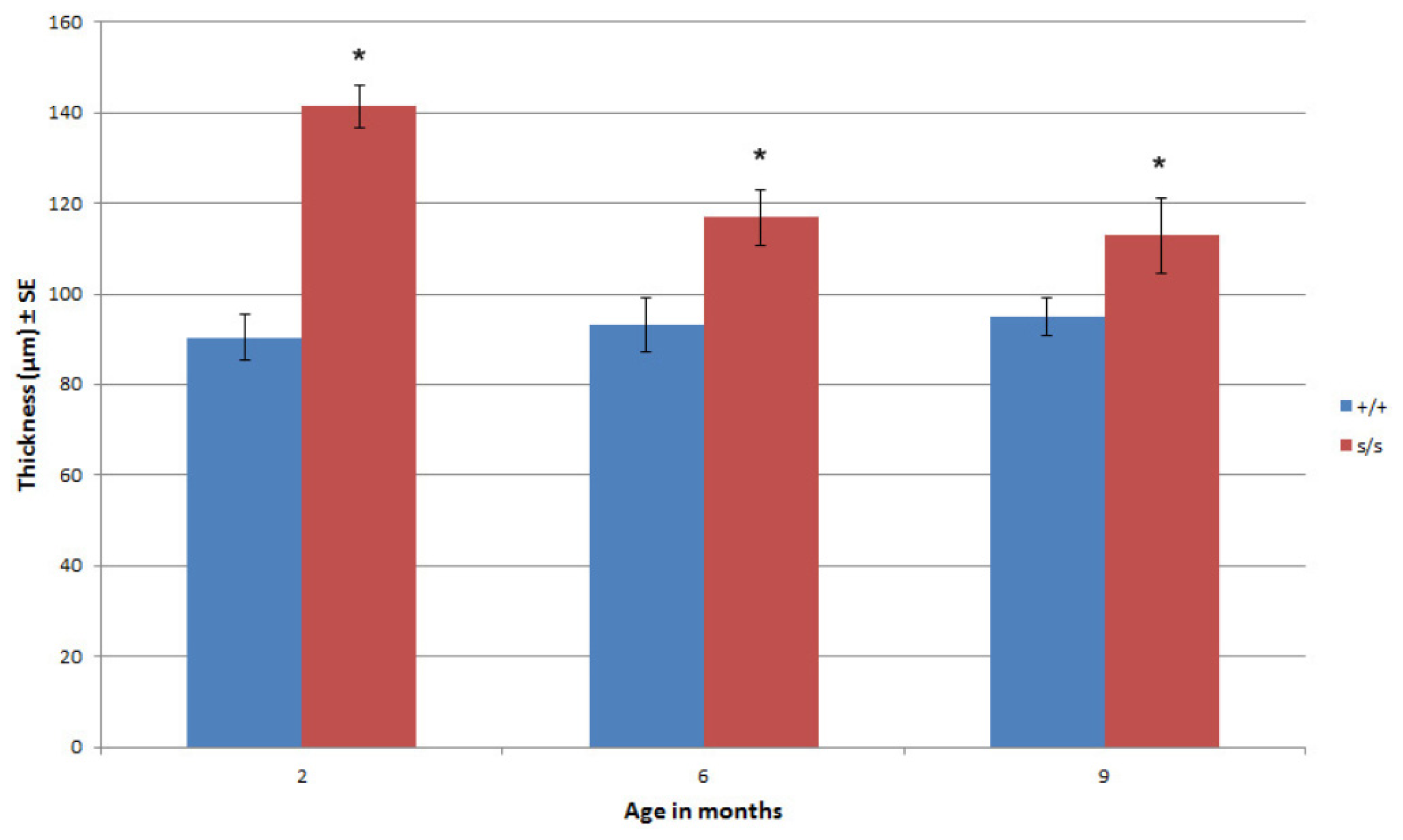
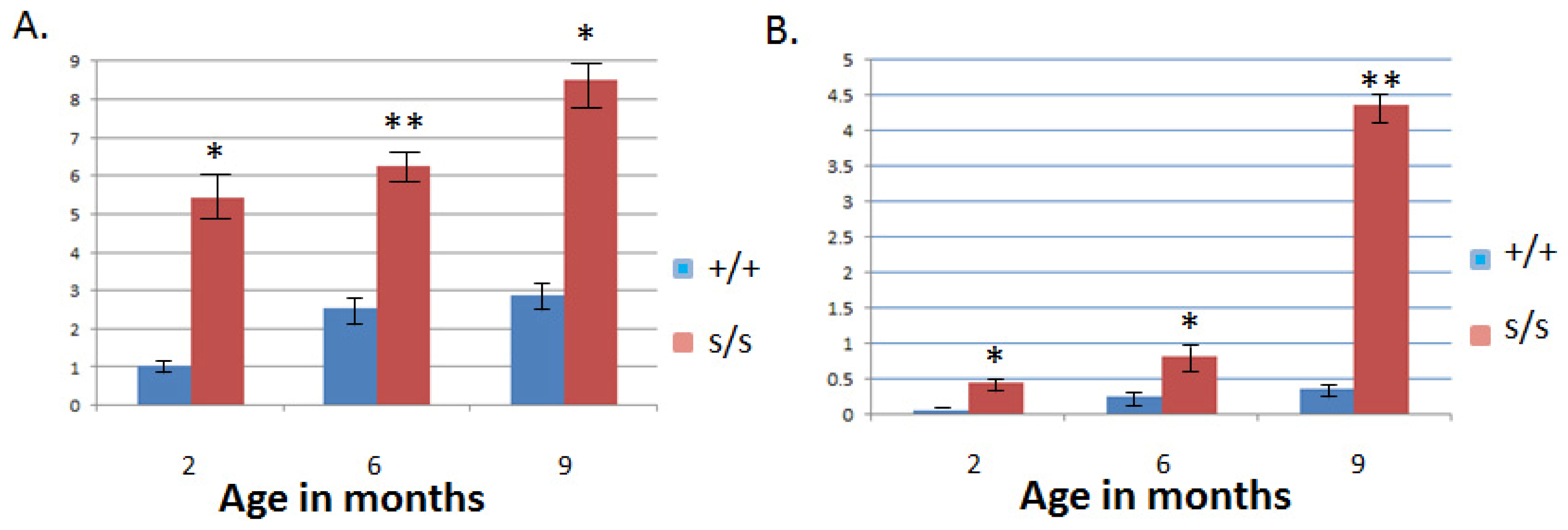
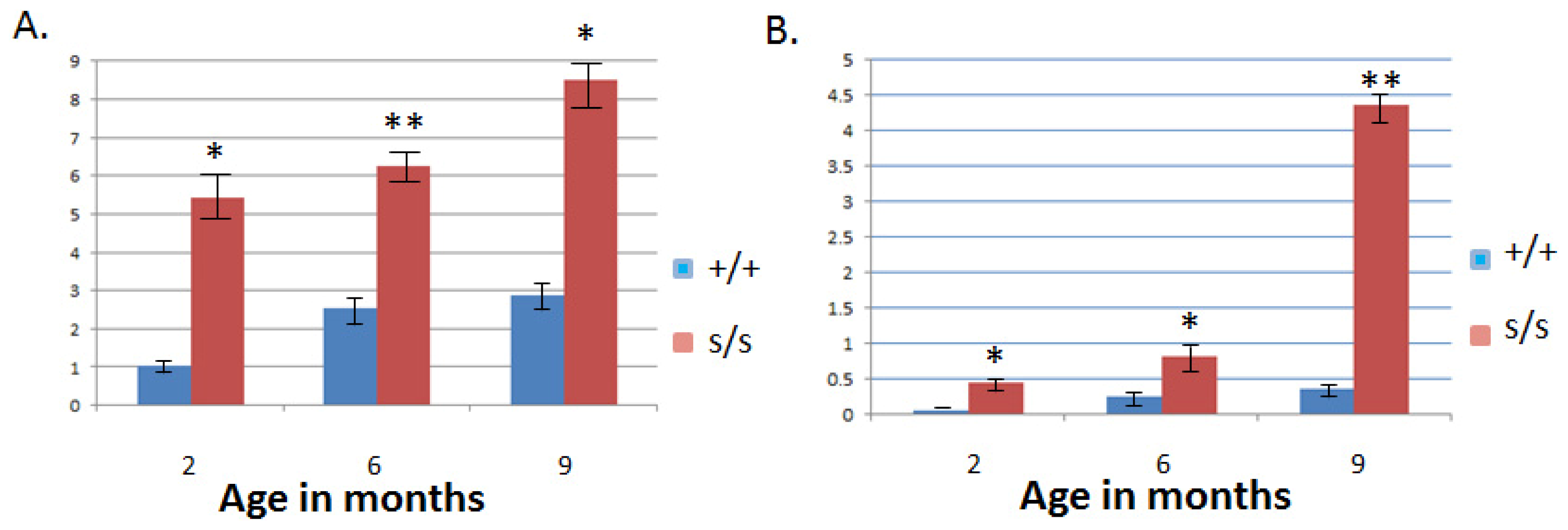
2.3. The Homozygous Mutant Shows Early Onset and Increasing Severity of OA in the Knee Joint
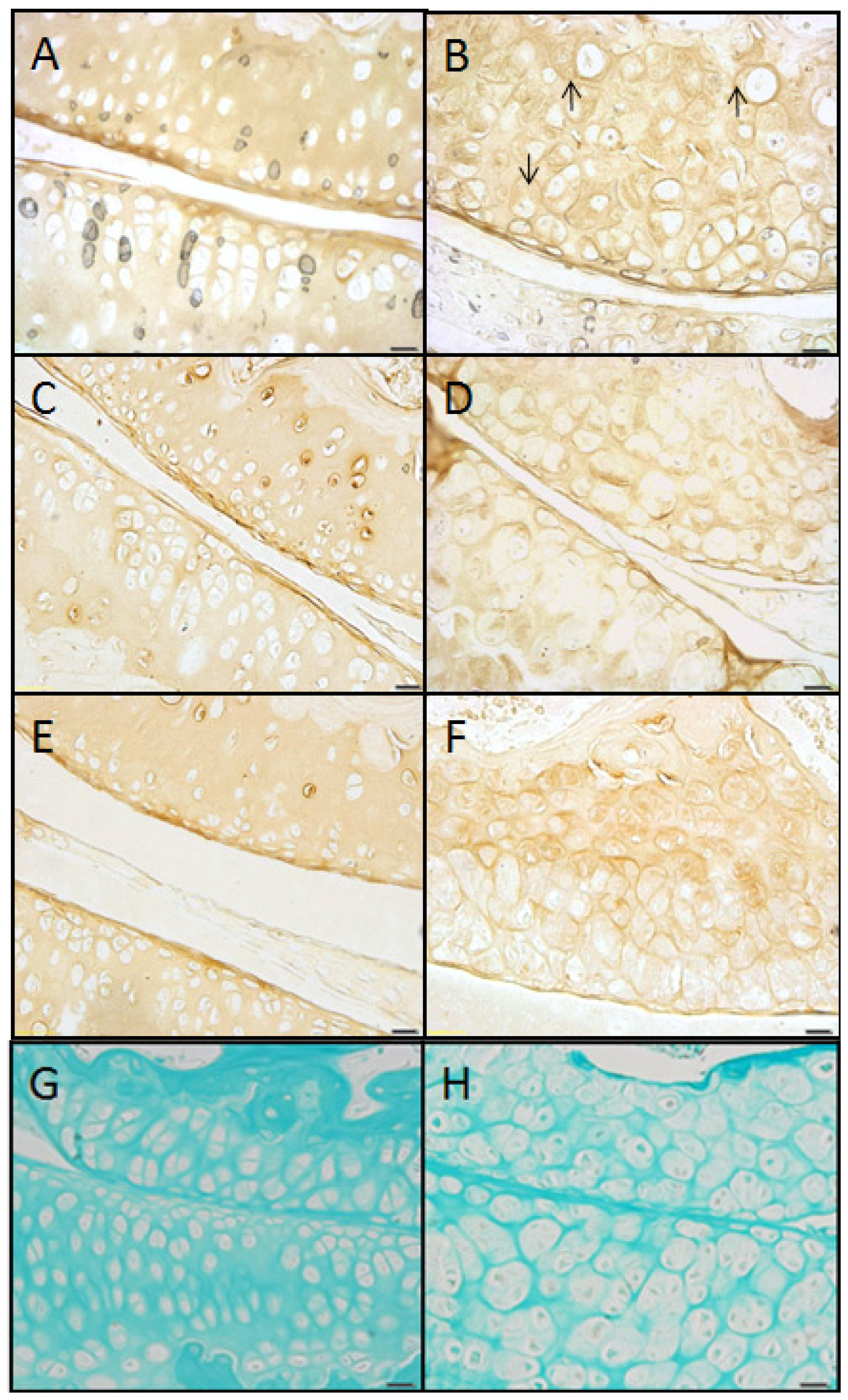
2.4. Type II Collagen Is Present in sedc/sedc Articular Cartilage in both ECM and PCS
Wildtype control (+/+)
Mutant (sedc/sedc)
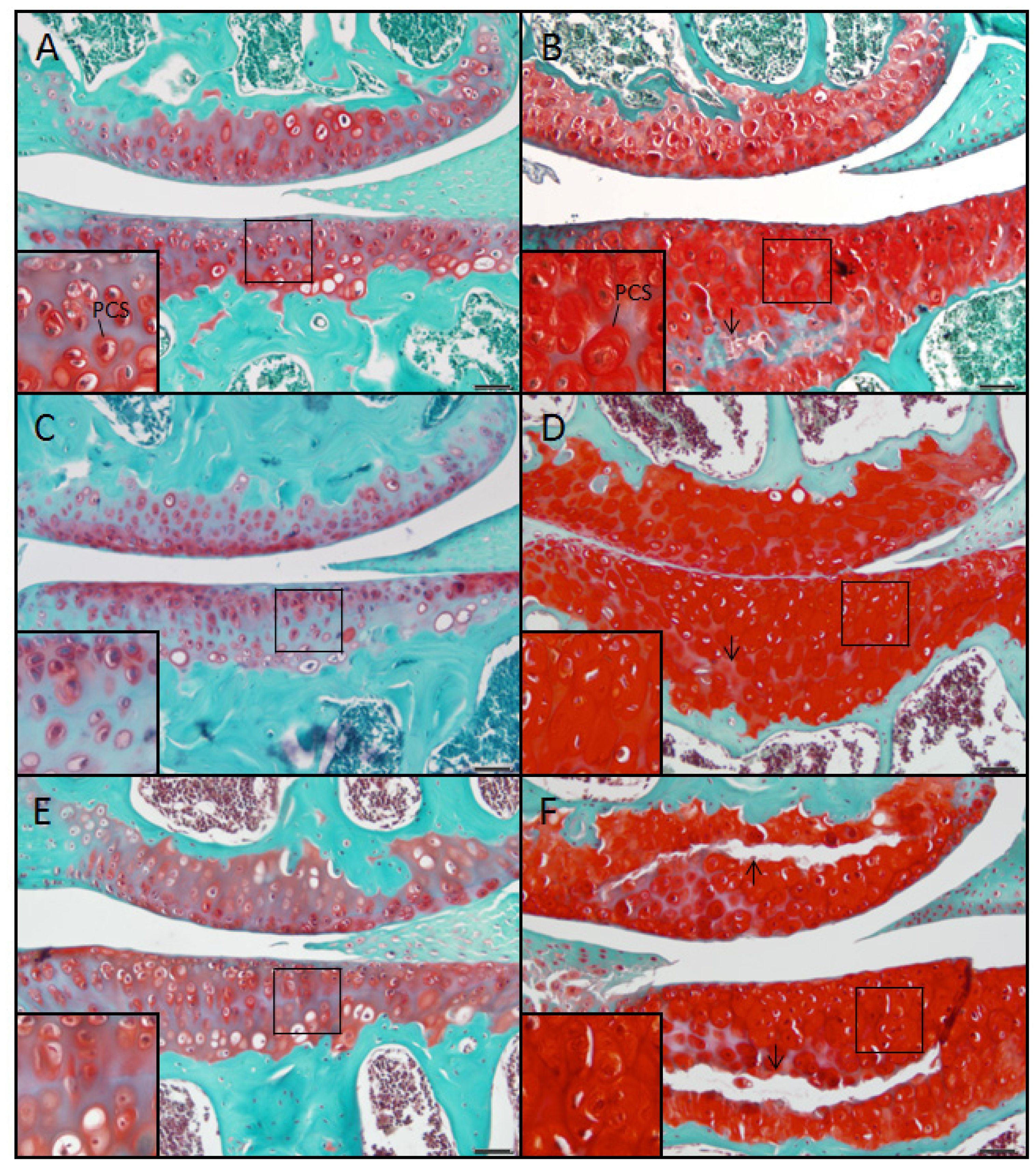
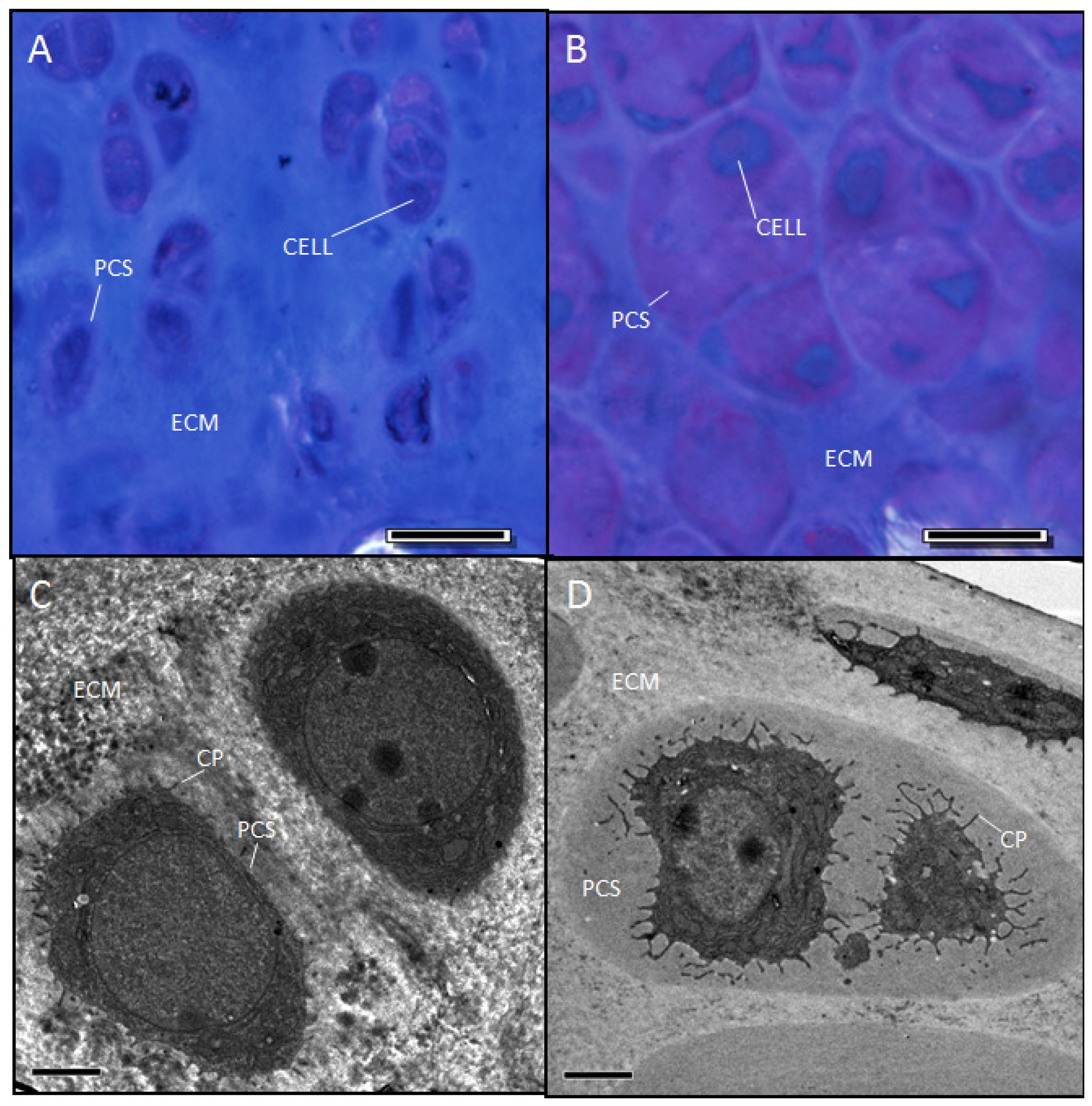
2.5. Evidence Confirming the Presence of Proteoglycan in the Expanded PCS of sedc/sedc Articular Cartilage
Wildtype control (+/+)
Mutant (sedc/sedc)
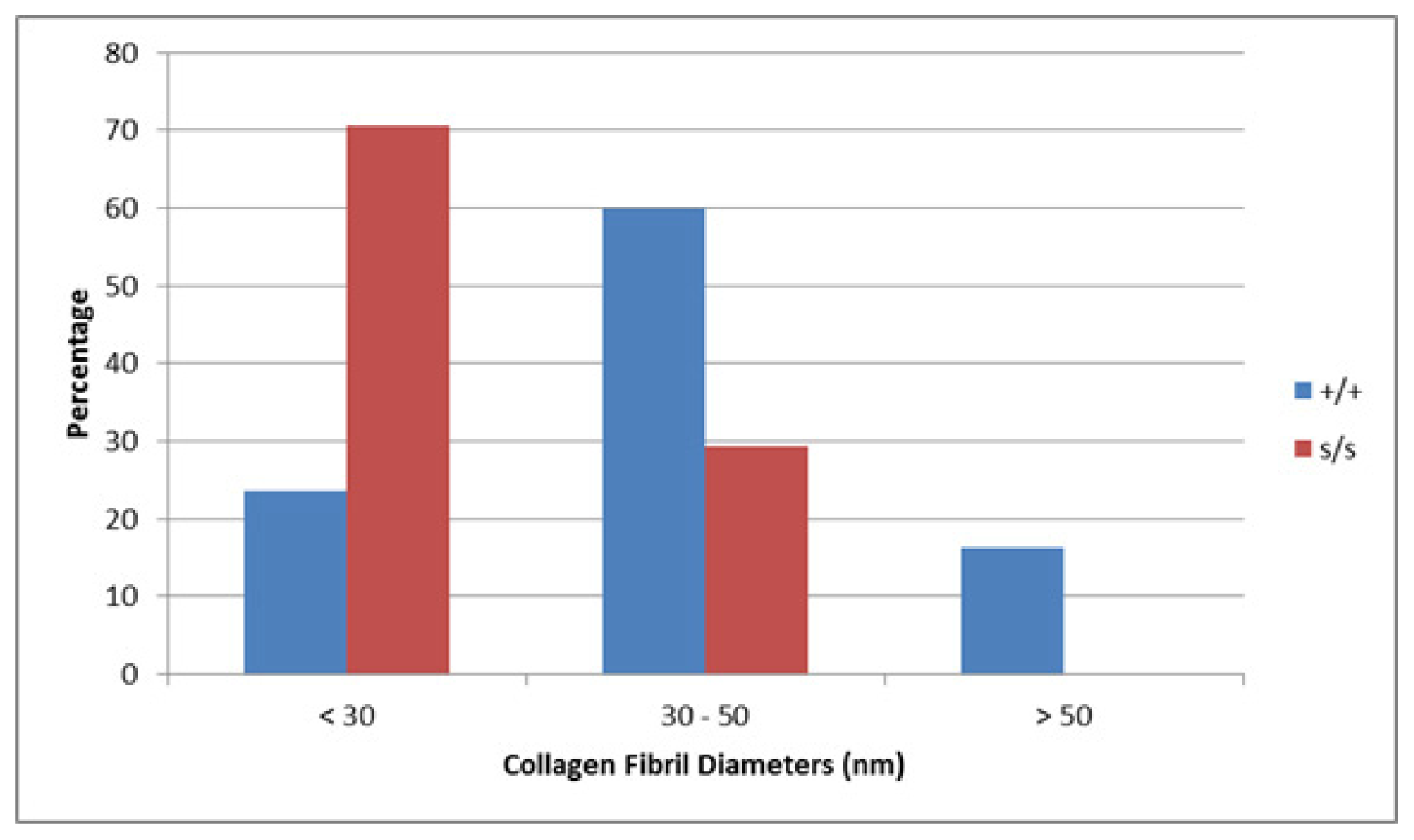
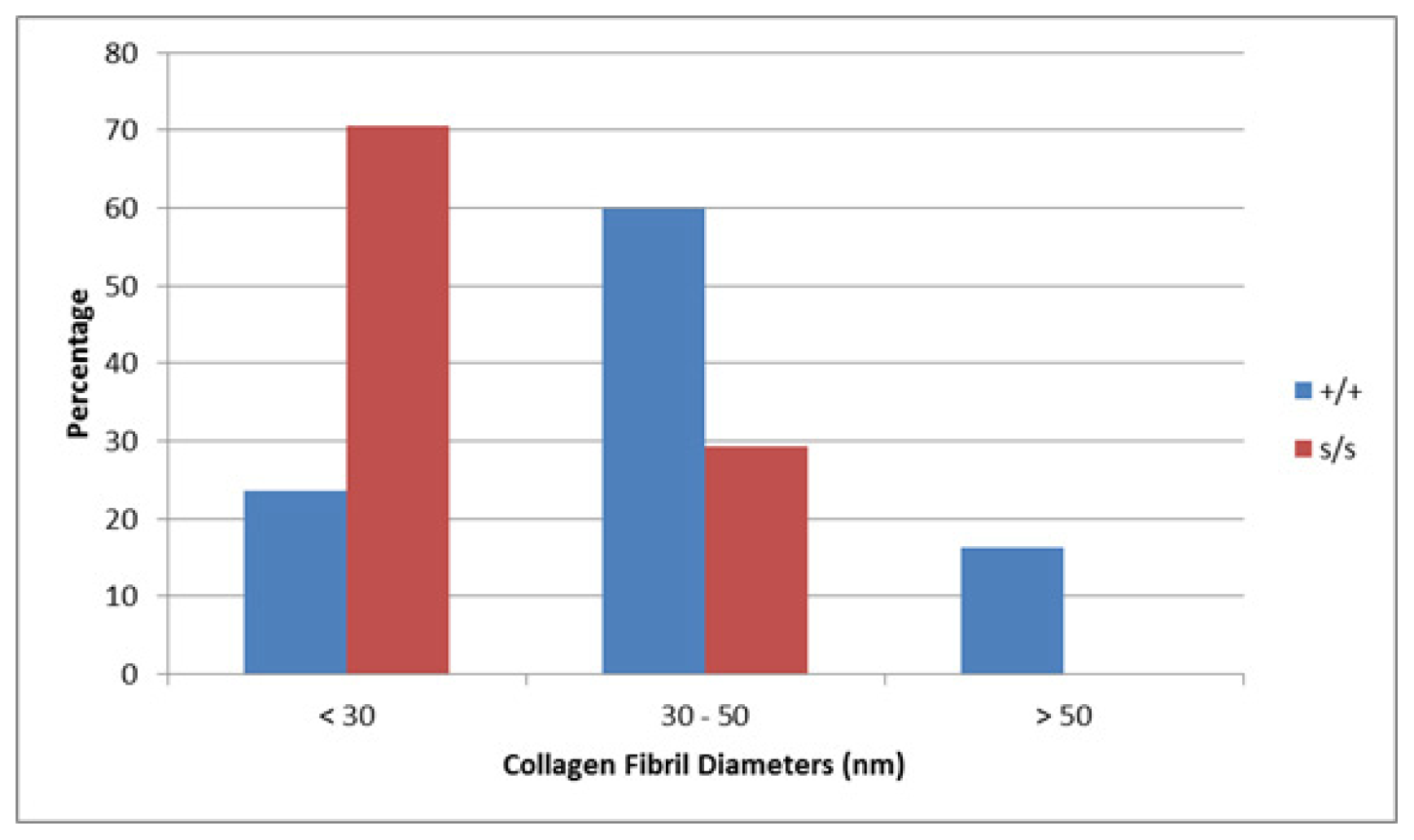
2.6. Collagen Fibril Diameter Is Smaller in sedc/sedc Articular Cartilage
Wildtype control (+/+)
Mutant (sedc/sedc)
3. Experimental Section
3.1. Experimental Animals and Determination of Genotype
3.2. Tissue Processing
3.3. Histological Evaluation of Articular Cartilage
3.4. Collagen Described by Immunohistochemistry
3.5. Localization of Proteoglycan by Light and Electron Microscopy
3.6. Determination of Collagen Fibril Diameter
3.7. Statistical Analysis
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Hollander, A.P.; Heathfield, T.F.; Webber, C.; Iwata, Y.; Bourne, R.; Rorabeck, C.; Poole, A.R. Increased damage to type II collagen in osteoarthritic articular cartilage detected by a new immunoassay. J. Clin. Invest 1994, 93, 1722–1732. [Google Scholar]
