Novel Hybrid Virtual Screening Protocol Based on Molecular Docking and Structure-Based Pharmacophore for Discovery of Methionyl-tRNA Synthetase Inhibitors as Antibacterial Agents

Abstract
:1. Introduction
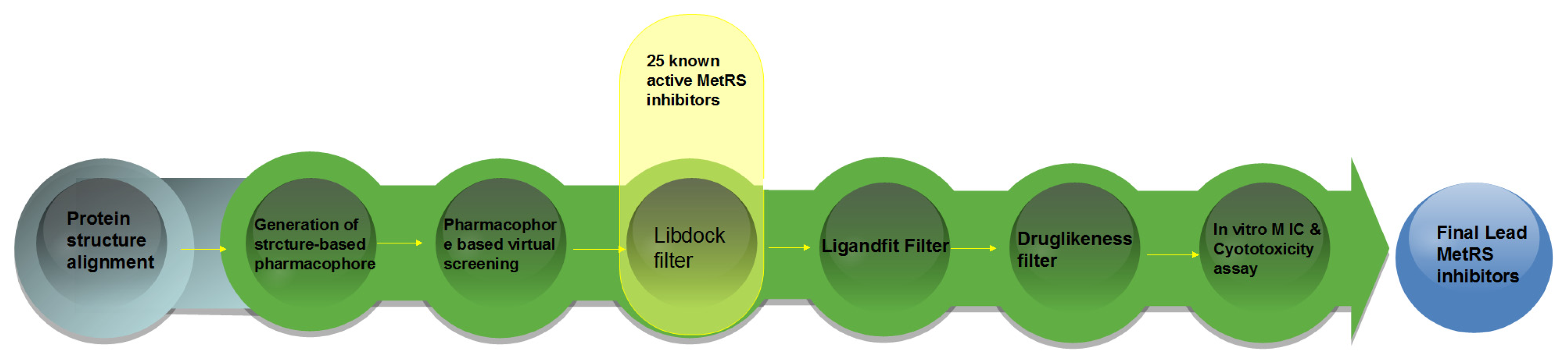
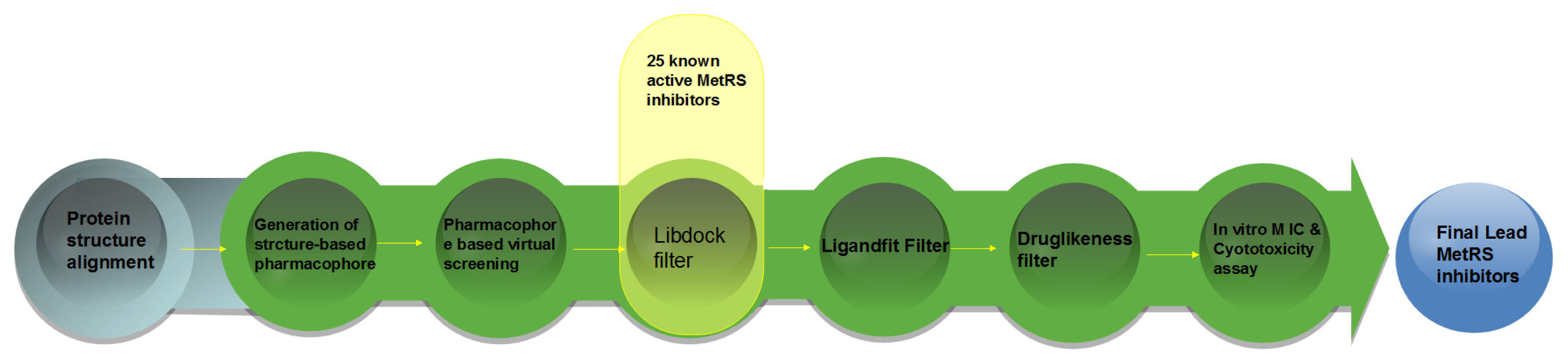
2. Result and Discussion
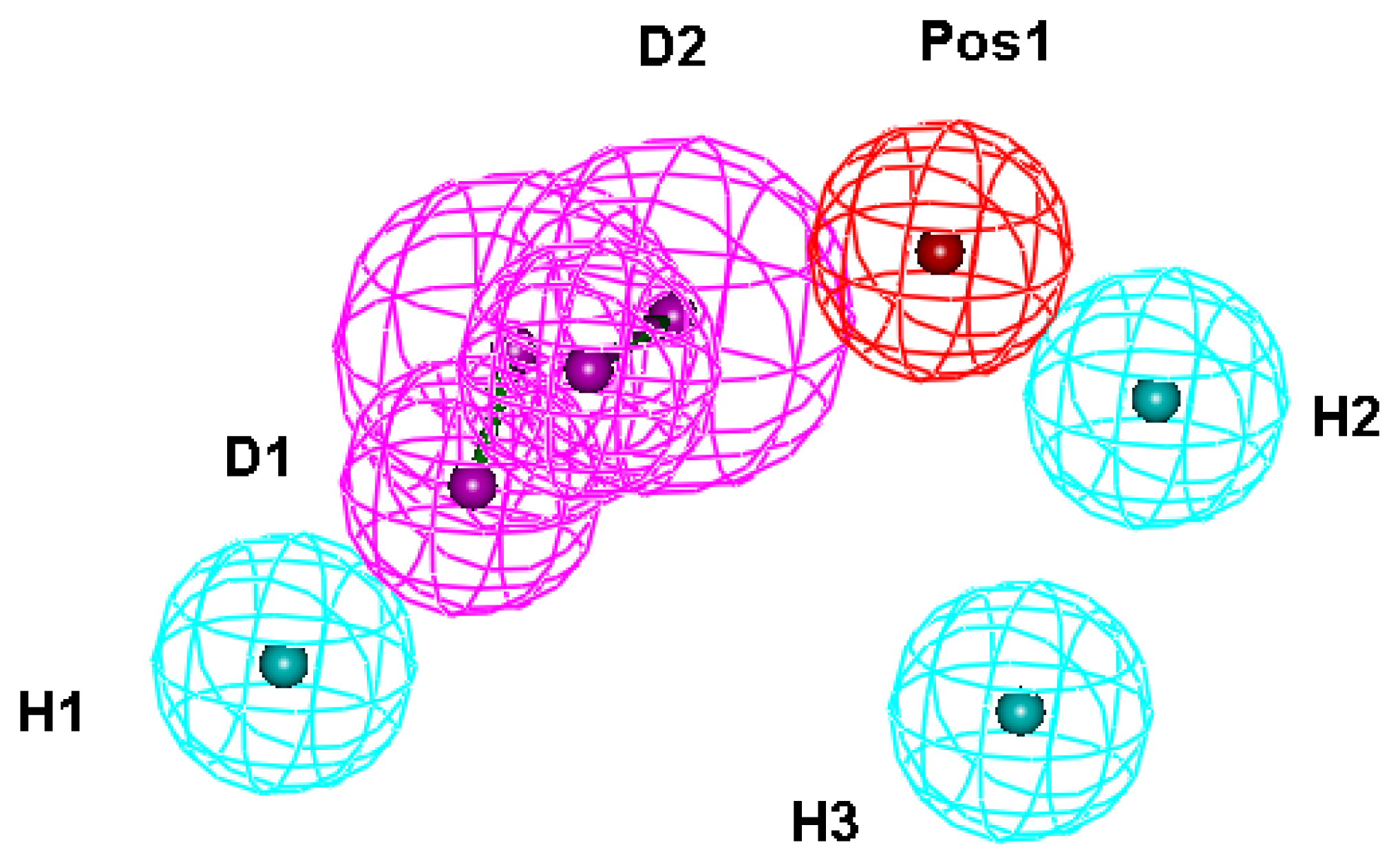
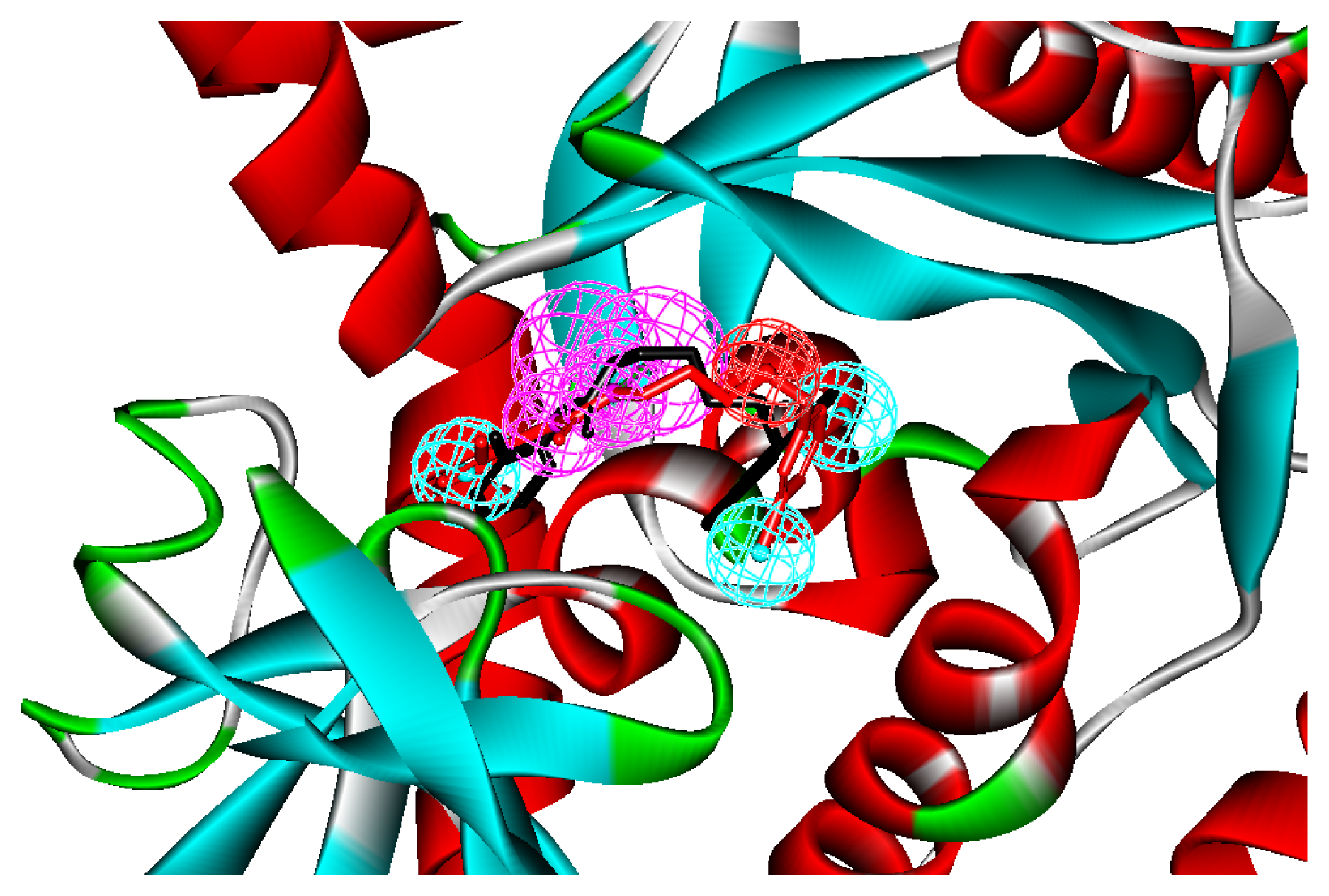
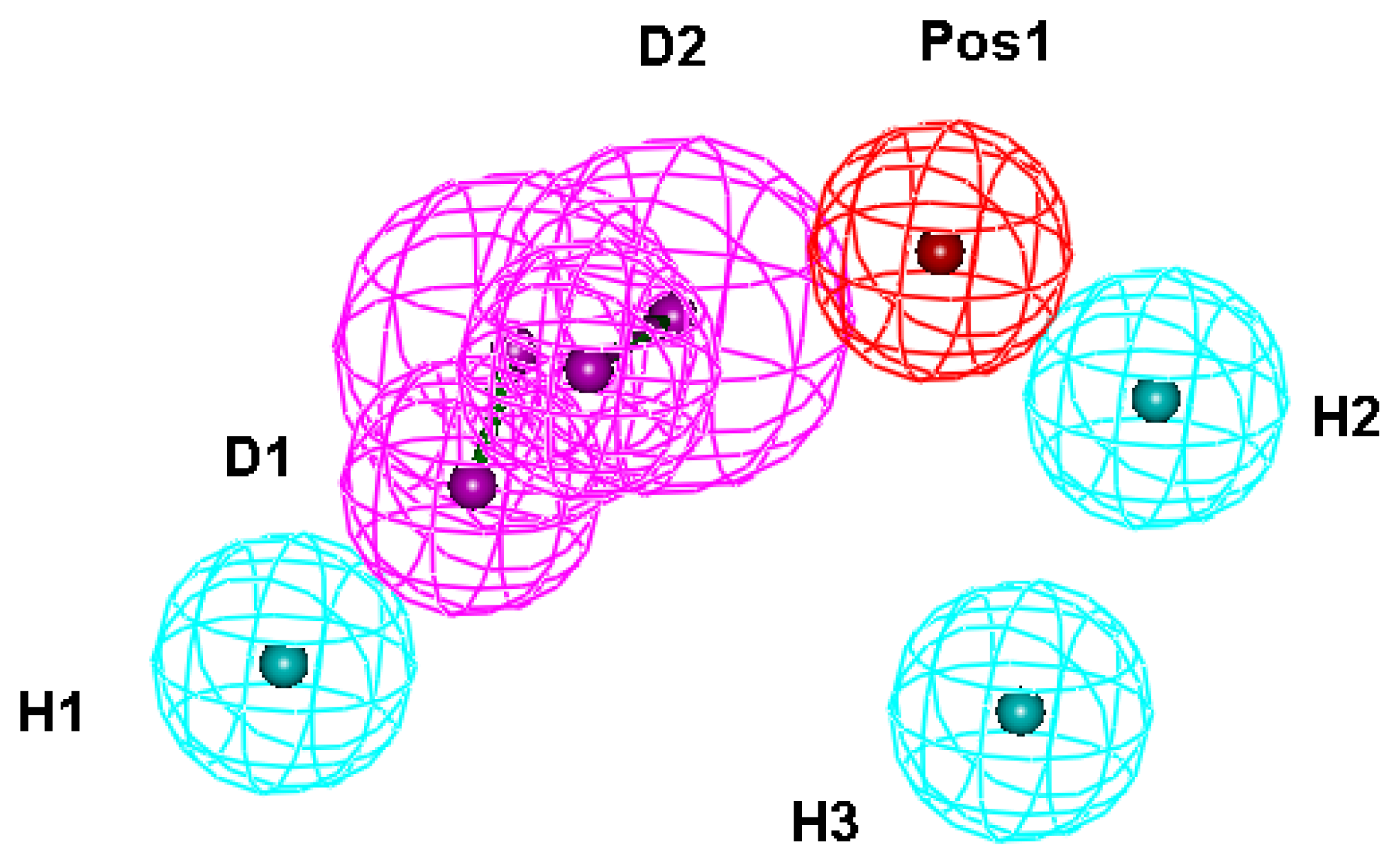
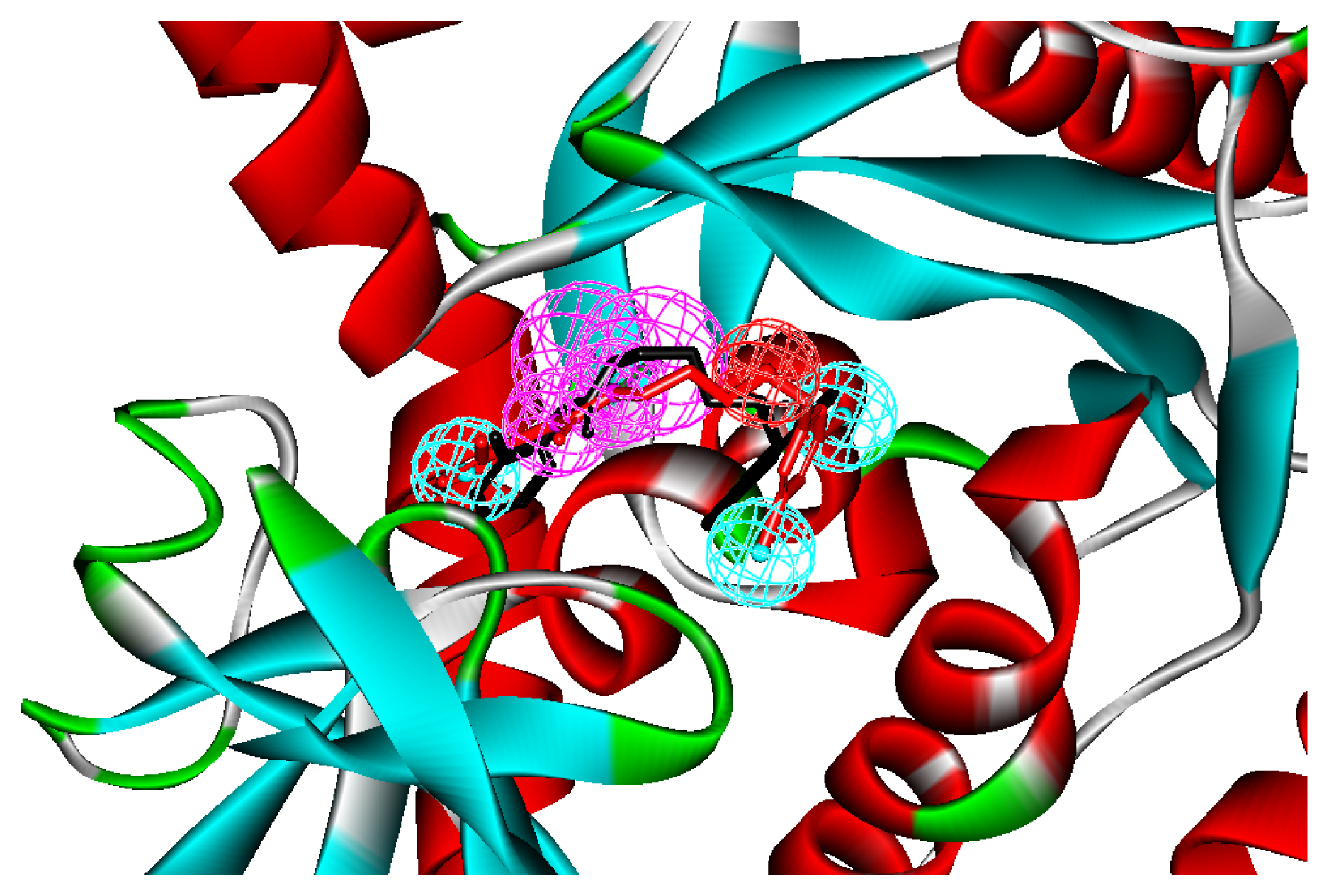
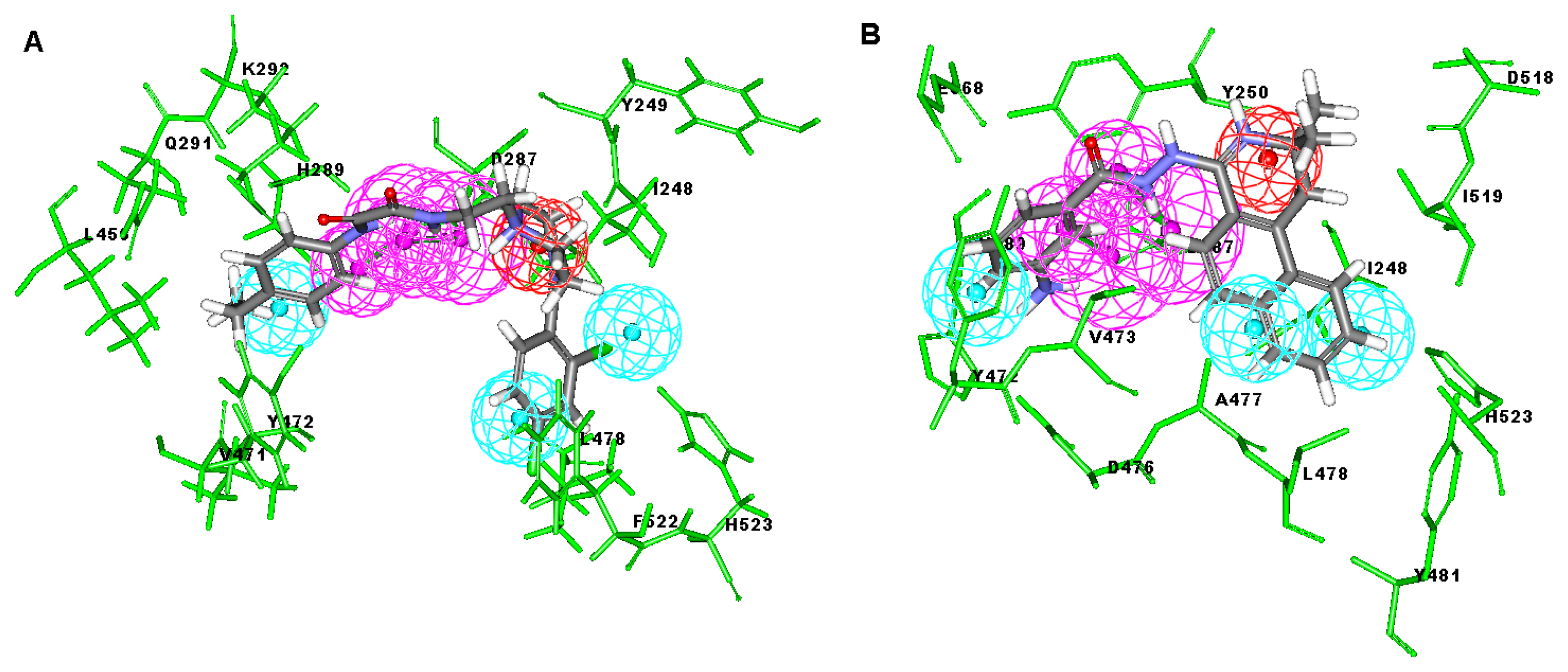
2.1. Generation and Validation of Structure-Based Pharmacophore
2.2. 3D-Model Structure of SaMetRS and hMetRS Were Built by Homology Modeling
2.3. Molecular Docking
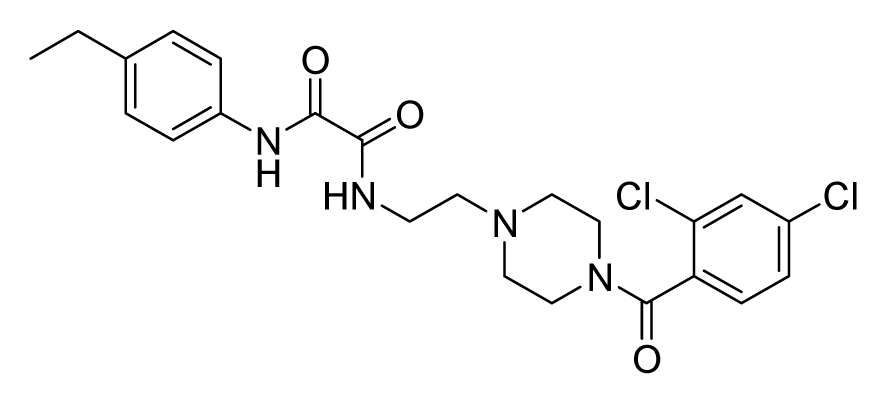
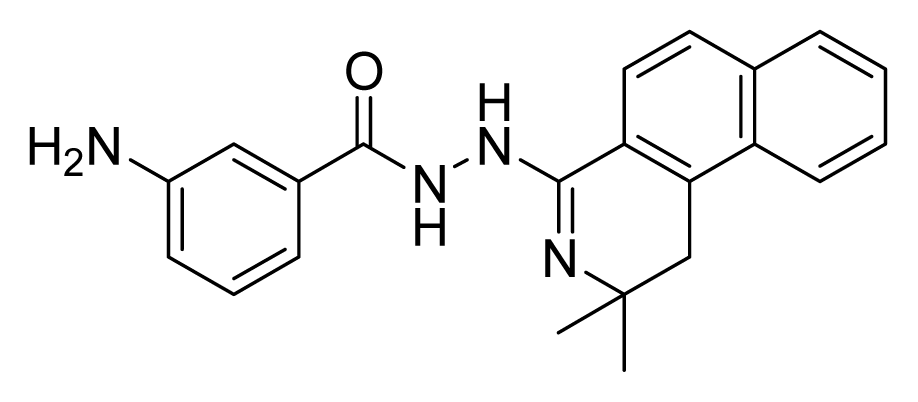
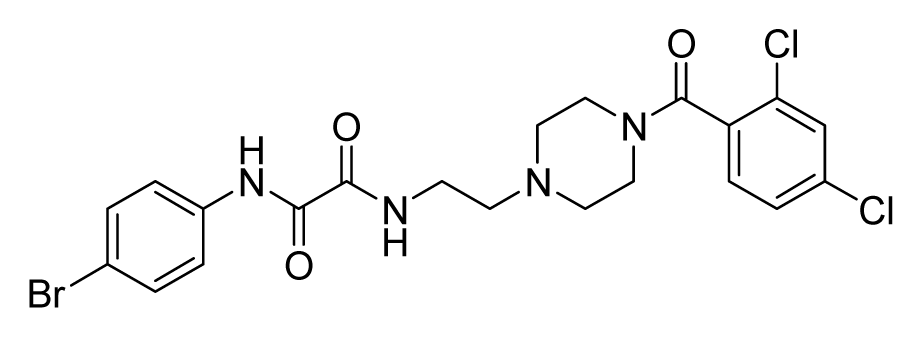
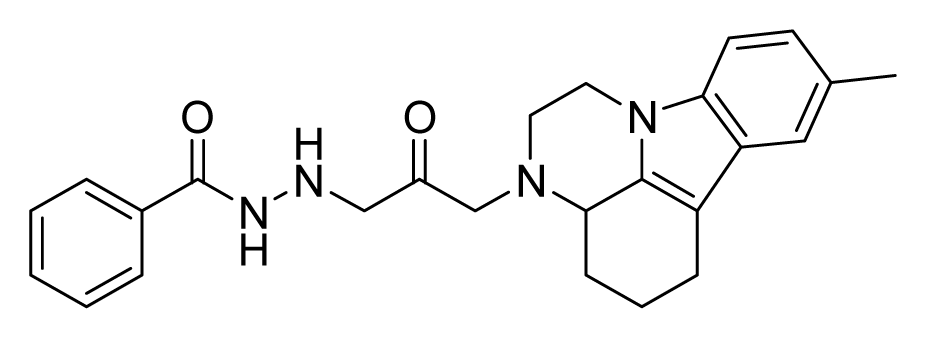
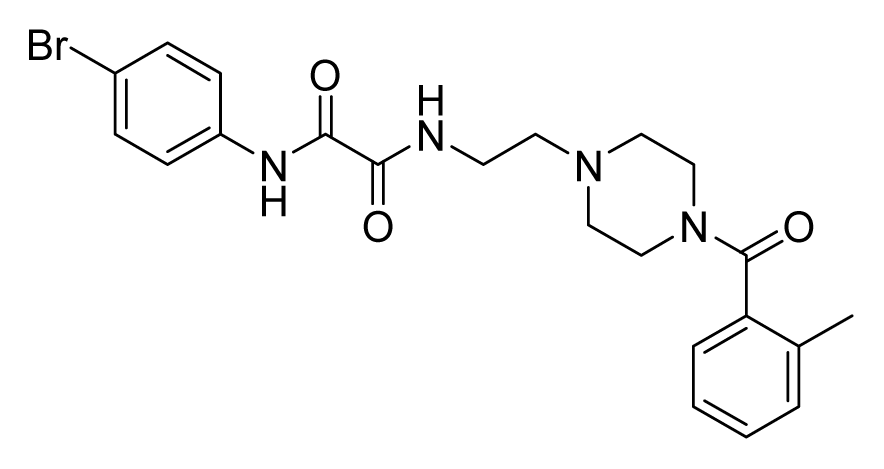
2.4. Database Screening
2.5. In Vitro Minimum Inhibitory Concentration and Cytotoxicity Assay
3. Computational and Experimental Methods
3.1. Generation of Structure-Based Pharmacophore Models
3.2. Pharmacophore-Based Virtual Screening
3.3. Homology Modeling
3.4. Molecular Docking Study
3.5. In Vitro Minimum Inhibitory Concentration Assay
3.6. In Vitro Cytotoxicity Assay
4. Conclusions
Supplementary Information
ijms-14-14225-s001.pdfAcknowledgments
Conflict of Interest
References
- Li, T.; Froeyen, M.; Herdewijn, P. Computational alanine scanning and free energy decomposition for E-coli type I signal peptidase with lipopeptide inhibitor complex. J. Mol. Graph 2008, 26, 813–823. [Google Scholar]
- Lee, S.W.; Cho, B.H.; Park, S.G.; Kim, S. Aminoacyl-tRNA synthetase complexes: Beyond translation. J. Cell Sci 2004, 117, 3725–3734. [Google Scholar]
