1. Introduction
Ovarian carcinoma is the most lethal gynecological malignancy in American [
1] and Japanese women. Among ovarian carcinomas, high-grade serous ovarian carcinoma mostly presents at an advanced stage and has a low overall survival rate. Standard treatment involves aggressive cytoreductive surgery followed by platinum- and taxane-based chemotherapy [
2,
3]. Although initial response rates to platinum-based chemotherapy exceed 75%, most tumors will eventually recur and become refractory to treatment, with a median survival of less than five years [
2,
4]. Although there are well-established surgical and chemotherapeutic treatments for ovarian cancer, there is significant opportunity to develop drugs targeting specific molecular pathways and to reduce rates of metastasis or relapse. Drugs of this type would be particularly useful for recurrent disease that shows chemoresistance. Thus, there is an initial, preclinical need to improve our understanding of the molecular pathways underlying ovarian carcinogenesis.
Recent morphological, immunohistochemical, and molecular genetic studies have led to the development of a new paradigm for the pathogenesis and origin of epithelial ovarian cancer based on a dualistic model of carcinogenesis that divides epithelial ovarian cancer into two broad categories, designated as types I and II. Type I tumors include low-grade serous, low-grade endometrioid, clear cell, and mucinous carcinomas; these tumors develop in a stepwise fashion from well-established precursor lesions, such as borderline tumors and endometriosis. Type I tumors are relatively genetically stable and typically display a variety of somatic mutations in
KRAS,
BRAF,
PTEN,
PIK3CA,
CTNNB1, and
ARID1A, but very rarely
P53 [
5]. Type II tumors comprise high-grade serous, high-grade endometrioid, malignant mixed mesodermal tumors (carcinosarcomas), and undifferentiated carcinomas. They are aggressive, present at advanced stages, and have a very high frequency of
TP53 mutations, but rarely harbor the mutations detected in type I tumors [
5].
Recent genome-wide analysis by TCGA, single nucleotide polymorphism arrays identified
KRAS and
MAPK1 as two of the most frequently amplified genes in high-grade serous ovarian carcinomas with the prototypic type in type II ovarian carcinomas [
6]. Gene amplification represents one of the molecular genetic hallmarks of human cancer. Elucidating the molecular mechanisms of how amplified genes initiate and maintain malignant phenotypes and propel tumor progression is fundamental to understanding the molecular etiology of human cancer and its therapeutic implications. Gene amplification is an important mechanism that allows cancer cells to increase expression of driver genes, such as oncogenes, that are involved in growth regulation and genes responsible for drug resistance. Therefore, detection of gene amplification in tumors may be of diagnostic, prognostic, and/or therapeutic relevance for patient management.
Based on the above findings, we hypothesized that KRAS or MAPK1 amplification may play an important role in type II ovarian carcinoma progression and that amplification may correlate more strongly with clinical parameters. Furthermore, type II ovarian carcinomas with KRAS or MAPK1 amplification may be sensitive to a potential targeted therapeutic agent. To test this hypothesis, we undertook the current study to assess gene copy numbers of KRAS or MAPK1 and to evaluate any prognostic significance in patients with type II ovarian carcinoma. In addition, we compared phenotypes in cultured type II ovarian carcinoma cell lines with various KRAS or MAPK1 copy numbers after treatment using a selective MEK inhibitor.
3. Discussion
Type II ovarian carcinomas are the most common and fatal form of ovarian cancer [
9]. While most tumors are highly sensitive to cytoreductive surgery and platinum-and taxane-based chemotherapy, the majority of patients experience recurrence of treatment-resistant tumors. Therefore, a new potential molecular targeted therapy for type II ovarian carcinomas needs to be developed. We reported earlier that
KRAS or
BRAF mutations were quite common in low-grade serous ovarian carcinomas with prototypic histology of type I ovarian carcinoma but rare in high-grade serous ovarian carcinomas [
8,
10]. Our present results showing low frequencies of
KRAS mutations in type II ovarian carcinoma are consistent with our earlier reports [
8,
10].