- Buckwalter, J.A.; Mankin, H.J. Articular cartilage: Tissue design and chondrocyte-matrix interactions. Instr. Course Lect 1998, 47, 477–486. [Google Scholar]
- Newman, A.P. Articular cartilage repair. Am. J. Sports Med 1998, 26, 309–324. [Google Scholar]
- Ofek, G.; Revell, C.M.; Hu, J.C.; Allison, D.D.; Grande-Allen, K.J.; Athanasiou, K.A. Matrix development in self-assembly of articular cartilage. PLoS One 2008, 3, e2795. [Google Scholar]
- Gentili, C.; Cancedda, R. Cartilage and bone extracellular matrix. Curr. Pharm. Des 2009, 15, 1334–1348. [Google Scholar]
- Knudson, C.B.; Knudson, W. Cartilage proteoglycans. Semin. Cell Dev. Biol 2001, 12, 69–78. [Google Scholar]
- Hunziker, E.B.; Quinn, T.M.; Hauselmann, H.J. Quantitative structural organization of normal adult human articular cartilage. Osteoarthritis Cartilage 2002, 10, 564–572. [Google Scholar]
- Schumacher, B.L.; Su, J.L.; Lindley, K.M.; Kuettner, K.E.; Cole, A.A. Horizontally oriented clusters of multiple chondrons in the superficial zone of ankle, but not knee articular cartilage. Anat. Rec 2002, 266, 241–248. [Google Scholar]
- Aszodi, A.; Hunziker, E.B.; Brakebusch, C.; Fassler, R. Beta1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev 2003, 17, 2465–2479. [Google Scholar]
