Epigenetics Meets Radiation Biology as a New Approach in Cancer Treatment

Abstract
:1. Introduction
2. Epigenetic Regulation in Cancer
3. Radiation Biology in Cancer
4. Epigenetic Regulation and Radiation
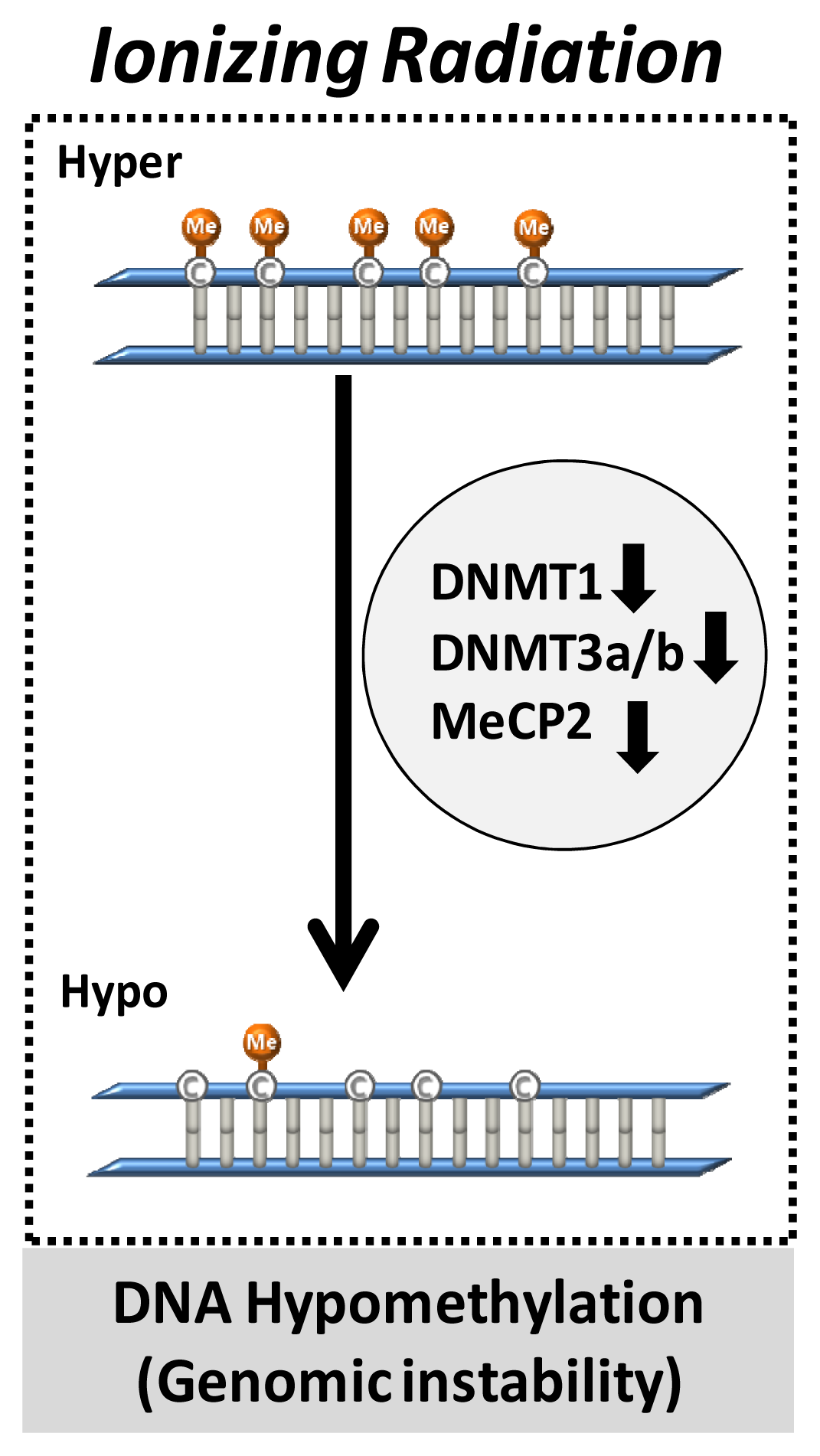
4.1. DNA Methylation and Radiation
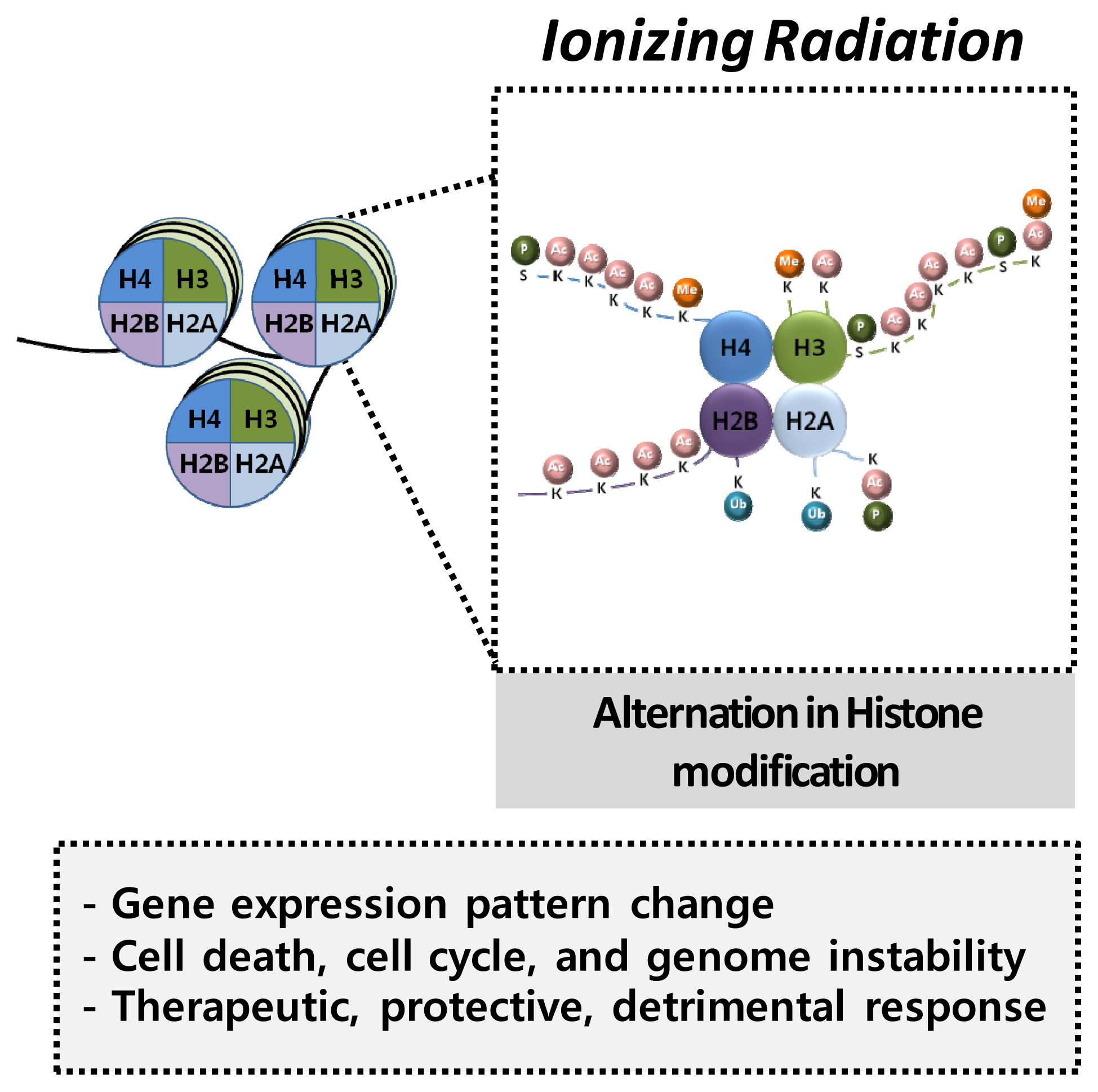
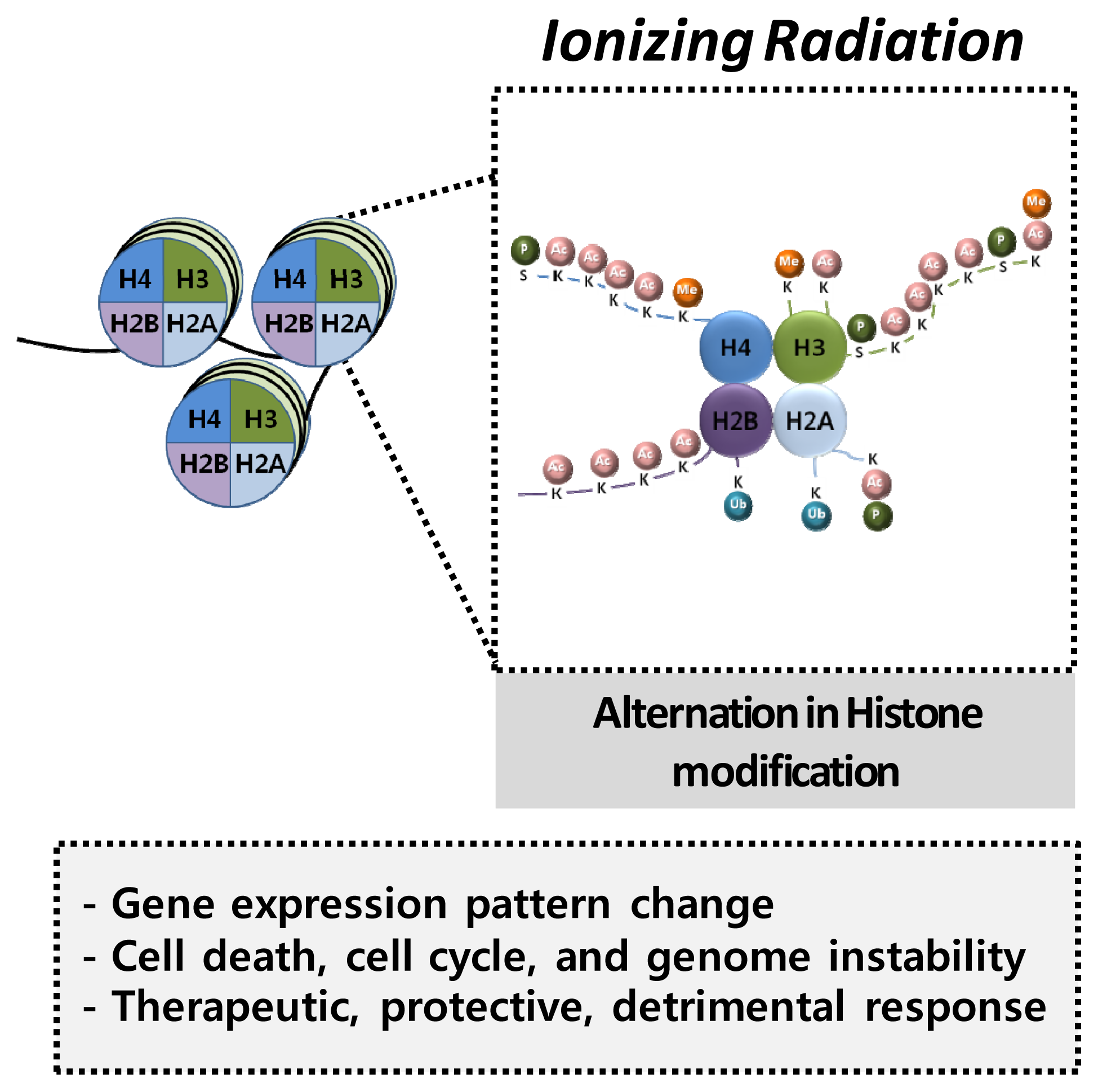
4.2. Histone Modification and Radiation
4.3. Small RNAs and Radiation
5. Colon Cancer: Radiation and Epigenetics
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Jaenisch, R.; Young, R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008, 132, 567–582. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar]
- Little, J.B. Induction of genetic instability by ionizing radiation. C. R. Acad. Sci. III 1999, 322, 127–134. [Google Scholar]
- Little, J.B. Radiation carcinogenesis. Carcinogenesis 2000, 21, 397–404. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet 2002, 3, 415–428. [Google Scholar]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar]
- Cheung, P.; Lau, P. Epigenetic regulation by histone methylation and histone variants. Mol. Endocrinol 2005, 19, 563–573. [Google Scholar]
- Kim, J.G.; Yi, J.M.; Park, J.S.; Son, T.G.; Yang, K.; Yoo, M.A.; Heo, K. Histone demethylase JMJD2B-mediated cell proliferation regulated by hypoxia and radiation in gastric cancer cell. Biochim. Biophys. Acta 2012, 1819, 1200–1207. [Google Scholar]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med 2003, 349, 2042–2054. [Google Scholar]
- Ng, H.H.; Bird, A. DNA methylation and chromatin modification. Curr. Opin. Genet. Dev 1999, 9, 158–163. [Google Scholar]
- Antequera, F.; Bird, A. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr. Biol 1999, 9, R661–R667. [Google Scholar]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar]
- Laird, P.W. Oncogenic mechanisms mediated by DNA methylation. Mol. Med. Today 1997, 3, 223–229. [Google Scholar]
- Wajed, S.A.; Laird, P.W.; DeMeester, T.R. DNA methylation: An alternative pathway to cancer. Ann. Surg 2001, 234, 10–20. [Google Scholar]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med 2008, 358, 1148–1159. [Google Scholar]
- Jones, P.A.; Laird, P.W. Cancer epigenetics comes of age. Nat. Genet 1999, 21, 163–167. [Google Scholar]
- Lee, B.B.; Lee, E.J.; Jung, E.H.; Chun, H.K.; Chang, D.K.; Song, S.Y.; Park, J.; Kim, D.H. Aberrant methylation of APC, MGMT, RASSF2A, and Wif-1 genes in plasma as a biomarker for early detection of colorectal cancer. Clin. Cancer Res 2009, 15, 6185–6191. [Google Scholar]
