Toll-Like Receptors in Atherosclerosis

Abstract
:1. Introduction
2. TLRs: Key Pattern Recognition Receptors (PRRs)
3. Role of MyD88 in Atherosclerosis
4. Functional Diversity of Extracellular TLRs in Atherosclerosis
5. Endosomal TLRs: Friends or Foes of the Arterial Wall?
6. TLRs in Cardiovascular Risk Factors
7. Promising Perspectives for Treating TLRs in Atherosclerosis
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar]
- Robbins, J.M.; Webb, D.A.; Sciamanna, C.N. Cardiovascular comorbidities among public health clinic patients with diabetes: The Urban. Diabetics Study. BMC Public Health 2005, 5, 15. [Google Scholar]
- Libby, P.; Okamoto, Y.; Rocha, V.Z.; Folco, E. Inflammation in atherosclerosis: Transition from theory to practice. Circ. J 2010, 74, 213–220. [Google Scholar]
- Laberge, M.A.; Moore, K.J.; Freeman, M.W. Atherosclerosis and innate immune signaling. Ann. Med 2005, 37, 130–140. [Google Scholar]
- Frantz, S.; Ertl, G.; Bauersachs, J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med 2007, 4, 444–454. [Google Scholar]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect Biol. 2012. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med 2000, 192, 565–570. [Google Scholar]
- Seneviratne, A.N.; Sivagurunathan, B.; Monaco, C. Toll-like receptors and macrophage activation in atherosclerosis. Clin. Chim. Acta 2012, 413, 3–14. [Google Scholar]
- Shimizu, T.; Kida, Y.; Kuwano, K. Triacylated lipoproteins derived from Mycoplasma pneumoniae activate nuclear factor-kappaB through toll-like receptors 1 and 2. Immunology 2007, 121, 473–483. [Google Scholar]
- Wyllie, D.H.; Kiss-Toth, E.; Visintin, A.; Smith, S.C.; Boussouf, S.; Segal, D.M.; Duff, G.W.; Dower, S.K. Evidence for an accessory protein function for Toll-like receptor 1 in anti-bacterial responses. J. Immunol 2000, 165, 7125–7132. [Google Scholar]
- Nijhuis, M.M.; Pasterkamp, G.; Sluis, N.I.; de Kleijn, D.P.; Laman, J.D.; Ulfman, L.H. Peptidoglycan increases firm adhesion of monocytes under flow conditions and primes monocyte chemotaxis. J. Vasc. Res 2007, 44, 214–222. [Google Scholar]
- Andreakos, E.; Foxwell, B.; Feldmann, M. Is targeting Toll-like receptors and their signaling pathway a useful therapeutic approach to modulating cytokine-driven inflammation? Immunol. Rev 2004, 202, 250–265. [Google Scholar]
- Hajishengallis, G.; Wang, M.; Bagby, G.J.; Nelson, S. Importance of TLR2 in early innate immune response to acute pulmonary infection with Porphyromonas gingivalis in mice. J. Immunol 2008, 181, 4141–4149. [Google Scholar]