- Jadin, K.D.; Wong, B.L.; Bae, W.C.; Li, K.W.; Williamson, A.K.; Schumacher, B.L.; Price, J.H.; Sah, R.L. Depth-varying density and organization of chondrocytes in immature and mature bovine articular cartilage assessed by 3d imaging and analysis. J. Histochem. Cytochem 2005, 53, 1109–1119. [Google Scholar]
- Lamande, S.R.; Bateman, J.F. Procollagen folding and assembly: The role of endoplasmic reticulum enzymes and molecular chaperones. Semin. Cell Dev. Biol 1999, 10, 455–464. [Google Scholar]
- Seegmiller, R.E.; Bomsta, B.D.; Bridgewater, L.C.; Niederhauser, C.M.; Montano, C.; Sudweeks, S.; Eyre, D.R.; Fernandes, R.J. The heterozygous disproportionate micromelia (dmm) mouse: Morphological changes in fetal cartilage precede postnatal dwarfism and compared with lethal homozygotes can explain the mild phenotype. J. Histochem. Cytochem 2008, 56, 1003–1011. [Google Scholar]
- Tasab, M.; Batten, M.R.; Bulleid, N.J. Hsp47: A molecular chaperone that interacts with and stabilizes correctly-folded procollagen. EMBO J 2000, 19, 2204–2211. [Google Scholar]
- Kuivaniemi, H.; Tromp, G.; Prockop, D.J. Mutations in fibrillar collagens (types I, II, III, and XI), fibril-associated collagen (type IX), and network-forming collagen (type X) cause a spectrum of diseases of bone, cartilage, and blood vessels. Hum. Mutat 1997, 9, 300–315. [Google Scholar]