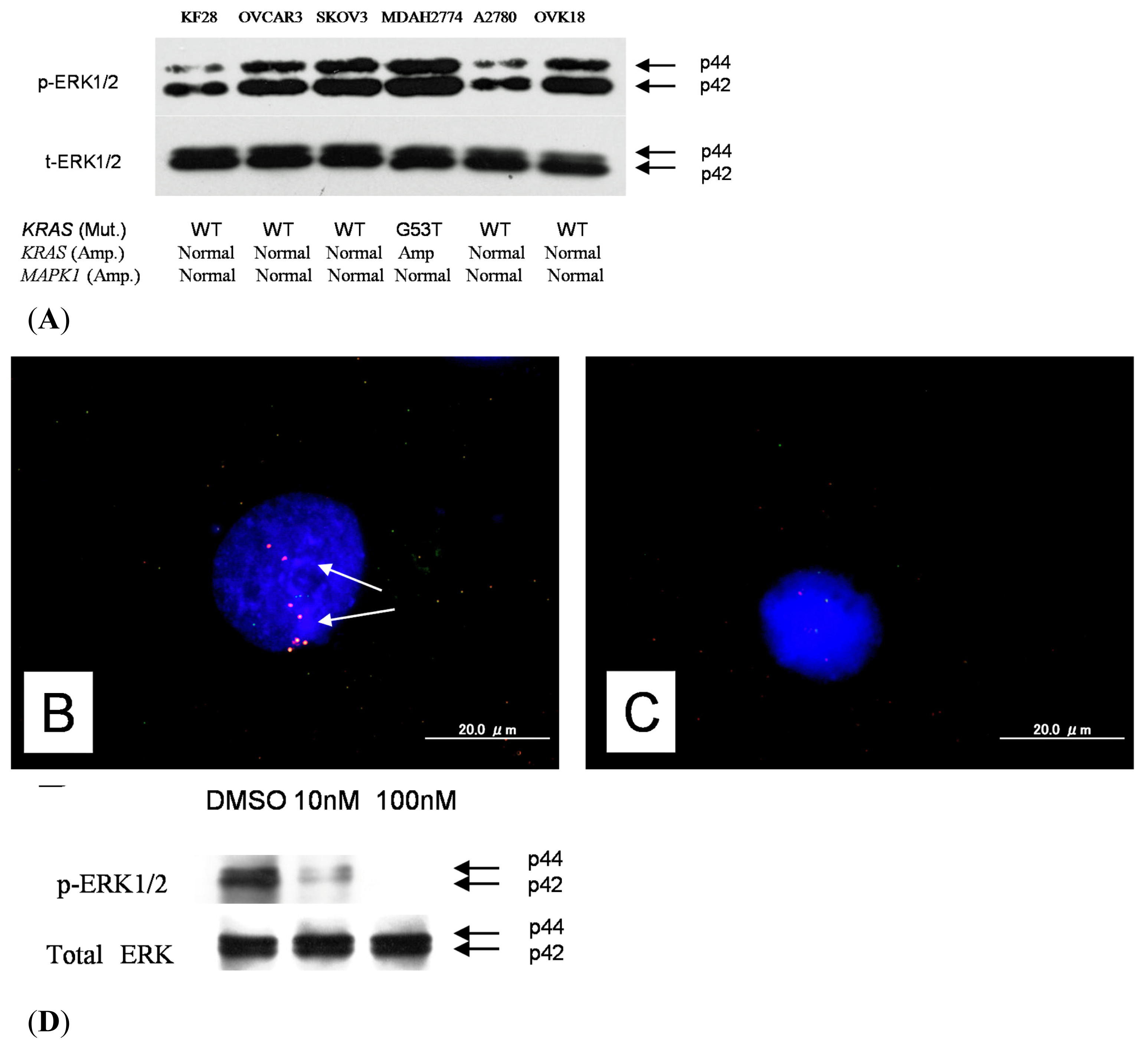
Based on our combined FISH and immunohistochemical analysis, we found that p-ERK was not consistently expressed in all
KRAS or
MAPK1 gene amplification tumors. Why does this discrepancy happen? This may be due to expression of the mitogen-activated protein kinase (MAPK) phosphatase (MKP)-1 in type II ovarian carcinomas. MKP-1 is overexpressed in several types of cancers [
11,
12]. MKP-1 is the prototypic member of a family of dual-specificity phosphatases that dephosphorylate tyrosine and threonine residues on target proteins. This phosphatase is best known for its specificity toward p44/42 MAPK [
13]. Further examination is needed to clarify the relationship
KRAS/MAPK1 amplification and MKP-1 expression. Furthermore, many tumors exhibited positive p-ERK signals in the absence of
KRAS or
MAPK1 amplification. Therefore, gene amplification is probably just one of multiple mechanisms for p-ERK expression. Additionally, the low frequency of