- Jarvest, R.L.; Berge, J.M.; Berry, V.; Boyd, H F.; Brown, M.J.; Elder, J.S.; Forrest, A.K.; Fosberry, A.P.; Gentry, D.R.; Hibbs, M.J.; et al. Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against Gram-positive pathogens. J. Med. Chem. 2002, 45, 1959–1962. [Google Scholar]
- Wu, X.Q.; Gross, H.J. The long extra arms of human tRNA((Ser)Sec) and tRNA(Ser) function as major identify elements for serylation in an orientation-dependent, but not sequence-specific manner. Nucleic acids Res 1993, 21, 5589–5594. [Google Scholar]
- Ochsner, U.A.; Sun, X.; Jarvis, T.; Critchley, I.; Janjic, N. Aminoacyl-tRNA synthetases: Essential and still promising targets for new anti-infective agents. Expert Opin. Investig. Drugs 2007, 16, 573–593. [Google Scholar]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother 2005, 49, 4821–4833. [Google Scholar]
- Vondenhoff, G.H.; Gadakh, B.; Severinov, K.; van Aerschot, A. Microcin C and albomycin analogues with aryl-tetrazole substituents as nucleobase isosters are selective inhibitors of bacterial aminoacyl tRNA synthetases but lack efficient uptake. ChemBioChem 2012, 13, 1959–1969. [Google Scholar]
- Gadakh, B.; van Aerschot, A. Aminoacyl-tRNA synthetase inhibitors as antimicrobial agents: A patent review from 2006 till present. Expert Opin. Ther. Pat 2012, 22, 1453–1465. [Google Scholar]
- Vondenhoff, G.H.M.; van Aerschot, A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur. J. Med. Chem. 2011, 46, 5227–5236. [Google Scholar]
- Kobitski, A.Y.; Hengesbach, M.; Seidu-Larry, S.; Dammertz, K.; Chow, C.S.; van Aerschot, A.; Nienhaus, G.U.; Helm, M. Single-molecule FRET reveals a cooperative effect of two methyl group modifications in the folding of human mitochondrial tRNA(Lys). Chem. Biol 2011, 18, 928–936. [Google Scholar]