- Pace, J.M.; Kuslich, C.D.; Willing, M.C.; Byers, P.H. Disruption of one intra-chain disulphide bond in the carboxyl-terminal propeptide of the proalpha1(I) chain of type I procollagen permits slow assembly and secretion of overmodified, but stable procollagen trimers and results in mild osteogenesis imperfecta. J. Med. Genet 2001, 38, 443–449. [Google Scholar]
- Eyre, D. Collagen of articular cartilage. Arthritis. Res 2002, 4, 30–35. [Google Scholar]
- Hulmes, D.J. Building collagen molecules, fibrils, and suprafibrillar structures. J. Struct. Biol 2002, 137, 2–10. [Google Scholar]
- Boudko, S.P.; Engel, J. Structure formation in the C terminus of type III collagen guides disulfide cross-linking. J. Mol. Biol 2004, 335, 1289–1297. [Google Scholar]
- Matthews, B.F. Collagen-chondroitin sulphate ratio of human articular cartilage related to function. Br. Med. J 1952, 2, 1295. [Google Scholar]
- Lane, J.M.; Weiss, C. Review of articular cartilage collagen research. Arthritis Rheum 1975, 18, 553–562. [Google Scholar]
- Hardingham, T.E.; Fosang, A.J. Proteoglycans: Many forms and many functions. FASEB J 1992, 6, 861–870. [Google Scholar]
- Campbell, S.C.; Schwartz, N.B. Kinetics of intracellular processing of chondroitin sulfate proteoglycan core protein and other matrix components. J. Cell Biol 1988, 106, 2191–2202. [Google Scholar]
- Varki, A. Essentials of Glycobiology, 2nd ed; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; p. 784. [Google Scholar]