KRAS mutation or
BRAF mutations in type II ovarian carcinomas in the current and previous studies suggest that p-ERK expression may be affected by the other receptor tyrosine kinase regulation mechanisms (
i.e., EGFR, Her2,
etc.) [
8,
14,
15]. This is in consistent with a recent report showing that this pathway is frequently activated independent of the status of
KRAS and
BRAF in endometrioid-type endometrial cancer [
11]. The RAS–RAF–MEK–ERK pathway may be affected by multiple pathways in type II ovarian carcinogenesis and endometrial carcinogenesis. For example, alternative pathways for ERK activation, such as crosstalk with the PI3K pathway, exist in high-grade serous ovarian carcinomas. Indeed, enhanced PI3K signaling due to amplification occurs in 10%–18% of high-grade serous ovarian carcinomas, which is the dominant histology of type II ovarian carcinoma [
6,
10,
16].
Biomarkers that can predict clinical prognosis, including treatment response and overall survival, can have a substantial clinical impact on the management of patients with ovarian carcinoma [
17]. To further explore the clinical relevance of
KRAS or
MAPK1 alterations in type II ovarian carcinomas, we identified a correlation between
KRAS or
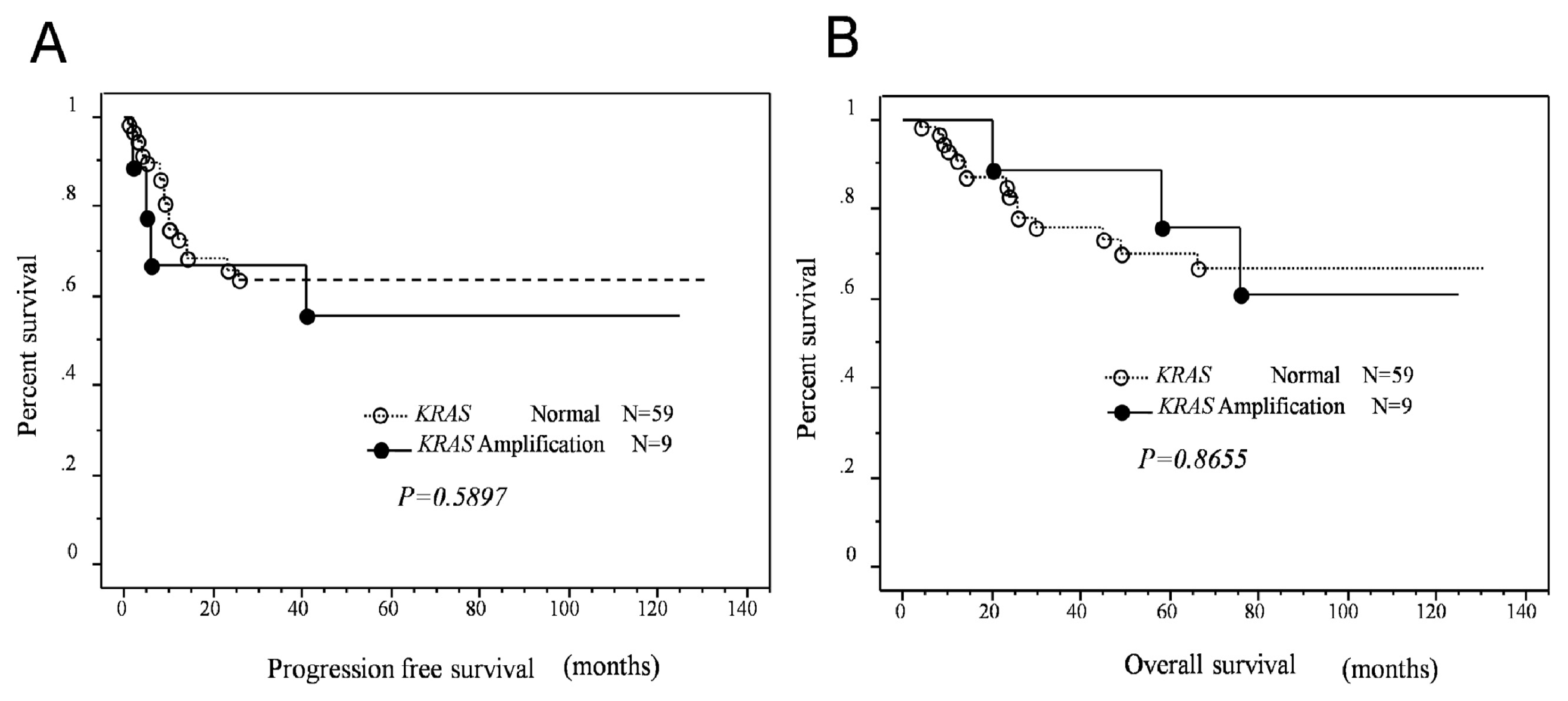
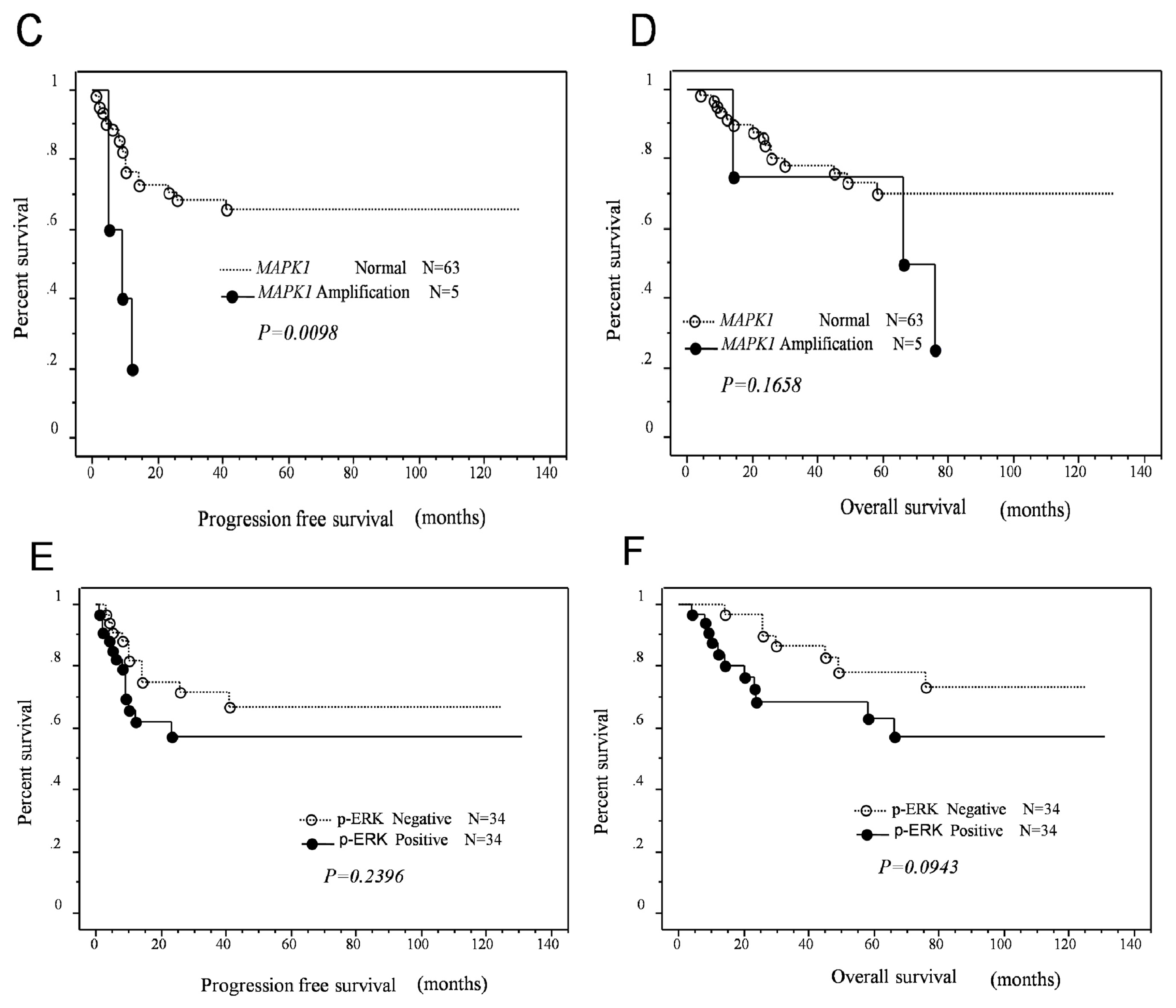
MAPK1 amplification or p-ERK protein expression and length of progression-free/overall survival in patients with type II ovarian carcinomas. Interestingly, a strong correlation between poor progression-free prognosis and
MAPK1 amplification was identified in patients who received taxane- and platinum-based chemotherapy. In contrast to
MAPK1 amplification,
KRAS amplification did not affect patient survival. However, p-ERK protein overexpression was associated with an insignificant trend toward poor progression-free/overall survival. The mechanism underlying the association between
MAPK1 amplification and shorter progression-free survival is not known; however, because mortality of patients with type II ovarian carcinomas is directly related to recurrence of disease after chemotherapy, it is conceivable that
MAPK1 amplification may confer resistance to chemotherapy and/or enhance cell proliferation in recurrent chemoresistant tumors. Current study has several limitations. First, differences in histological heterogeneity in type II ovarian carcinoma, including high-grade serous, high-grade endometriod, and carcinosarcoma used to assess amplification
KRAS/MAPK1 may produce significant variability among results. Second, the sample sizes of this study are relatively small. Larger sample studies are required to definitively establish the percentage of
KRAS/MAPK1 amplification on type II ovarian carcinomas.