- Van de Vijver, P.; vondenhoff, G.H.M.; Kazakov, T.S.; Semenova, E.; Kuznedelov, K.; Metlitskaya, A.; van Aerschot, A.; Severinov, K. Synthetic microcin C analogs targeting different aminoacyl-tRNA synthetases. J. Bacteriol 2009, 191, 6273–6280. [Google Scholar]
- Lacivita, E.; Patarnello, D.; Stroth, N.; Caroli, A.; Niso, M.; Contino, M.; de Giorgio, P.; di Pilato, P.; Colabufo, N.A.; Berardi, F.; et al. Investigations on the 1-(2-Biphenyl)piperazine motif: Identification of new potent and selective ligands for the serotonin(7) (5-HT7) receptor with agonist or antagonist action in vitro or ex vivo. J. Med. Chem 2012, 55, 6375–6380. [Google Scholar]
- Kim, S.Y.; Lee, Y.-S.; Kang, T.; Kim, S.; Lee, J. Pharmacophore-based virtual screening: The discovery of novel methionyl-tRNA synthetase inhibitors. Bioorgan. Med. Chem. Lett 2006, 16, 4898–4907. [Google Scholar]
- Tandon, M.; Coffen, D.L.; Gallant, P.; Keith, D.; Ashwell, M.A. Potent and selective inhibitors of bacterial methionyl tRNA synthetase derived from an oxazolone-dipeptide scaffold. Bioorgan. Med. Chem. Lett 2004, 14, 1909–1911. [Google Scholar]
- Lee, J.; Kang, S.U.; Kang, M.K.; Chun, M.W.; Jo, Y.J.; Kwak, J.H.; Kim, S. Methionyl adenylate analogues as inhibitors of methionyl-tRNA synthetase. Bioorgan. Med. Chem. Lett 1999, 9, 1365–1370. [Google Scholar]
- Finn, J.; Stidham, M.; Hilgers, M.; Kedar, G.C. Identification of novel inhibitors of methionyl-tRNA synthetase (MetRS) by virtual screening. Bioorgan. Med. Chem. Lett 2008, 18, 3932–3937. [Google Scholar]
- Ouyang, L.; Huang, Y.; Zhao, Y.; He, G.; Xie, Y.; Liu, J.; He, J.; Liu, B.; Wei, Y. Preparation, antibacterial evaluation and preliminary structure-activity relationship (SAR) study of benzothiazol-and benzoxazol-2-amine derivatives. Bioorgan. Med. Chem. Lett 2012, 22, 3044–3049. [Google Scholar]