- Borinstein, S.C.; Conerly, M.; Dzieciatkowski, S.; Biswas, S.; Washington, M.K.; Trobridge, P.; Henikoff, S.; Grady, W.M. Aberrant DNA methylation occurs in colon neoplasms arising in the azoxymethane colon cancer model. Mol. Carcinoq 2010, 49, 94–103. [Google Scholar]
- Nishio, M.; Sakakura, C.; Nagata, T.; Komiyama, S.; Miyashita, A.; Hamada, T.; Kuryu, Y.; Ikoma, H.; Kubota, T.; Kimura, A.; et al. RUNX3 promoter methylation in colorectal cancer: Its relationship with microsatellite instability and its suitability as a novel serum tumor marker. Anticancer. Res 2010, 30, 2673–2682. [Google Scholar]
- Wang, Z.; Yuan, X.; Jiao, N.; Zhu, H.; Zhang, Y.; Tong, J. CDH13 and FLBN3 gene methylation are associated with poor prognosis in colorectal cancer. Pathol. Oncol. Res 2012, 18, 263–270. [Google Scholar]
- McGivern, A.; Wynter, C.V.; Whitehall, V.L.; Kambara, T.; Spring, K.J.; Walsh, M.D.; Barker, M.A.; Arnold, S.; Simms, L.A.; Leggett, B.A.; et al. Promoter hypermethylation frequency and BRAF mutations distinguish hereditary non-polyposis colon cancer from sporadic MSI-H colon cancer. Fam. Cancer 2004, 3, 101–107. [Google Scholar]
- Bernet, A.; Mazelin, L.; Coissieux, M.M.; Gadot, N.; Ackerman, S.L.; Scoazec, J.Y.; Mehlen, P. Inactivation of the UNC5C Netrin-1 receptor is associated with tumor progression in colorectal malignancies. Gastroenterology 2007, 133, 1840–1848. [Google Scholar]
- Asting, A.G.; Carén, H.; Andersson, M.; Lönnroth, C.; Lagerstedt, K.; Lundholm, K. COX-2 gene expression in colon cancer tissue related to regulating factors and promoter methylation status. BMC. Cancer 2011, 11. [Google Scholar] [CrossRef]
- Graziano, F.; Humar, B.; Guilford, P. The role of the E-cadherin gene (CDH1) in diffuse gastric cancer susceptibility: From the laboratory to clinical practice. Ann. Oncol 2003, 14, 1705–1713. [Google Scholar]
- Moinova, H.R.; Chen, W.D.; Shen, L.; Smiraglia, D.; Olechnowicz, J.; Ravi, L.; Kasturi, L.; Myeroff, L.; Plass, C.; Parsons, R.; et al. HLTF gene silencing in human colon cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 4562–4567. [Google Scholar]
- Isaksson-Mettävainio, M.; Palmqvist, R.; Dahlin, A.M.; Van Guelpen, B.; Rutegard, J.; Oberg, A.; Henriksson, M.L. High SMAD4 levels appear in microsatellite instability and hypermethylated colon cancers, and indicate a better prognosis. Int. J. Cancer 2012, 131, 779–788. [Google Scholar]