In the current study, type II ovarian carcinomas with both
KRAS mutations and
KRAS amplification were highly sensitive to growth inhibition by the selective MEK inhibitor, PD0325901. This observation suggested that type II ovarian carcinoma with concomitant amplification and mutation of
KRAS was more highly dependent on the activation of the RAS/RAF/MEK/ERK pathway for cell proliferation and survival than those without such amplification or mutation. Thus, inactivation of ERK1/2 resulted in marked growth inhibition in ovarian carcinomas with amplification/mutations in
KRAS in comparison with only a slight effect on wild-type tumors. The above observations lend strong support to the view of ‘oncogene addiction’ [
18] by which the activating amplifications in the kinase pathway confer susceptibility of the tumors to an inhibitor [
8,
19,
20]. Recently, Wagner
et al. reported that concomitant
KRAS amplification and mutation enhances the aggressiveness of non-small cell lung carcinoma [
21]. In contrast, in the current study no cases with concomitant
KRAS or
MAPK1 amplification and
KRAS mutation were identified. This discrepancy may be due to differences in organ-specific oncogenic pathways and mechanisms of carcinogenesis. The potential for organ-specific differences in
KRAS amplification and
KRAS mutations in malignant tissues is the subject of ongoing investigation. Although our
in vitro results showed that concomitant
KRAS amplification and mutation was more highly dependent on the activation of the RAS/RAF/MEK/ERK pathway for cell proliferation than those without such amplification or mutation. Considering the above findings, concomitant
KRAS amplification/mutation and function in neoplastic tissues may be dependent on cellular context. In order to fully understand the role of the complex concomitant amplification and mutation of
KRAS in type II ovarian carcinoma physiology, larger studies including functional analyses are required.
4. Experimental Section
4.1. Tissue Samples