- Nakamura, N.; Yoshida, M.; Umeda, M.; Huang, Y.; Kitajima, S.; Inoue, Y.; Ishikawa, I.; Iwai, T. Extended exposure of lipopolysaccharide fraction from Porphyromonas gingivalis facilitates mononuclear cell adhesion to vascular endothelium via Toll-like receptor-2 dependent mechanism. Atherosclerosis 2008, 196, 59–67. [Google Scholar]
- Hreggvidsdottir, H.S.; Ostberg, T.; Wahamaa, H.; Schierbeck, H.; Aveberger, A.C.; Klevenvall, L.; Palmblad, K.; Ottosson, L.; Andersson, U.; Harris, H.E. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J. Leukoc. Biol 2009, 86, 655–662. [Google Scholar]
- Peter, K.; Bobik, A. HMGB1 signals danger in acute coronary syndrome: Emergence of a new risk marker for cardiovascular death? Atherosclerosis 2012, 221, 317–318. [Google Scholar]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar]
- Holvoet, P.; Davey, P.C.; de Keyzer, D.; Doukoure, M.; Deridder, E.; Bochaton-Piallat, M.L.; Gabbiani, G.; Beaufort, E.; Bishay, K.; Andrieux, N.; et al. Oxidized low-density lipoprotein correlates positively with toll-like receptor 2 and interferon regulatory factor-1 and inversely with superoxide dismutase-1 expression: Studies in hypercholesterolemic swine and THP-1 cells. Arterioscler. Thromb. Vasc. Biol 2006, 26, 1558–1565. [Google Scholar]
- Cheng, N.; He, R.; Tian, J.; Ye, P.P.; Ye, R.D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol 2008, 181, 22–26. [Google Scholar]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar]
- Takeuchi, O.; Kawai, T.; Muhlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol 2001, 13, 933–940. [Google Scholar]
- Anwar, M.A.; Basith, S.; Choi, S. Negative regulatory approaches to the attenuation of Toll-like receptor signaling. Exp. Mol. Med 2013, 45, e11. [Google Scholar]
- Okusawa, T.; Fujita, M.; Nakamura, J.; Into, T.; Yasuda, M.; Yoshimura, A.; Hara, Y.; Hasebe, A.; Golenbock, D.T.; Morita, M.; et al. Relationship between structures and biological activities of mycoplasmal diacylated lipopeptides and their recognition by toll-like receptors 2 and 6. Infect. Immun 2004, 72, 1657–1665. [Google Scholar]
- Vabulas, R.M.; Ahmad-Nejad, P.; da Costa, C.; Miethke, T.; Kirschning, C.J.; Hacker, H.; Wagner, H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem 2001, 276, 31332–31339. [Google Scholar]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar]
- Kariko, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem 2004, 279, 12542–12550. [Google Scholar]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol 1999, 162, 3749–3752. [Google Scholar]