- McDevitt, C.A. Biochemistry of articular cartilage. Nature of proteoglycans and collagen of articular cartilage and their role in ageing and in osteoarthrosis. Ann. Rheum. Dis 1973, 32, 364–378. [Google Scholar]
- Fraser, J.R.; Laurent, T.C.; Laurent, U.B. Hyaluronan: Its nature, distribution, functions and turnover. J. Intern. Med 1997, 242, 27–33. [Google Scholar]
- Watanabe, H.; Yamada, Y.; Kimata, K. Roles of aggrecan, a large chondroitin sulfate proteoglycan, in cartilage structure and function. J. Biochem 1998, 124, 687–693. [Google Scholar]
- Hunziker, E.B.; Rosenberg, L.C. Repair of partial-thickness defects in articular cartilage: Cell recruitment from the synovial membrane. J. Bone Joint Surg 1996, 78, 721–733. [Google Scholar]
- Gelse, K.; von der Mark, K.; Aigner, T.; Park, J.; Schneider, H. Articular cartilage repair by gene therapy using growth factor-producing mesenchymal cells. Arthritis Rheum 2003, 48, 430–441. [Google Scholar]
- Donahue, L.R.; Chang, B.; Mohan, S.; Miyakoshi, N.; Wergedal, J.E.; Baylink, D.J.; Hawes, N.L.; Rosen, C.J.; Ward-Bailey, P.; Zheng, Q.Y.; et al. A missense mutation in the mouse Col2a1 gene causes spondyloepiphyseal dysplasia congenita, hearing loss, and retinoschisis. J. Bone Miner. Res 2003, 18, 1612–1621. [Google Scholar]
- Holt, D.W.; Henderson, M.L.; Stockdale, C.E.; Farrell, J.T.; Kooyman, D.L.; Bridgewater, L.C.; Seegmiller, R.E. Osteoarthritis-like changes in the heterozygous sedc mouse associated with the HtrA1-Ddr2-Mmp-13 degradative pathway: A new model of osteoarthritis. Osteoarthritis Cartilage 2012, 20, 430–439. [Google Scholar]