Formalin-fixed, paraffin-embedded tissue samples from 68 type II ovarian carcinomas, including 43 high-grade serous, 16 high-grade endometrioid, and 5 carcinosarcoma tumors, were used in this study. Samples were obtained from the Department of Obstetrics and Gynecology at the Shimane University Hospital. Diagnosis was based on conventional morphological examination of sections stained with hematoxylin and eosin (H & E), and tumors were classified according to the WHO classification. Tumor staging was performed according to the International Federation of Gynecology and Obstetrics (FIGO) classification. All patients were primarily treated with cytoreductive surgery and adjuvant platinum and taxane chemotherapy (CBDCA AUC5 with paclitaxel 175 mg/m2 or docetaxel 70 mg/m2). All patients received 6–12 courses of this combination regimen. The acquisition of tumor tissues was approved by the Shimane University Institutional Review Board. Paraffin tissue blocks were organized into tissue microarrays, each made by removing tumor cores measuring 3 mm in diameter from the block. Selection of the area to core was made by a gynecologic oncologist (K.N.) and pathology technician (K.I.) and was based on review of the H & E slides.
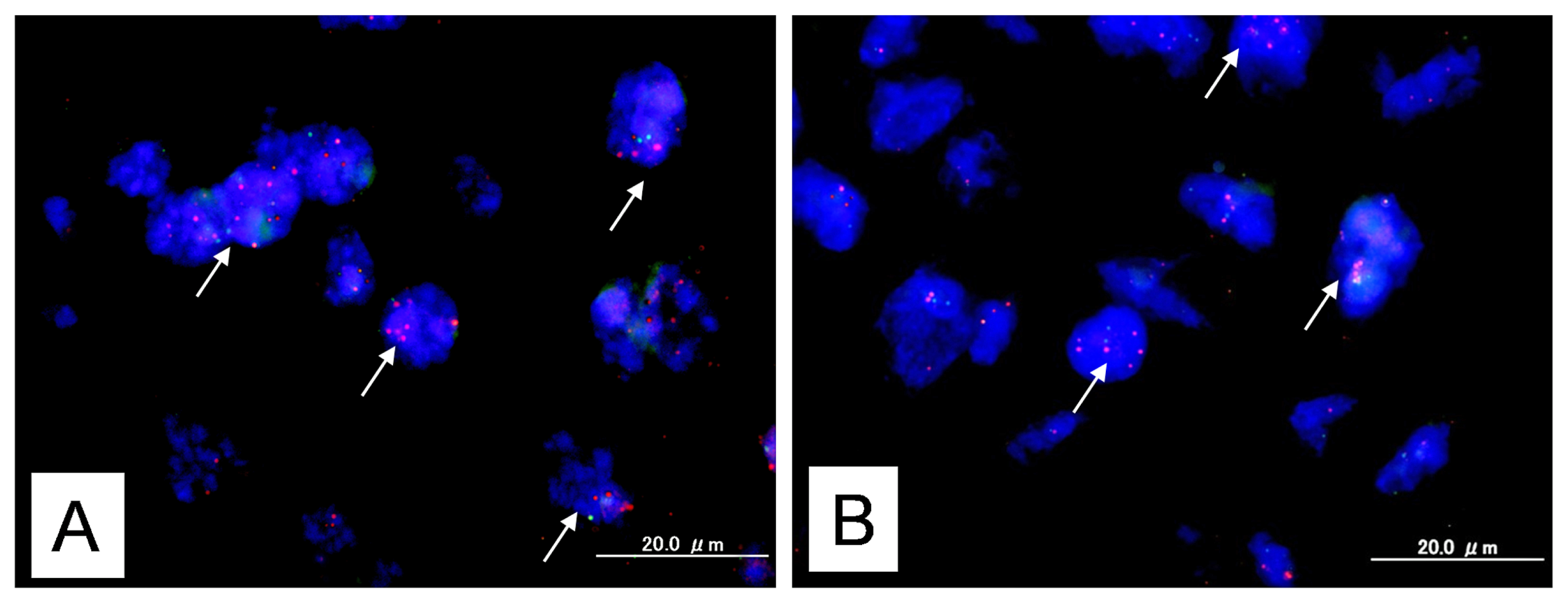
4.2. Fluorescence in Situ Hybridization
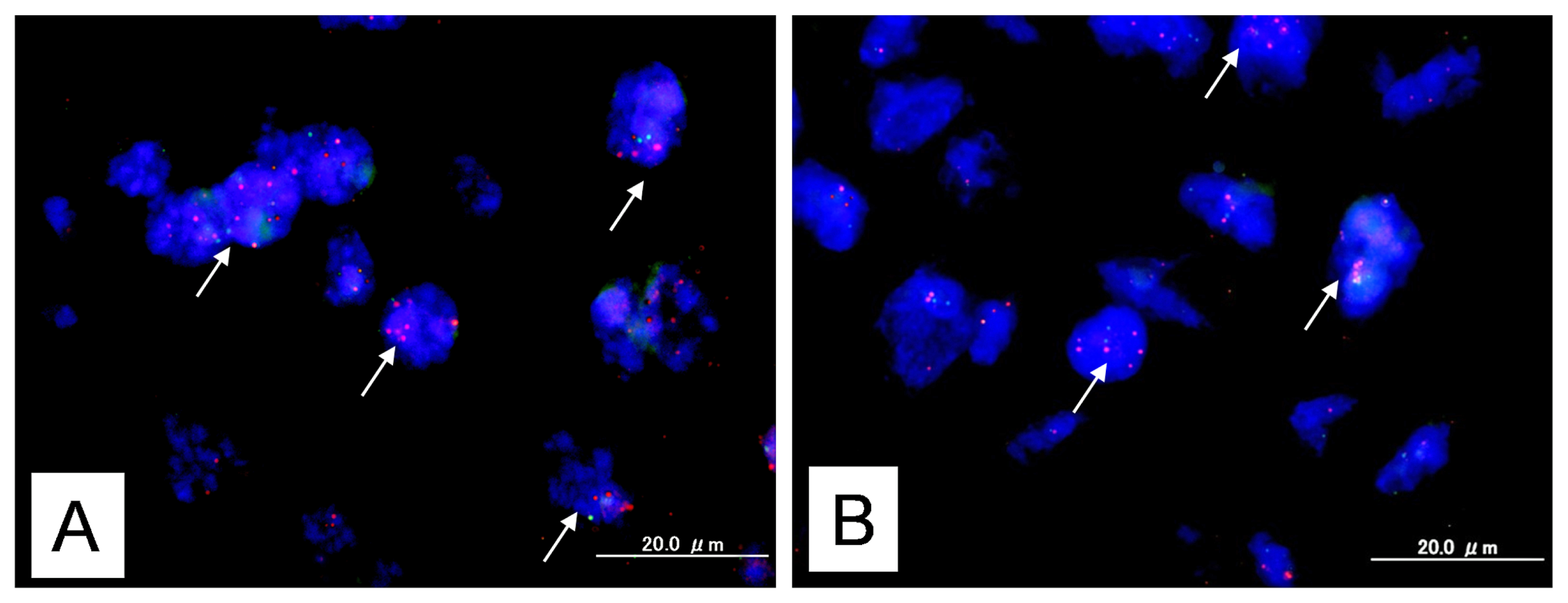
BAC clones (CTD2174F1 and CTD-2536C1) containing the genomic sequences of the 12p12.1 chromosome at the KRAS locus were purchased from Bacpac Resources (Children’s Hospital, Oakland, CA, USA) and Invitrogen (Carlsbad, CA, USA). Bac clones located at Chr12q23.3 (RP11-482D24) were used as reference probes. BAC clones (RP11-647D11 and RP11-1109D18) containing the genomic sequences of the 22q11.21–22q11.22 chromosome at the MAPK1 locus were purchased from Bacpac Resources and Invitrogen. Bac clones located at Chr22q13.32-13.33 (RP11-29C18) were used as reference probes.