- Rassa, J.C.; Meyers, J.L.; Zhang, Y.; Kudaravalli, R.; Ross, S.R. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. USA 2002, 99, 2281–2286. [Google Scholar]
- Bulut, Y.; Faure, E.; Thomas, L.; Karahashi, H.; Michelsen, K.S.; Equils, O.; Morrison, S.G.; Morrison, R.P.; Arditi, M. Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J. Immunol 2002, 168, 1435–1440. [Google Scholar]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med 2009, 15, 774–780. [Google Scholar]
- Xu, X.H.; Shah, P.K.; Faure, E.; Equils, O.; Thomas, L.; Fishbein, M.C.; Luthringer, D.; Xu, X.P.; Rajavashisth, T.B.; Yano, J.; et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 2001, 104, 3103–3108. [Google Scholar]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol 2010, 11, 155–161. [Google Scholar]
- Roelofs, M.F.; Boelens, W.C.; Joosten, L.A.; Abdollahi-Roodsaz, S.; Geurts, J.; Wunderink, L.U.; Schreurs, B.W.; van den Berg, W.B.; Radstake, T.R. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J. Immunol 2006, 176, 7021–7027. [Google Scholar]
- Fang, H.; Wu, Y.; Huang, X.; Wang, W.; Ang, B.; Cao, X.; Wan, T. Toll-like receptor 4 (TLR4) is essential for Hsp70-like protein 1 (HSP70L1) to activate dendritic cells and induce Th1 response. J. Biol. Chem 2011, 286, 30393–30400. [Google Scholar]
- Chase, M.A.; Wheeler, D.S.; Lierl, K.M.; Hughes, V.S.; Wong, H.R.; Page, K. Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-κB-dependent mechanism. J. Immunol 2007, 179, 6318–6324. [Google Scholar]
- Yang, Y.; Liu, B.; Dai, J.; Srivastava, P.K.; Zammit, D.J.; Lefrancois, L.; Li, Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 2007, 26, 215–226. [Google Scholar]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med 2005, 11, 1173–1179. [Google Scholar]
- Erridge, C.; Kennedy, S.; Spickett, C.M.; Webb, D.J. Oxidized phospholipid inhibition of toll-like receptor (TLR) signaling is restricted to TLR2 and TLR4: Roles for CD14, LPS-binding protein, and MD2 as targets for specificity of inhibition. J. Biol. Chem 2008, 283, 24748–24759. [Google Scholar]
- Biragyn, A.; Ruffini, P.A.; Leifer, C.A.; Klyushnenkova, E.; Shakhov, A.; Chertov, O.; Shirakawa, A.K.; Farber, J.M.; Segal, D.M.; Oppenheim, J.J.; et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 2002, 298, 1025–1029. [Google Scholar]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar]