- Micheli, L.; Curtis, C.; Shervin, N. Articular cartilage repair in the adolescent athlete: Is autologous chondrocyte implantation the answer? Clin. J. Sport Med 2006, 16, 465–470. [Google Scholar]
- Schmal, H.; Pestka, J.M.; Salzmann, G.; Strohm, P.C.; Sudkamp, N.P.; Niemeyer, P. Autologous chondrocyte implantation in children and adolescents. Knee Surg. Sport Tr. A 2013, 21, 671–677. [Google Scholar]
- Ahmad, N.N.; Dimascio, J.; Knowlton, R.G.; Tasman, W.S. Stickler syndrome. A mutation in the nonhelical 3′ end of type II procollagen gene. Arch. Ophthalmol 1995, 113, 1454–1457. [Google Scholar]
- Richards, A.J.; Morgan, J.; Bearcroft, P.W.; Pickering, E.; Owen, M.J.; Holmans, P.; Williams, N.; Tysoe, C.; Pope, F.M.; Snead, M.P.; et al. Vitreoretinopathy with phalangeal epiphyseal dysplasia, a type II collagenopathy resulting from a novel mutation in the C-propeptide region of the molecule. J. Med. Genet 2002, 39, 661–665. [Google Scholar]
- Zabel, B.; Hilbert, K.; Stoss, H.; SupertiFurga, A.; Spranger, J.; Winterpacht, A. A specific collagen type II gene (COL2A1) mutation presenting as spondyloperipheral dysplasia. Am J Med Genet 1996, 63, 123–128. [Google Scholar]