- He, G.; Qiu, M.; Li, R.; Ouyang, L.; Wu, F.; Song, X.; Cheng, L.; Xiang, M.; Yu, L. Multicomplex-based pharmacophore-guided 3D-QSAR studies of N-Substituted 2′-(aminoaryl)benzothiazoles as Aurora-A inhibitors. Chem. Biol. Drug Des 2012, 79, 960–971. [Google Scholar]
- He, G.; Qiu, M.H.; Li, R.; Song, X.R.; Zheng, X.; Shi, J.Y.; Xu, G.B.; Han, J.; Yu, L.T.; Yang, S.Y.; et al. Molecular docking-based 3D-QSAR studies of pyrrolo 3,4-c pyrazole derivatives as Aurora-A inhibitors. Mol. Simul 2011, 37, 31–42. [Google Scholar]
- Ouyang, L.; He, G.; Huang, W.; Song, X.; Wu, F.; Xiang, M. Combined structure-based pharmacophore and 3D-QSAR studies on phenylalanine series compounds as TPH1 inhibitors. Int. J. Mol. Sci 2012, 13, 5348–5363. [Google Scholar]
- Wu, F.; Xu, T.; He, G.; Ouyang, L.; Han, B.; Peng, C.; Song, X.; Xiang, M. Discovery of novel focal adhesion kinase inhibitors using a hybrid protocol of virtual screening approach based on multicomplex-based pharmacophore and molecular docking. Int. J. Mol. Sci 2012, 13, 15668–15678. [Google Scholar]
- Xiang, M.; Lin, Y.; He, G.; Chen, L.; Yang, M.; Yang, S.; Mo, Y. Correlation between biological activity and binding energy in systems of integrin with cyclic RGD-containing binders: A QM/MM molecular dynamics study. J. Mol. Model 2012, 18, 4917–4927. [Google Scholar]
- Zhong, H.; Huang, W.; He, G.; Peng, C.; Wu, F.; Ouyang, L. Molecular dynamics simulation of tryptophan hydroxylase-1: Binding modes and free energy analysis to phenylalanine derivative inhibitors. Int. J. Mol. Sci 2013, 13, 9947–9962. [Google Scholar]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct 2000, 29, 291–325. [Google Scholar]