- Grützmann, R.; Molnar, B.; Pilarsky, C.; Habermann, J.K.; Schlag, P.M.; Saeger, H.D.; Miehlke, S.; Stolz, T.; Model, F.; Roblick, U.J.; et al. Sensitive detection of colorectal cancer in peripheral blood by septin 9 DNA methylation assay. PLoS One 2008, 3, e3759. [Google Scholar]
- deVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grützmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating methylated SEPT9 DNA in plasma is a biomarker for colorectal cancer. Clin. Chem 2009, 55, 1337–1346. [Google Scholar]
- Hall, E.J.; Worgul, B.V.; Smilenov, L.; Elliston, C.D.; Brenner, D.J. The relative biological effectiveness of densely ionizing heavy-ion radiation for inducing ocular cataracts in wild type versus mice heterozygous for the ATM gene. N. Engl. J. Med 2006, 45, 99–104. [Google Scholar]
- De Potter, P.; Levecq, L.; Godfraind, C.; Renard, L. Primary orbital melanoma treated with iodine-125 plaque radiotherapy. Am. J. Ophthalmol 2006, 142, 864–866. [Google Scholar]
- Erven, K.; van Limbergen, E. Regional lymph node irradiation in breast cancer. Future Oncol 2007, 3, 343–352. [Google Scholar]
- Bismar, M.M.; Sinicrope, F.A. Radiation enteritis. Curr. Gastroenterol. Rep 2002, 4, 361–365. [Google Scholar]
- Barcellos-Hoff, M.H.; Park, C.; Wright, E.G. Radiation and the microenvironment-tumorigenesis and therapy. Nat. Rev. Cancer 2005, 5, 867–875. [Google Scholar]
- Park, H.J.; Griffin, R.J.; Hui, S.; Levitt, S.H.; Song, C.W. Radiation-induced vascular damage in tumors: Implications of vascular damage in ablative hypofractionated radiotherapy (SBRT and SRS). Radiat. Res 2012, 177, 311–327. [Google Scholar]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar]
- Brown, M.; Bristow, R.; Glazer, P.; Hill, R.; McBride, W.; McKenna, G.; Muschel, R. Comment on “tumor response to radiotherapy regulated by endothelial cell apoptosis” (II). Science 2003, 302. [Google Scholar] [CrossRef]
- Dungey, F.A.; Caldecott, K.W.; Chalmers, A.J. Enhanced radiosensitization of human glioma cells by combining inhibition of poly (ADP-ribose) polymerase with inhibition of heat shock protein 90. Mol. Cancer Ther 2009, 8, 2243–2254. [Google Scholar]
- Rosen, J.B.; Jordan, C.T. The increasing complexity of the cancer stem cell paradigm. Science 2009, 324, 1670–1673. [Google Scholar]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar]