- Buwitt-Beckmann, U.; Heine, H.; Wiesmuller, K.H.; Jung, G.; Brock, R.; Akira, S.; Ulmer, A.J. Toll-like receptor 6-independent signaling by diacylated lipopeptides. Eur. J. Immunol 2005, 35, 282–289. [Google Scholar]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RN. A. Science 2004, 303, 1529–1531. [Google Scholar]
- Boule, M.W.; Broughton, C.; Mackay, F.; Akira, S.; Marshak-Rothstein, A.; Rifkin, I.R. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J. Exp. Med 2004, 199, 1631–1640. [Google Scholar]
- Lau, C.M.; Broughton, C.; Tabor, A.S.; Akira, S.; Flavell, R.A.; Mamula, M.J.; Christensen, S.R.; Shlomchik, M.J.; Viglianti, G.A.; Rifkin, I.R.; et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med 2005, 202, 1171–1177. [Google Scholar]
- Leadbetter, E.A.; Rifkin, I.R.; Hohlbaum, A.M.; Beaudette, B.C.; Shlomchik, M.J.; Marshak-Rothstein, A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002, 416, 603–607. [Google Scholar]
- Chiang, C.Y.; Engel, A.; Opaluch, A.M.; Ramos, I.; Maestre, A.M.; Secundino, I.; de Jesus, P.D.; Nguyen, Q.T.; Welch, G.; Bonamy, G.M.; et al. Cofactors required for TLR7- and TLR9-dependent innate immune responses. Cell Host Microbe 2012, 11, 306–318. [Google Scholar]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar]
- Parroche, P.; Lauw, F.N.; Goutagny, N.; Latz, E.; Monks, B.G.; Visintin, A.; Halmen, K.A.; Lamphier, M.; Olivier, M.; Bartholomeu, D.C.; et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc. Natl. Acad. Sci. USA 2007, 104, 1919–1924. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar]
- Cole, J.E.; Mitra, A.T.; Monaco, C. Treating atherosclerosis: The potential of Toll-like receptors as therapeutic targets. Exp. Rev. Cardiovasc. Ther 2010, 8, 1619–1635. [Google Scholar]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol 2005, 17, 1–14. [Google Scholar]
- Bjorkbacka, H.; Kunjathoor, V.V.; Moore, K.J.; Koehn, S.; Ordija, C.M.; Lee, M.A.; Means, T.; Halmen, K.; Luster, A.D.; Golenbock, D.T.; et al. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat. Med 2004, 10, 416–421. [Google Scholar]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar]
- Subramanian, M.; Thorp, E.; Hansson, G.K.; Tabas, I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J. Clin. Invest 2013, 123, 179–188. [Google Scholar]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar]
- Lin, Z.; Lu, J.; Zhou, W.; Shen, Y. Structural insights into TIR domain specificity of the bridging adaptor Mal. in TLR4 signaling. PLoS One 2012, 7, e34202. [Google Scholar]