- Mortier, G.R.; Weis, M.; Nuytinck, L.; King, L.M.; Wilkin, D.J.; de Paepe, A.; Lachman, R.S.; Rimoin, D.L.; Eyre, D.R.; Cohn, D.H. Report of five novel and one recurrent COL2A1 mutations with analysis of genotype-phenotype correlation in patients with a lethal type II collagen disorder. J. Med. Genet 2000, 37, 263–271. [Google Scholar] [Green Version]
- Unger, S.L.; Briggs, M.D.; Holden, P.; Zabel, B.; Ala-Kokko, L.; Paassilta, P.; Lohiniva, J.; Rimoin, D.L.; Lachman, R.S.; Cohn, D.H. Multiple epiphyseal dysplasia: Radiographic abnormalities correlated with genotype. Pediatr. Radiol 2001, 31, 10–18. [Google Scholar]
- Bomsta, B.D.; Bridgewater, L.C.; Seegmiller, R.E. Premature osteoarthritis in the Disproportionate micromelia (Dmm) mouse. Osteoarthritis Cartilage 2006, 14, 477–485. [Google Scholar]
- Pritzker, K.P.; Gay, S.; Jimenez, S.A.; Ostergaard, K.; Pelletier, J.P.; Revell, P.A.; Salter, D.; van den Berg, W.B. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthritis Cartilage 2006, 14, 13–29. [Google Scholar]
- Xu, L.; Servais, J.; Polur, I.; Kim, D.; Lee, P.L.; Chung, K.; Li, Y. Attenuation of osteoarthritis progression by reduction of the discoidin domain receptor 2 in mice. Arthritis Rheum 2010, 62, 2736–2744. [Google Scholar]
- Eyre, D.R.; Pietka, T.; Weis, M.A.; Wu, J.J. Covalent cross-linking of the NC1 domain of collagen type IX to collagen type II in cartilage. J. Biol. Chem 2004, 279, 2568–2574. [Google Scholar]