- Zhang, M.; Atkinson, R.L.; Rosen, J.M. Selective targeting of radiation-resistant tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3522–3527. [Google Scholar]
- Jiang, L.; Li, J.; Song, L. Bmi-1, stem cells and cancer. Acta Biochim. Biophys. Sin. (Shanghai) 2009, 41, 527–534. [Google Scholar]
- You, H.; Ding, W.; Rountree, C.B. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology 2010, 51, 1635–1644. [Google Scholar]
- Subramaniam, D.; Ramalingam, S.; Houchen, C.W.; Anant, S. Cancer stem cells: A novel paradigm for cancer prevention and treatment. Mini Rev. Med. Chem 2010, 10, 359–371. [Google Scholar]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar]
- Han, K.; Lee, J.; Km, H.S.; Yang, K.M.; Yi, J.M. DNA methylation of mobile genetic elements in human cancers. Genes Genom 2013, 35, 265–271. [Google Scholar]
- Pogribny, I.; Raiche, J.; Slovack, M.; Kovalchuk, O. Dose-dependence, sex- and tissue-specificity, and persistence of radiation-induced genomic DNA methylation changes. Biochem. Biophys. Res. Commun 2004, 320, 1253–1261. [Google Scholar]
- Loree, J.; Koturbash, I.; Kutanzi, K.; Baker, M.; Pogribny, I.; Kovalchuk, O. Radiation-induced molecular changes in rat mammary tissue: Possible implications for radiation-induced carcinogenesis. Int. J. Radiat. Biol 2006, 82, 805–815. [Google Scholar]
- Raiche, J.; Rodriguez-Juarez, R.; Pogribny, I.; Kovalchuk, O. Sex- and tissue-specific expression of maintenance and de novo DNA methyltransferases upon low dose X-irradiation in mice. Biochem. Biophys. Res. Commun 2004, 325, 39–47. [Google Scholar]
- Hofstetter, B.; Niemierko, A.; Forrer, C.; Benhattar, J.; Albertini, V.; Pruschy, M.; Bosman, F.T.; Catapano, C.V.; Ciernik, I.F. Impact of genomic methylation on radiation sensitivity of colorectal carcinoma. Int. J. Radiat. Oncol. Biol. Phys 2010, 76, 1512–1519. [Google Scholar]
- Cho, H.J.; Kim, S.Y.; Kim, K.H.; Kang, W.K.; Kim, J.I.; Oh, S.T.; Kim, J.S.; An, C.H. The combination effect of sodium butyrate and 5-aza-2′-deoxycytidine on radiosensitivity in RKO colorectal cancer and MCF-7 breast cancer cell lines. World J. Surg. Oncol. 2009, 7. [Google Scholar] [CrossRef]
- Kuhmann, C.; Weichenhan, D.; Rehli, M.; Plass, C.; Schmezer, P.; Popanda, O. DNA methylation changes in cells regrowing after fractioned ionizing radiation. Radiother. Oncol 2011, 101, 116–121. [Google Scholar]