- Kawagoe, T.; Sato, S.; Matsushita, K.; Kato, H.; Matsui, K.; Kumagai, Y.; Saitoh, T.; Kawai, T.; Takeuchi, O.; Akira, S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol 2008, 9, 684–691. [Google Scholar]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar]
- Dunzendorfer, S.; Lee, H.K.; Tobias, P.S. Flow-dependent regulation of endothelial Toll-like receptor 2 expression through inhibition of SP1 activity. Circ. Res 2004, 95, 684–691. [Google Scholar]
- Mullick, A.E.; Tobias, P.S.; Curtiss, L.K. Modulation of atherosclerosis in mice by Toll-like receptor 2. J. Clin. Invest 2005, 115, 3149–3156. [Google Scholar]
- Mullick, A.E.; Soldau, K.; Kiosses, W.B.; Bell, T.A., 3rd; Tobias, P.S.; Curtiss, L.K. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J. Exp. Med. 2008, 205, 373–383. [Google Scholar]
- Madan, M.; Amar, S. Toll-like receptor-2 mediates diet and/or pathogen associated atherosclerosis: Proteomic findings. PLoS One 2008, 3, e3204. [Google Scholar]
- Monaco, C.; Gregan, S.M.; Navin, T.J.; Foxwell, B.M.; Davies, A.H.; Feldmann, M. Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation 2009, 120, 2462–2469. [Google Scholar]
- Tabas, I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Arterioscler. Thromb. Vasc. Biol 2005, 25, 2255–2264. [Google Scholar]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell MeTable 2010, 12, 467–482. [Google Scholar]
- Higashimori, M.; Tatro, J.B.; Moore, K.J.; Mendelsohn, M.E.; Galper, J.B.; Beasley, D. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol 2011, 31, 50–57. [Google Scholar]
- Curtiss, L.K.; Black, A.S.; Bonnet, D.J.; Tobias, P.S. Atherosclerosis induced by endogenous and exogenous toll-like receptor (TLR)1 or TLR6 agonists. J. Lipid Res 2012, 53, 2126–2132. [Google Scholar]
- Choi, S.H.; Harkewicz, R.; Lee, J.H.; Boullier, A.; Almazan, F.; Li, A.C.; Witztum, J.L.; Bae, Y.S.; Miller, Y.I. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ. Res 2009, 104, 1355–1363. [Google Scholar]
- Hayashi, C.; Papadopoulos, G.; Gudino, C.V.; Weinberg, E.O.; Barth, K.R.; Madrigal, A.G.; Chen, Y.; Ning, H.; LaValley, M.; Gibson, F.C., 3rd; et al. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J. Immunol 2012, 189, 3681–3688. [Google Scholar]
- Cole, J.E.; Navin, T.J.; Cross, A.J.; Goddard, M.E.; Alexopoulou, L.; Mitra, A.T.; Davies, A.H.; Flavell, R.A.; Feldmann, M.; Monaco, C. Unexpected protective role for Toll-like receptor 3 in the arterial wall. Proc. Natl. Acad. Sci. USA 2011, 108, 2372–2377. [Google Scholar]