- Zou, J.; Xie, H.-Z.; Yang, S.-Y.; Chen, J.-J.; Ren, J.-X.; Wei, Y.-Q. Towards more accurate pharmacophore modeling: Multicomplex-based comprehensive pharmacophore map and most-frequent-feature pharmacophore model of CDK2. J. Mol. Graph 2008, 27, 430–438. [Google Scholar]
- Nadarajan, S.P.; Mathew, S.; Deepankumar, K.; Yun, H. An in silico approach to evaluate the polyspecificity of methionyl-tRNA synthetases. J. Mol. Graph. 2013, 39, 79–86. [Google Scholar]
- Singh, P.; Verma, P.; Yadav, B.; Komath, S.S. Synthesis and evaluation of indole-based new scaffolds for antimicrobial activities-identification of promising candidates. Bioorgan. Med. Chem. Lett 2011, 21, 3367–3372. [Google Scholar]
- Wu, G.; Ouyang, L.; Liu, J.; Zeng, S.; Huang, W.; Han, B.; Wu, F.; He, G.; Xiang, M. Synthesis of novel spirooxindolo-pyrrolidines, pyrrolizidines, and pyrrolothiazoles via a regioselective three-component [3+2] cycloaddition and their preliminary antimicrobial evaluation. Mol. Divers. 2013, 17, 271–283. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar]
- Crepin, T.; Schmitt, E.; Mechulam, Y.; Sampson, P.B.; Vaughan, M.D.; Honek, J.F.; Blanquet, S. Use of analogues of methionine and methionyl adenylate to sample conformational changes during catalysis in Escherichia coli methionyl-tRNA synthetase. J. Mol. Biol 2003, 332, 59–72. [Google Scholar]
- Koh, C.Y.; Kim, J.E.; Shibata, S.; Ranade, R.M.; Yu, M.; Liu, J.; Gillespie, J.R.; Buckner, F.S.; Verlinde, C.L.M.J.; Fan, E.; et al. Distinct states of methionyl-tRNA synthetase indicate inhibitor binding by conformational selection. Structure 2012, 20, 1681–1691. [Google Scholar]
- Discovery Studio, version 3.5; Accelrys, Inc: San Diego, CA, USA, 2012.