- Chaudhry, M.A.; Omaruddin, R.A. Differential DNA methylation alterations in radiation-sensitive and -resistant cells. DNA Cell. Biol 2012, 31, 908–916. [Google Scholar]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet 2005, 37, 391–400. [Google Scholar]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar]
- Nguyen, C.T.; Weisenberger, D.J.; Velicescu, M.; Gonzales, F.A.; Lin, J.C.; Liang, G.; Jones, P.A. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res 2002, 62, 6456–6461. [Google Scholar]
- McGarvey, K.M.; Fahrner, J.A.; Greene, E.; Martens, J.; Jenuwein, T.; Baylin, S.B. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res 2006, 66, 3541–3549. [Google Scholar]
- Song, J.S.; Kim, Y.S.; Kim, D.K.; Park, S.I.; Jang, S.J. Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathol. Int 2012, 62, 182–190. [Google Scholar]
- Barlési, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non small-cell lung cancer. J. Clin. Oncol 2007, 25, 4358–4364. [Google Scholar]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol 2009, 174, 1619–1628. [Google Scholar]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Rogenhofer, S.; Heukamp, L.C.; Gütgemann, I.; Walter, B.; Hofstädter, F.; Büttner, R.; et al. Global levels of histone modifications predict prostate cancer recurrence. Prostate 2010, 70, 61–69. [Google Scholar]
- Behbahani, T.E.; Kahl, P.; von der Gathen, J.; Heukamp, L.C.; Baumann, C.; Gütgemann, I.; Walter, B.; Hofstädter, F.; Bastian, P.J.; von Ruecker, A.; et al. Alterations of global histone H4K20 methylation during prostate carcinogenesis. BMC Urol 2012, 12, 5–10. [Google Scholar]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol. Biomarkers Prev 2010, 19, 2611–2622. [Google Scholar]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 2009, 69, 3802–3809. [Google Scholar]
- Leszinski, G.; Gezer, U.; Siegele, B.; Stoetzer, O.; Holdenrieder, S. Relevance of histone marks H3K9me3 and H4K20me3 in cancer. Anticancer Res 2012, 32, 2199–2205. [Google Scholar]
- Müller-Tidow, C.; Klein, H.U.; Hascher, A.; Isken, F.; Tickenbrock, L.; Thoennissen, N.; Agrawal-Singh, S.; Tschanter, P.; Disselhoff, C.; Wang, Y.; et al. Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood 2010, 116, 3564–3571. [Google Scholar]
- Park, Y.S.; Jin, M.Y.; Kim, Y.J.; Yook, J.H.; Kim, B.S.; Jang, S.J. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann. Surg. Oncol 2008, 15, 1968–1976. [Google Scholar]