- Richards, M.R.; Black, A.S.; Bonnet, D.J.; Barish, G.D.; Woo, C.W.; Tabas, I.; Curtiss, L.K.; Tobias, P.S. The LPS2 mutation in TRIF is atheroprotective in hyperlipidemic low density lipoprotein receptor knockout mice. Innate Immun 2013, 19, 20–29. [Google Scholar]
- Zimmer, S.; Steinmetz, M.; Asdonk, T.; Motz, I.; Coch, C.; Hartmann, E.; Barchet, W.; Wassmann, S.; Hartmann, G.; Nickenig, G. Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ. Res 2011, 108, 1358–1366. [Google Scholar]
- Lundberg, A.M.; Ketelhuth, D.F.; Johansson, M.E.; Gerdes, N.; Liu, S.; Yamamoto, M.; Akira, S.; Hansson, G.K. Toll-like receptor 3 and 4 signalling through the TRIF and TRAM adaptors in haematopoietic cells promotes atherosclerosis. Cardiovasc. Res 2013. [Google Scholar] [CrossRef]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol 2002, 3, 196–200. [Google Scholar]
- Jurk, M.; Heil, F.; Vollmer, J.; Schetter, C.; Krieg, A.M.; Wagner, H.; Lipford, G.; Bauer, S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol 2002, 3, 499. [Google Scholar]
- Salagianni, M.; Galani, I.E.; Lundberg, A.M.; Davos, C.H.; Varela, A.; Gavriil, A.; Lyytikainen, L.P.; Lehtimaki, T.; Sigala, F.; Folkersen, L.; et al. Toll-like receptor 7 protects from atherosclerosis by constraining “inflammatory” macrophage activation. Circulation 2012, 126, 952–962. [Google Scholar]
- Janssens, S.; Burns, K.; Tschopp, J.; Beyaert, R. Regulation of interleukin-1- and lipopolysaccharide-induced NF-κB activation by alternative splicing of MyD88. Curr. Biol. 2002, 12, 467–471. [Google Scholar]
- Goossens, P.; Gijbels, M.J.; Zernecke, A.; Eijgelaar, W.; Vergouwe, M.N.; van der Made, I.; Vanderlocht, J.; Beckers, L.; Buurman, W.A.; Daemen, M.J.; et al. Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell MeTable 2010, 12, 142–153. [Google Scholar]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar]
- Caricilli, A.M.; Saad, M.J. The role of gut microbiota on insulin resistance. Nutrients 2013, 5, 829–851. [Google Scholar]
- Jang, H.J.; Kim, H.S.; Hwang, D.H.; Quon, M.J.; Kim, J.A. Toll-like receptor 2 mediates high-fat diet-induced impairment of vasodilator actions of insulin. Am. Physiological. Soc 2013, 304, E1077–E1088. [Google Scholar]
- Tinsley, J.H.; Chiasson, V.L.; Mahajan, A.; Young, K.J.; Mitchell, B.M. Toll-like receptor 3 activation during pregnancy elicits preeclampsia-like symptoms in rats. Am. J. Hypertens 2009, 22, 1314–1319. [Google Scholar]