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Potential virtual lead identification in the discovery of renin inhibitors: Application of ligand and structure-based pharmacophore modeling approaches. Eur. J. Med. Chem 2011, 46, 2469–2476. [Google Scholar]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [Google Scholar]
- Specs database. Available online: www.specs.net (on accessed 10 April 2013).
- Wu, Y.; Ma, Q.; Song, X.; Zheng, Y.; Ren, W.; Zhang, J.; Ouyang, L.; Wu, F.; He, G. Biocompatible PEG-poly(γ-cholesterol-l-glutamate) copolymers: Synthesis, characterization and in vitro studies. J. Polym. Sci 2012, 50, 4532–4537. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Feature name | ID | Count | Statistical frequency (%) | Structure-based pharmacophore model | Related amino acid residues |
|---|---|---|---|---|---|---|
| 1 | HBA-F 1 | A1 | 1 | 7 | ||
| 2 | HBA-F 2 | A2 | 1 | 7 | ||
| 3 | HBD 1 | D1 | 11 | 79 | ✓ | H21, D296, I297 |
| 4 | HBD 2 | D2 | 9 | 64 | ✓ | H24, D296, I297 |
| 5 | HBD 3 | D3 | 2 | 14 | ||
| 6 | HBD 4 | D4 | 1 | 7 | ||
| 7 | HBD 5 | D5 | 1 | 7 | ||
| 8 | HBD 6 | D6 | 1 | 7 | ||
| 9 | HBD 7 | D7 | 1 | 7 | ||
| 10 | HBD 8 | D8 | 1 | 7 | ||
| 11 | Hydrophobic 1 | H1 | 12 | 86 | ✓ | V326, M333 |
| 12 | Hydrophobic 2 | H2 | 10 | 71 | ✓ | A12, L13, P257, Y260 |
| 13 | Hydrophobic 3 | H3 | 9 | 64 | ✓ | W253 |
| 14 | Hydrophobic 4 | H4 | 5 | 36 | ||
| 15 | Hydrophobic 5 | H5 | 2 | 14 | ||
| 16 | Hydrophobic 6 | H6 | 1 | 7 | ||
| 17 | Positive ionizable 1 | Pos1 | 9 | 64 | ✓ | D296, Y325 |
| 18 | Positive ionizable 1 | Pos1 | 2 | 14 | ||