- Zhang, L.; Zhong, K.; Dai, Y.; Zhou, H. Genome-wide analysis of histone H3 lysine 27 trimethylation by ChIP-chip in gastric cancer patients. J. Gastroenterol 2009, 44, 305–312. [Google Scholar]
- Tzao, C.; Tung, H.J.; Jin, J.S.; Sun, G.H.; Hsu, H.S.; Chen, B.H.; Yu, C.P.; Lee, S.C. Prognostic significance of global histone modifications in resected squamous cell carcinoma of the esophagus. Mod. Pathol 2009, 22, 252–260. [Google Scholar]
- Cohen, I.; Poręba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes. Cancer 2011, 2, 631–647. [Google Scholar]
- Rogenhofer, S.; Kahl, P.; Mertens, C.; Hauser, S.; Hartmann, W.; Büttner, R.; Müller, S.C.; von Ruecker, A.; Ellinger, J. Global histone H3 lysine 27 (H3K27) methylation levels and their prognostic relevance in renal cell carcinoma. BJU Int 2012, 109, 459–465. [Google Scholar]
- He, C.; Xu, J.; Zhang, J.; Xie, D.; Ye, H.; Xiao, Z.; Cai, M.; Xu, K.; Zeng, Y.; Li, H.; et al. High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum. Pathol 2012, 43, 1425–1435. [Google Scholar]
- Cai, M.Y.; Hou, J.H.; Rao, H.L.; Luo, R.Z.; Li, M.; Pei, X.Q.; Lin, M.C.; Guan, X.Y.; Kung, H.F.; Zeng, Y.X.; et al. High expression of H3K27me3 in human hepatocellular carcinomas correlates closely with vascular invasion and predicts worse prognosis in patients. Mol. Med 2011, 17, 12–20. [Google Scholar]
- Manuyakorn, A.; Paulus, R.; Farrell, J.; Dawson, N.A.; Tze, S.; Cheung-Lau, G.; Hines, O.J.; Reber, H.; Seligson, D.B.; Horvath, S.; et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: Results from RTOG 9704. J. Clin. Oncol 2010, 28, 1358–1365. [Google Scholar]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar]
- Pilch, D.R.; Sedelnikova, O.A.; Redon, C.; Celeste, A.; Nussenzweig, A.; Bonner, W.M. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem. Cell Biol 2003, 81, 123–129. [Google Scholar]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem 1998, 273, 5858–5868. [Google Scholar]
- Celeste, A.; Fernandez-Capetillo, O.; Kruhlak, M.J.; Pilch, D.R.; Staudt, D.W.; Lee, A.; Bonner, R.F.; Bonner, W.M.; Nussenzweig, A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol 2003, 5, 675–679. [Google Scholar]