- Chatterjee, P.; Weaver, L.E.; Doersch, K.M.; Kopriva, S.E.; Chiasson, V.L.; Allen, S.J.; Narayanan, A.M.; Young, K.J.; Jones, K.A.; Kuehl, T.J.; et al. Placental Toll-like receptor 3 and Toll-like receptor 7/8 activation contributes to preeclampsia in humans and mice. PLoS One 2012, 7, e41884. [Google Scholar]
- Goulopoulou, S.; Matsumoto, T.; Bomfim, G.F.; Webb, R.C. Toll-like receptor 9 activation: A novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin. Sci 2012, 123, 429–435. [Google Scholar]
- Bomfim, G.F.; Dos Santos, R.A.; Oliveira, M.A.; Giachini, F.R.; Akamine, E.H.; Tostes, R.C.; Fortes, Z.B.; Webb, R.C.; Carvalho, M.H. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin. Sci 2012, 122, 535–543. [Google Scholar]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Krankel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 2013, 38, 754–768. [Google Scholar]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar]
- Creely, S.J.; McTernan, P.G.; Kusminski, C.M.; Fisher f, M.; Da Silva, N.F.; Khanolkar, M.; Evans, M.; Harte, A.L.; Kumar, S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am. J. Physiol 2007, 292, E740–E747. [Google Scholar]
- Paul-Clark, M.J.; McMaster, S.K.; Sorrentino, R.; Sriskandan, S.; Bailey, L.K.; Moreno, L.; Ryffel, B.; Quesniaux, V.F.; Mitchell, J.A. Toll-like receptor 2 is essential for the sensing of oxidants during inflammation. Am. J. Respir. Crit. Care Med 2009, 179, 299–306. [Google Scholar]
- Bertocchi, C.; Traunwieser, M.; Dorler, J.; Hasslacher, J.; Joannidis, M.; Dunzendorfer, S. Atorvastatin inhibits functional expression of proatherogenic TLR2 in arterial endothelial cells. Cell Physiol. Biochem 2011, 28, 625–630. [Google Scholar]
- Arslan, F.; Smeets, M.B.; O’Neill, L.A.; Keogh, B.; McGuirk, P.; Timmers, L.; Tersteeg, C.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation 2010, 121, 80–90. [Google Scholar]
- Arslan, F.; Keogh, B.; McGuirk, P.; Parker, A.E. TLR2 and TLR4 in ischemia reperfusion injury. Mediat. Inflamm 2010, 2010, 704202. [Google Scholar]
- Arslan, F.; Houtgraaf, J.H.; Keogh, B.; Kazemi, K.; de Jong, R.; McCormack, W.J.; O’Neill, L.A.; McGuirk, P.; Timmers, L.; Smeets, M.B.; et al. Treatment with OPN-305, a humanized anti-Toll-Like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ. Cardiovasc. Interv 2012, 5, 279–287. [Google Scholar]
- Ta, N.N.; Schuyler, C.A.; Li, Y.; Lopes-Virella, M.F.; Huang, Y. DPP-4 (CD26) inhibitor alogliptin inhibits atherosclerosis in diabetic apolipoprotein E-deficient mice. J. Cardiovasc. Pharmacol. 2011, 58, 157–166. [Google Scholar]