- Fernandes, R.J.; Seegmiller, R.E.; Nelson, W.R.; Eyre, D.R. Protein consequences of the Col2a1 C-propeptide mutation in the chondrodysplastic Dmm mouse. Matrix Biol 2003, 22, 449–453. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Macdonald, D.W.; Squires, R.S.; Avery, S.A.; Adams, J.; Baker, M.; Cunningham, C.R.; Heimann, N.B.; Kooyman, D.L.; Seegmiller, R.E. Structural Variations in Articular Cartilage Matrix Are Associated with Early-Onset Osteoarthritis in the Spondyloepiphyseal Dysplasia Congenita (Sedc) Mouse. Int. J. Mol. Sci. 2013, 14, 16515-16531. https://doi.org/10.3390/ijms140816515
Macdonald DW, Squires RS, Avery SA, Adams J, Baker M, Cunningham CR, Heimann NB, Kooyman DL, Seegmiller RE. Structural Variations in Articular Cartilage Matrix Are Associated with Early-Onset Osteoarthritis in the Spondyloepiphyseal Dysplasia Congenita (Sedc) Mouse. International Journal of Molecular Sciences. 2013; 14(8):16515-16531. https://doi.org/10.3390/ijms140816515
Chicago/Turabian StyleMacdonald, David W., Ryan S. Squires, Shaela A. Avery, Jason Adams, Melissa Baker, Christopher R. Cunningham, Nicholas B. Heimann, David L. Kooyman, and Robert E. Seegmiller. 2013. "Structural Variations in Articular Cartilage Matrix Are Associated with Early-Onset Osteoarthritis in the Spondyloepiphyseal Dysplasia Congenita (Sedc) Mouse" International Journal of Molecular Sciences 14, no. 8: 16515-16531. https://doi.org/10.3390/ijms140816515