- Pogribny, I.; Koturbash, I.; Tryndyak, V.; Hudson, D.; Stevenson, S.M.; Sedelnikova, O.; Bonner, W.; Kovalchuk, O. Fractionated low-dose radiation exposure leads to accumulation of DNA damage and profound alterations in DNA and histone methylation in the murine thymus. Mol. Cancer Res 2005, 3, 553–561. [Google Scholar]
- Falk, M.; Lukasova, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar]
- Bernstein, E.; Allis, C.D. RNA meets chromatin. Genes Dev 2005, 19, 1635–1655. [Google Scholar]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar]
- Nakamura, T.; Canaani, E.; Croce, C.M. Oncogenic All1 fusion proteins target Drosha-mediated microRNA processing. Proc. Natl. Acad. Sci. USA 2007, 104, 10980–10985. [Google Scholar]
- Liu, L.; Chen, L.; Xu, Y.; Li, R.; Du, X. microRNA-195 promotes apoptosis and suppresses tumorigenicity of human colorectal cancer cells. Biochem. Biophys. Res. Commun 2010, 400, 236–240. [Google Scholar]
- Borralho, P.M.; Kren, B.T.; Castro, R.E.; da Silva, I.B.; Steer, C.J.; Rodrigues, C.M. Micro RNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J 2009, 276, 6689–6700. [Google Scholar]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar]
- Hu, G.; Chen, D.; Li, X.; Yang, K.; Wang, H.; Wu, W. miR-133b regulates the MET proto-oncogene and inhibits the growth of colorectal cancer cells in vitro and in vivo. Cancer Biol. Ther 2010, 10, 190–197. [Google Scholar]
- Ilnytskyy, Y.; Zemp, F.J.; Koturbash, I.; Kovalchuk, O. Altered microRNA expression patterns in irradiated hematopoietic tissues suggest a sexspecific protective mechanism. Biochem. Biophys. Res. Commun 2008, 377, 41–45. [Google Scholar]
- Filkowski, J.N.; Ilnytskyy, Y.; Tamminga, J.; Koturbash, I.; Golubov, A.; Bagnyukova, T.; Pogribny, I.P.; Kovalchuk, O. Hypomethylation and genome instability in the germ line of exposed parents and their progeny is associated with altered miRNA expression. Carcinogenesis 2010, 31, 1110–1115. [Google Scholar]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res 2009, 15, 3938–3946. [Google Scholar]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol 2002, 20, 2429–2440. [Google Scholar]
- Kantarjian, H.; Issa, J.P.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 2006, 106, 1794–1803. [Google Scholar]
- Koukourakis, G.V. Role of radiation therapy in neoadjuvant era in patients with locally advanced rectal cancer. World J. Gastrointest. Oncol 2012, 4, 230–237. [Google Scholar]
- NIH consensus conference. Adjuvant therapy for patients with colon and rectal cancer. JAMA 1990, 264, 1444–1450.