- Lu, Z.; Zhang, X.; Li, Y.; Jin, J.; Huang, Y. TLR4 antagonist reduces early-stage atherosclerosis in diabetic apolipoprotein E-deficient mice. J. Endocrinol 2013, 216, 61–71. [Google Scholar]
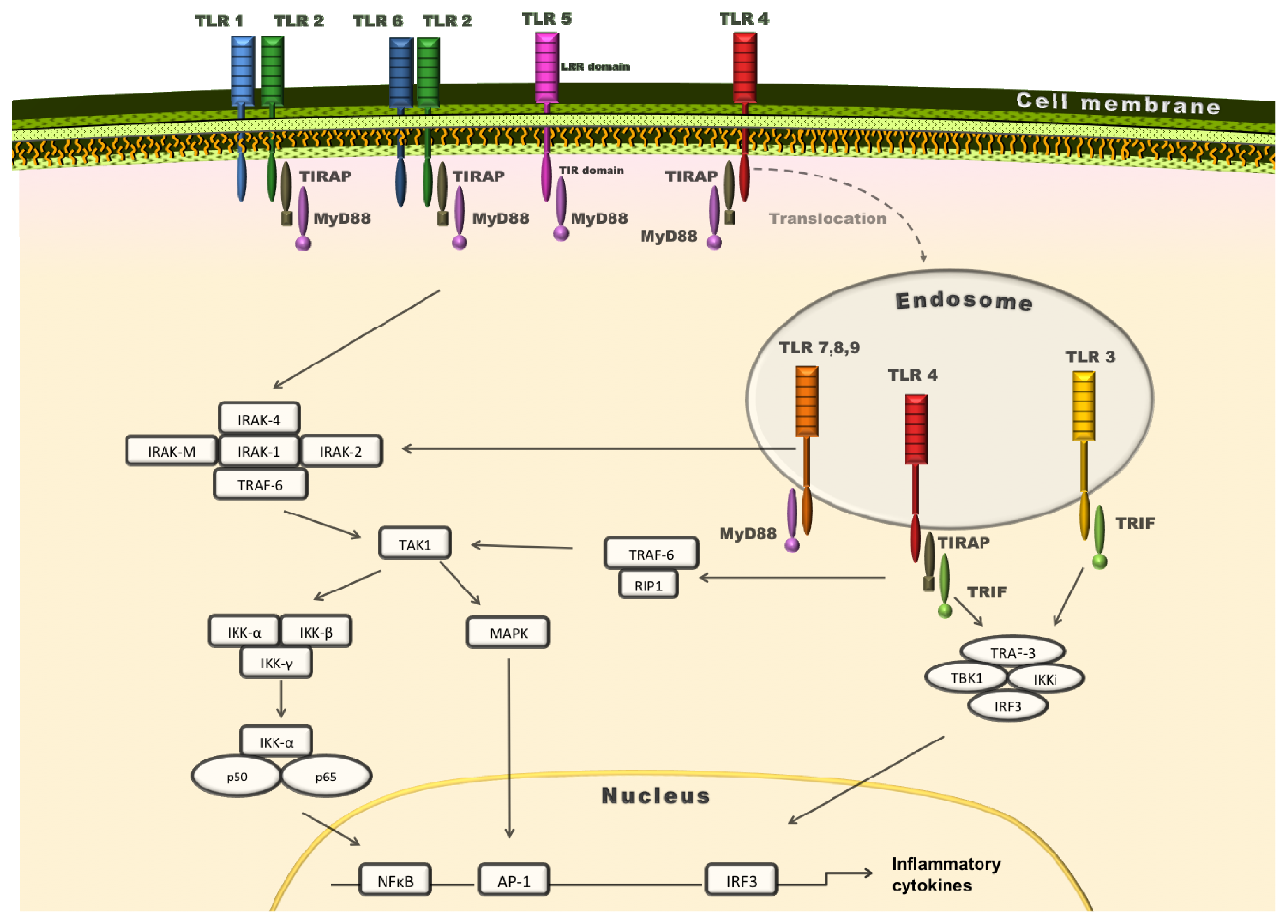

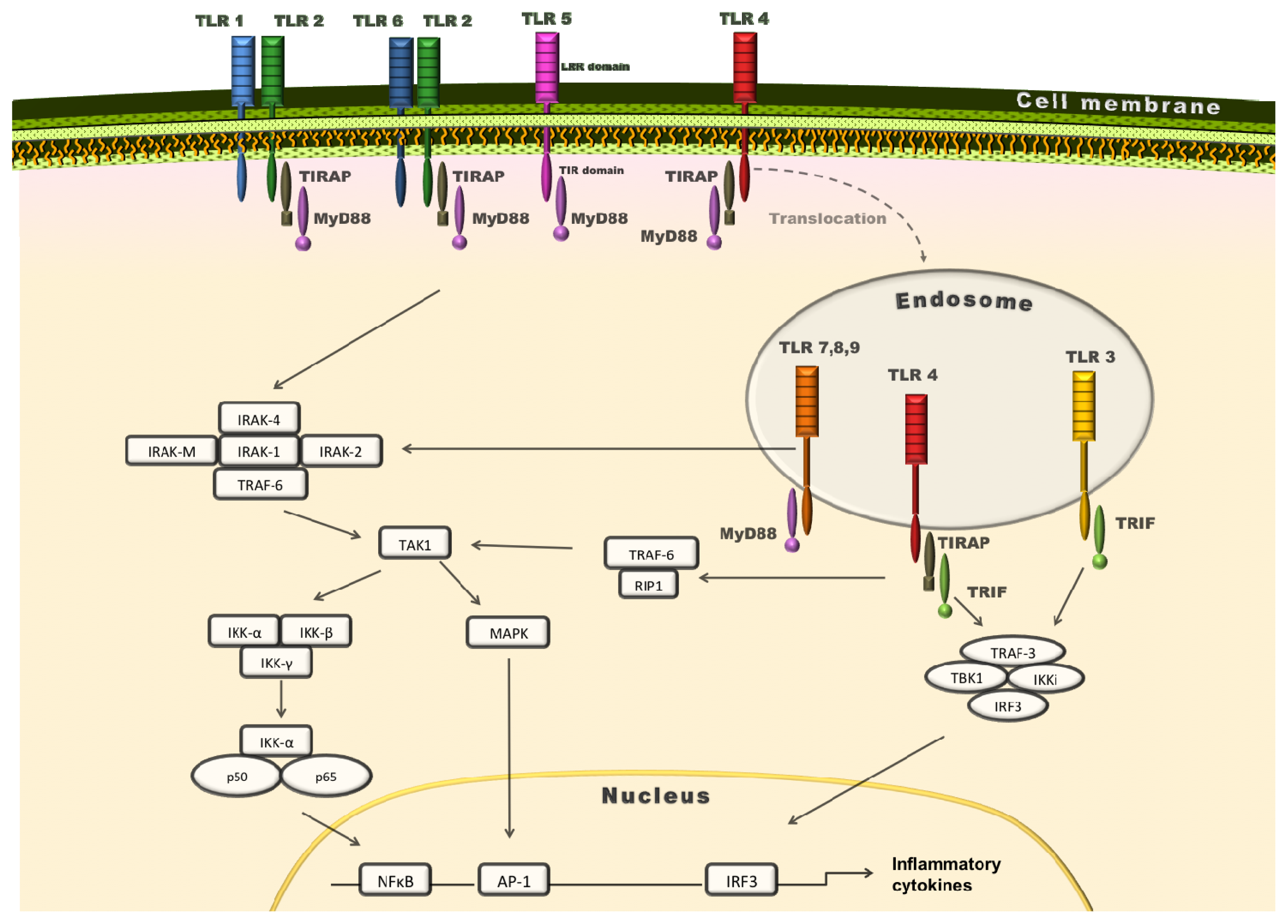{kind=link}
| TLR receptor | Exogenous ligands | Endogenous ligands | Exogenous source |
|---|---|---|---|
| TLR1 | Tri-acyl lipopeptides [9] | Not determined | Mycoplasma |
| TLR1/2 | Soluble factors [10]; PAM3 | Not determined | Gram-negative bacteria (Neisseria meningitidis) |
| TLR2 | Peptidoglycan [11]; Glycoinositolphospholipids; Glycolipids; Porins; Zymosan; Atypical LPS [12–14] | HMGB1 [15–17]; oxLDL [18]; Serum Amyloid A [19]; Amyloid beta [20] | Gram positive bacteria; Trypanozoma cruzi; Treponema maltophilum; Fungi; Leptospira interrogans; Porphyromonas gingivalis |
| TLR2/6 | Lipoproteins [21]; Zymosan; Lipoteichoic acids [22]; FSL-1 [23] | Heat-shock proteins such as HSP60 and 70 [24] Versican [25] | Gram positive Bacteria; Fungi |
| TLR3 | dsRNA; (PolyI:C) [26] | mRNA[27] | Virus |
| TLR4 | Lipopolysaccharide [28]; Glycoproteins [29]; Taxol, RSV Fusion Protein [12]; HSP60 [30] | Tenascin C [31]; oxLDL [32]; Amyloid beta [33]; HSP22 [34]; HSP70 [35]; HSP72 [36]; Gp96 [37]; ECM fragments [38]; Oxidized phospholipids [39]; Betadefensin-2 [40]; HMGB1 [15–17] | Virus; plant; Chlamydia pneumoniae |
| TLR5 | Flagellin [41] | Not determined | Bacteria |
| TLR6 | Di-acyl lipopeptides [42] | Not determined | Mycoplasma |
| TLR7 | Imidazoquinolines; loxoribine and Bropirine [12]; ssRNA [22] | ssRNA (immune complex) [22] | Virus |
| TLR7/8 | ssRNA; Imidazoquinolines [43] | ssRNA (immune complex) [22] | Virus |
| TLR7/9 | Not determined | Nucleic acid-containing immune complexes [44–47] | Not determined |
| TLR9 | CpG-DNA [48]; CpG oligonucleotide; Hemozoin [49] | Chromatin IgG complex [44] | Bacteria; Parasites (Plasmodium) |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Falck-Hansen, M.; Kassiteridi, C.; Monaco, C. Toll-Like Receptors in Atherosclerosis. Int. J. Mol. Sci. 2013, 14, 14008-14023. https://doi.org/10.3390/ijms140714008
Falck-Hansen M, Kassiteridi C, Monaco C. Toll-Like Receptors in Atherosclerosis. International Journal of Molecular Sciences. 2013; 14(7):14008-14023. https://doi.org/10.3390/ijms140714008
Chicago/Turabian StyleFalck-Hansen, Mika, Christina Kassiteridi, and Claudia Monaco. 2013. "Toll-Like Receptors in Atherosclerosis" International Journal of Molecular Sciences 14, no. 7: 14008-14023. https://doi.org/10.3390/ijms140714008