| 19 | Negative ionizable | Neg | 1 | 7 |
| Compounds | MIC (μg/mL) a | Cytotoxicity | ||||
|---|---|---|---|---|---|---|
| Gram-positive bacteria b | Gram-negative bacteria b | IC50 (μM) | ||||
| S. aureus | MRSA | E. coli | P. aeruginosa | K. pneumoniae | ||
ZINC06843697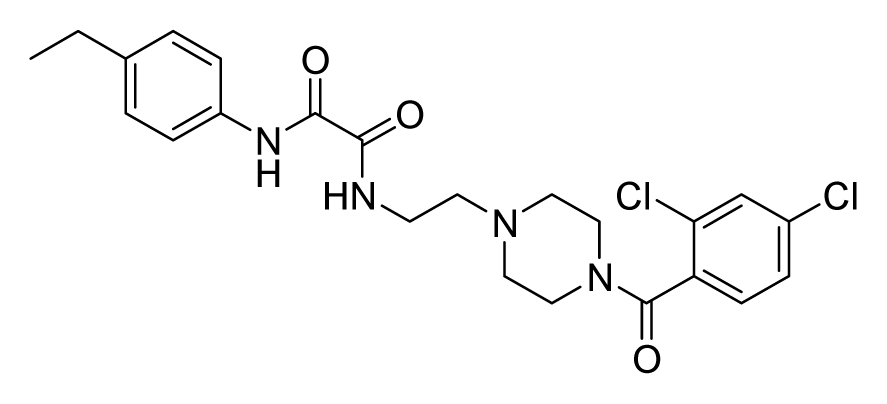 | 4 | 4 | 16 | 8 | 64 | >20 |
ZINC00729256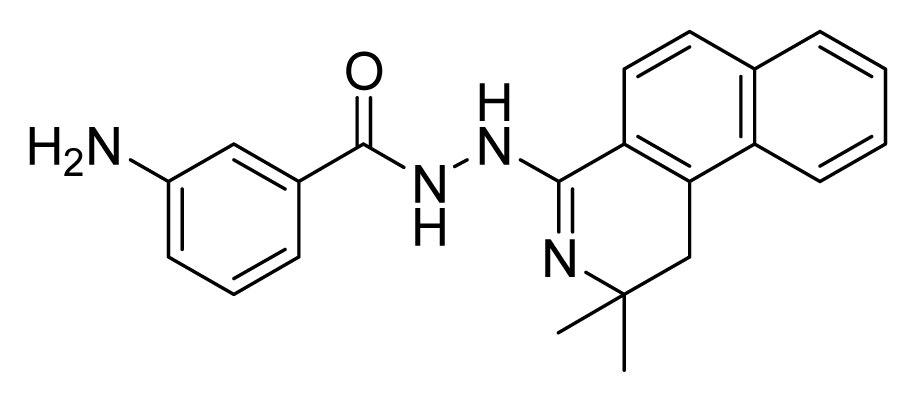 | 8 | 16 | 32 | 4 | 16 | >20 |
ZINC08451958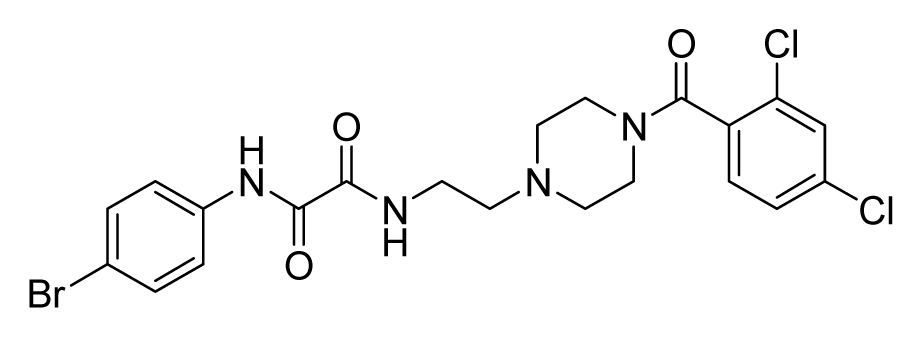 | 32 | 32 | 64 | 64 | 64 | >20 |
ZINC08430843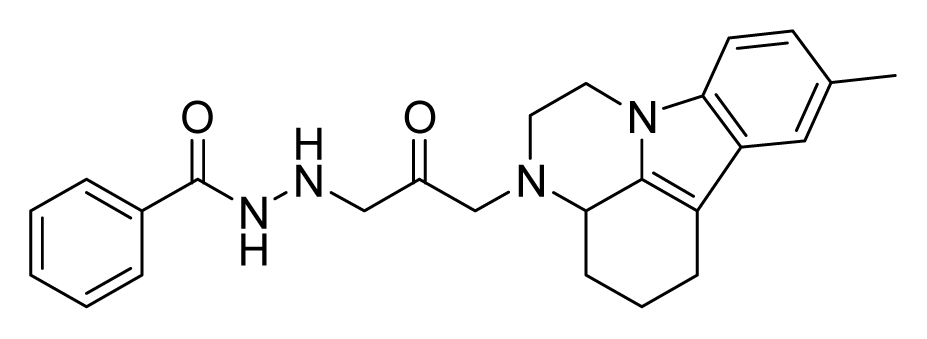 | 16 | 32 | 64 | 32 | 64 | >20 |
ZINC08452043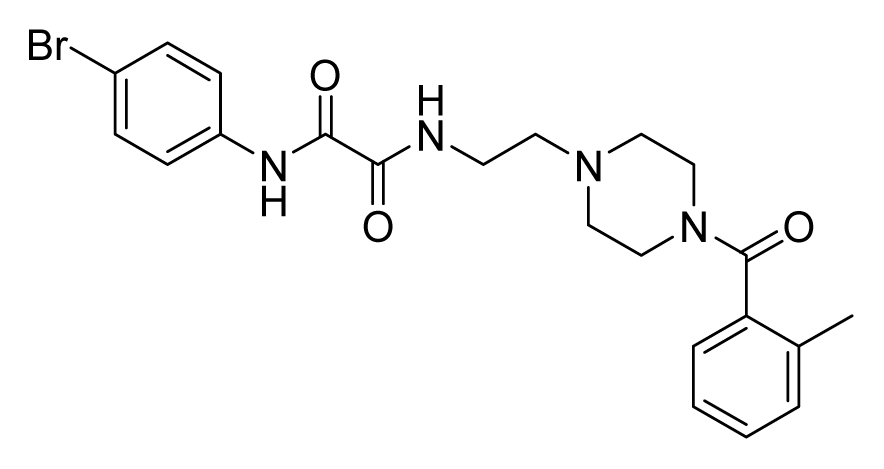 | 8 | 16 | 16 | 8 | 32 | 10.2 |
ZINC10312776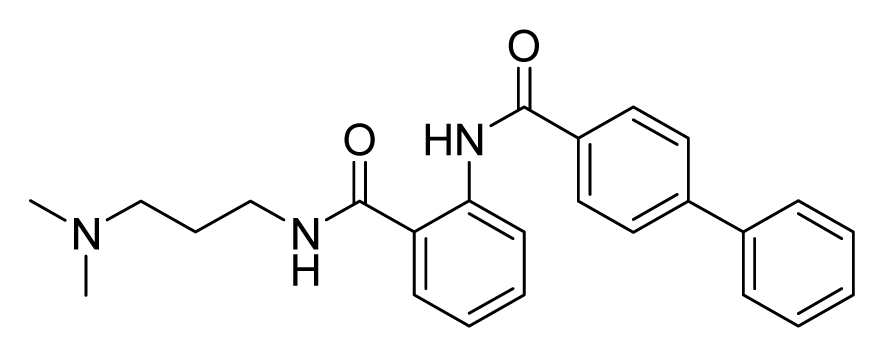 | 64 | 64 | 64 | 16 | 32 | >20 |
ZINC19797060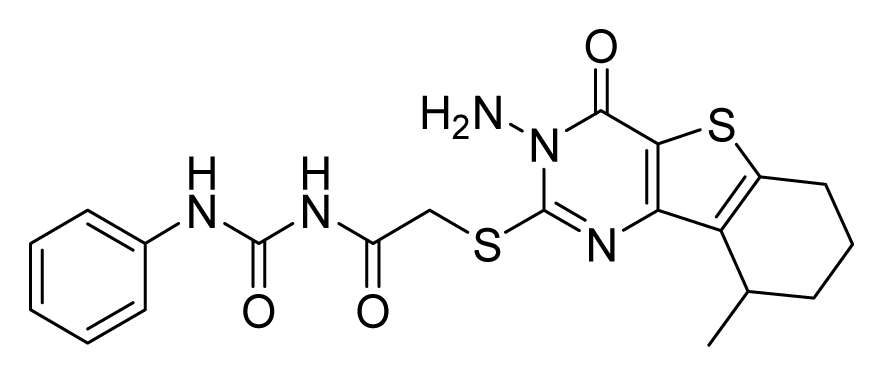 | 32 | 64 | 64 | 16 | 64 | >20 |
ZINC02086896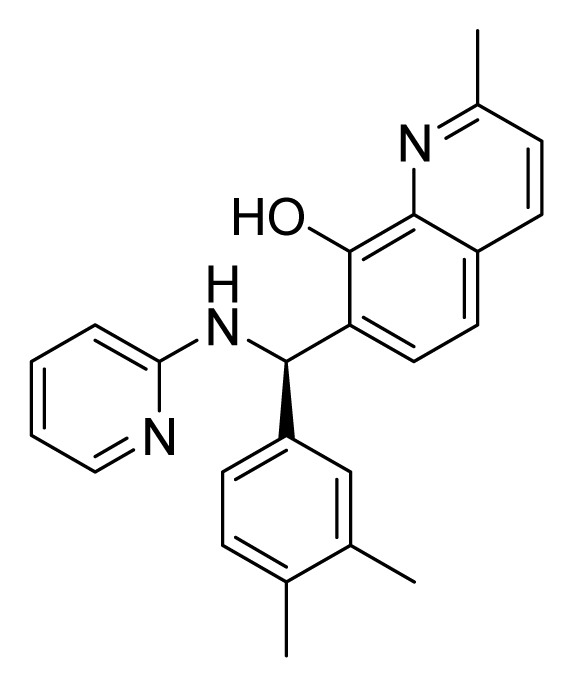 | 32 | 64 | 32 | 16 | 64 | 3.15 |
ZINC19922703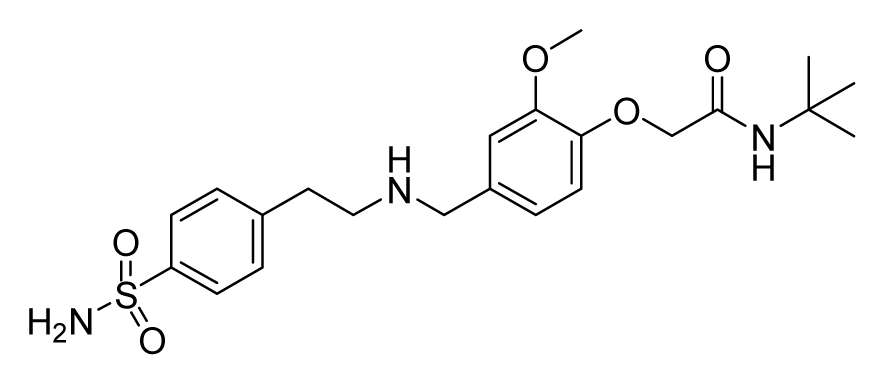 | 32 | 64 | 64 | 32 | 64 | >20 |
ZINC02181060 | 4 | 16 | 8 | 16 | 32 | 15.4 |
ZINC02424508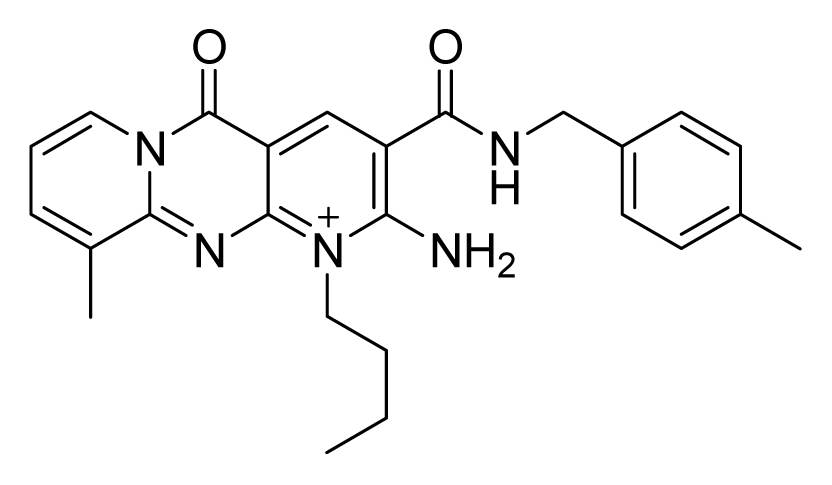 | 16 | 32 | 16 | 64 | 64 | >20 |
ZINC19797059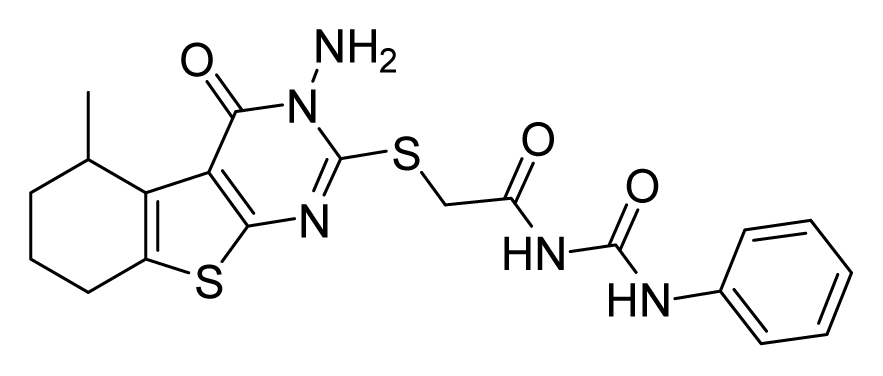 | 32 | 32 | 64 | 64 | 64 | >20 |
ZINC08384332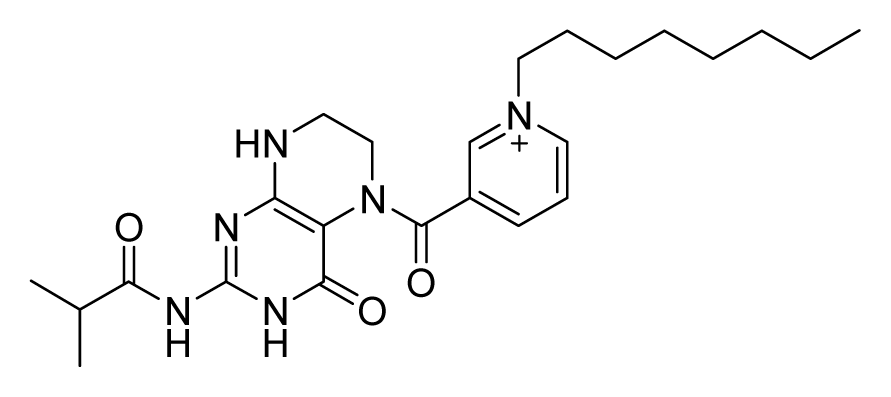 | 32 | 32 | 32 | 16 | 64 | >20 |
ZINC02709613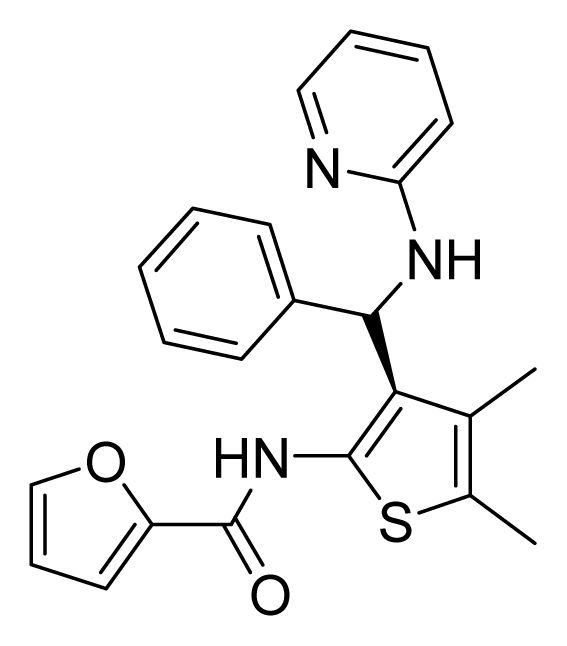 | 64 | 64 | 64 | 16 | 64 | >20 |
ZINC04380079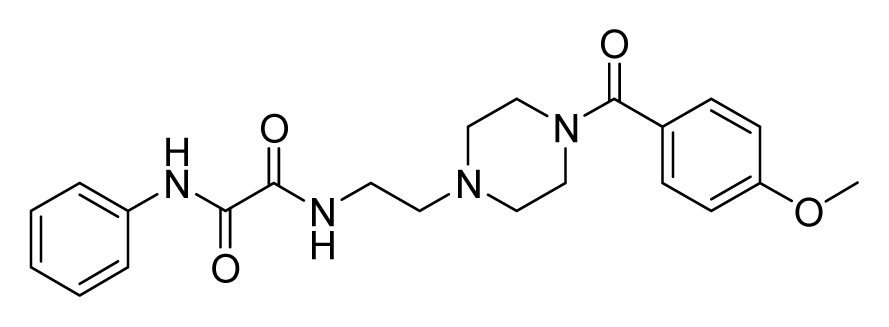 | 32 | 64 | 64 | 64 | 64 | 6.84 |
| No. | PDB | Resolution | Ligand | Release date |
|---|---|---|---|---|