{kind=link}
{kind=link}
| Gene | Biological function | References |
|---|---|---|
| RASSF1A, CDKN2A, CHFR, DLEC1, MYOD, RGC-32 | cell cycle arrest | Lee et al., 2009 [17]; Borinstein et al., 2010 [18] |
| MGMT, hMLH1, hMLH2 | DNA repair | Lee et al., 2009 [17] |
| APC, SFRP1 | Wnt pathway | Lee et al., 2009 [17] |
| RUNX3, TIMP3, DCC | apoptosis | Nishio et al., 2010 [19] |
| CDH13, ADAM23 | cell to cell interaction | Wang et al., 2011 [20] |
| MINT | Notch pathway | McGivern et al., 2004 [21] |
| UNC5C, DCC, EVL, VIM, FLNC | cytoskeleton remodeling and cell polarity | Bernet et al., 2007 [22] |
| COX2 | inflammation | Asting et al., 2011 [23] |
| CDH1 | invasion and metastasis | Graziano et al., 2004 [24] |
| HLTF | chromatin remodeling factor | Moinova et al., 2002 [25] |
| RAR-b, SMAD4, TWIST1 | growth and differentiation | Isaksson et al., 2012 [26] |
| Wif-1 | mesoderm segmentation | Lee et al., 2009 [17] |
| SOCS1, SEPT9 | cytokine signaling | Grützmann et al., 2008 [27]; deVos et al., 2009 [28] |
| Cancer type | Histone modification changes | References |
|---|---|---|
| Lung | H4K16ac, H3K18ac, H4K8ac, H4K5ac, H3K9ac, H4K12ac, H4K16ac | Song et al., 2012 [57]; Barlési et al., 2007 [58]; Seligson et al., 2009 [59] |
| H3K4me2, H3K9me3, H4K20me3 | ||
| Prostate | H3Ac, H4Ac, H3K18ac | Ellinger et al., 2010 [60]; Seligson et al., 2009 [59]; Behbahani et al., 2012 [61]; Bianoco-Miotto et al., 2010 [62] |
| H3K4me1, H3K9me2, H3K9me3, H3K4me2, H3K27me3, H4K20me1 | ||
| Breast | H3K18ac, H4K12ac,, H4K16ac | Elsheikh et al., 2009 [63]; Leszinski et al., 2012 [64] |
| H3K4me2, H3K9me3, H4K20me2, H4K20me3, H4R3me2 | ||
| Leukemia | H3K9me3 | Muller-Tidow et al., 2010 [65] |
| Stomach | H3K9me3, H3K27me3 | Park et al., 2008 [66]; Zhang et al., 2009 [67] |
| Esophagus | H3K18ac | Tzao et al., 2009 [68]; Cohen et al., 2011 [69] |
| H4R3me2,H3K27me3, H4R3me2 | ||
| Kidney | H3K4me1, H3K4me2, H3K4me3, H3K9me1, H3K27me1, H3K27me2, H3K27me3 | Ellinger et al., 2010 [60]; Rogenhofer et al., 2012 [70] |
| Liver | H3K4me3, H3K27me3 | He et al., 2012 [71]; Cai et al., 2011 [72] |
| Pancreas | H3K4me2, H3K9me2, H3K18ac | Manuyakorn et al., 2010 [73] |
| colon | H3K4me2, H3K9ac, H3K9me2, H3K27me3 | Seligson et al., 2009 [59] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, J.-G.; Park, M.-T.; Heo, K.; Yang, K.-M.; Yi, J.M. Epigenetics Meets Radiation Biology as a New Approach in Cancer Treatment. Int. J. Mol. Sci. 2013, 14, 15059-15073. https://doi.org/10.3390/ijms140715059
Kim J-G, Park M-T, Heo K, Yang K-M, Yi JM. Epigenetics Meets Radiation Biology as a New Approach in Cancer Treatment. International Journal of Molecular Sciences. 2013; 14(7):15059-15073. https://doi.org/10.3390/ijms140715059
Chicago/Turabian StyleKim, Joong-Gook, Moon-Taek Park, Kyu Heo, Kwang-Mo Yang, and Joo Mi Yi. 2013. "Epigenetics Meets Radiation Biology as a New Approach in Cancer Treatment" International Journal of Molecular Sciences 14, no. 7: 15059-15073. https://doi.org/10.3390/ijms140715059