| 1 | 1PFY | 1.93 | MSP | 5-27-2003 |
| 2 | 3KFL | 2.00 | ME8 | 10-27-2009 |
| 3 | 3TUN | 2.55 | C13 | 9-16-2011 |
| 4 | 3U1E | 2.31 | 387 | 9-29-2011 |
| 5 | 3U1F | 2.20 | 392 | 9-29-2011 |
| 6 | 3U1G | 2.35 | 415 | 9-29-2011 |
| 7 | 3U1Z | 2.90 | 43E | 9-30-2011 |
| 8 | 3U20 | 2.49 | 44F | 9-30-2011 |
| 9 | 4EG4 | 3.15 | 0OT | 3-30-2012 |
| 10 | 4EG5 | 3.10 | 0OU | 3-30-2012 |
| 11 | 4EG6 | 2.90 | 0P5 | 3-30-2012 |
| 12 | 4EG7 | 2.75 | 0P4 | 3-30-2012 |
| 13 | 4EG8 | 2.60 | 0P6 | 3-30-2012 |
| 14 | 4EGA | 2.70 | 0P8 | 3-30-2012 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, C.; He, G.; Jiang, Q.; Han, B.; Peng, C. Novel Hybrid Virtual Screening Protocol Based on Molecular Docking and Structure-Based Pharmacophore for Discovery of Methionyl-tRNA Synthetase Inhibitors as Antibacterial Agents. Int. J. Mol. Sci. 2013, 14, 14225-14239. https://doi.org/10.3390/ijms140714225
Liu C, He G, Jiang Q, Han B, Peng C. Novel Hybrid Virtual Screening Protocol Based on Molecular Docking and Structure-Based Pharmacophore for Discovery of Methionyl-tRNA Synthetase Inhibitors as Antibacterial Agents. International Journal of Molecular Sciences. 2013; 14(7):14225-14239. https://doi.org/10.3390/ijms140714225
Chicago/Turabian StyleLiu, Chi, Gu He, Qinglin Jiang, Bo Han, and Cheng Peng. 2013. "Novel Hybrid Virtual Screening Protocol Based on Molecular Docking and Structure-Based Pharmacophore for Discovery of Methionyl-tRNA Synthetase Inhibitors as Antibacterial Agents" International Journal of Molecular Sciences 14, no. 7: 14225-14239. https://doi.org/10.3390/ijms140714225