CTD2174F1, CTD-2536C1, RP11-647D11, and RP11-1109D18 were labeled by nick translation with biotin-dUTP; RP11-482D24 and RP11-29C18 were labeled similarly with digoxigenin-dUTP. To detect biotin-labeled and digoxigenin-labeled signals, slides were first incubated with FITC-avidin (Vector Laboratories, Burlingame, CA, USA) and anti-digoxigenin mouse antibodies (Roche Molecular Biochemicals, Mannheim, Germany). Slides were subsequently incubated with biotinylated anti-avidin antibodies (Vector Laboratories, Burlingame, CA, USA) and tetramethylrhodamine B isothiocyanate (TRITC)-conjugated rabbit anti-mouse antibodies (Sigma, St. Louis, MO, USA). The final incubation was with FITC-avidin and TRITC-conjugated goat anti-rabbit antibodies (Sigma, St. Louis, MO, USA). Slides were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI, Sigma, St. Louis, MO, USA).
Fluorescence in situ hybridization (FISH) signals were evaluated with an Olympus fluorescence microscope BX41 (Tokyo, Japan) by two individuals who were blinded to the treatment history of each patient. Separate narrow band pass filters were used for detection of TRITC, FITC, and DAPI signals. Using a 60× objective lens, approximately 100 tumor cells were examined for each specimen, and the number of fluorescent signals within tumor cells from KRAS or MAPK1 gene BAC probes and chromosome 12q23.3 or 22q13.32-13.33 reference BAC probe was recorded. Amplification of KRAS or MAPK1 was defined as a ratio of KRAS or MAPK1 BAC probe signals to chromosome 12 or chromosome 22 centromeric reference BAC probe signals of 2:1 or more.
4.3. Mutational Analysis of KRAS
Genomic DNA was purified from all cell lines and formalin-fixed, paraffin-embedded tissues using a Qiaquick polymerase chain reaction (PCR) purification kit (Qiagen, Valencia, CA, USA). PCR was then carried out followed by nucleotide sequencing using the iCycler (Bio-Rad, Hercules, CA, USA). Exon 1 of
KRAS was sequenced, as these mutational hot spots together harbor nearly all published mutations [
22,
23]. The primers for PCR and sequencing were manufactured by GeneLink (Hawthorne, NY, USA), and their sequences were described in an earlier report [
9]. The sequences were analyzed using the Lasergene program, DNASTAR (Madison, WI, USA).
4.4. Immunohistochemistry
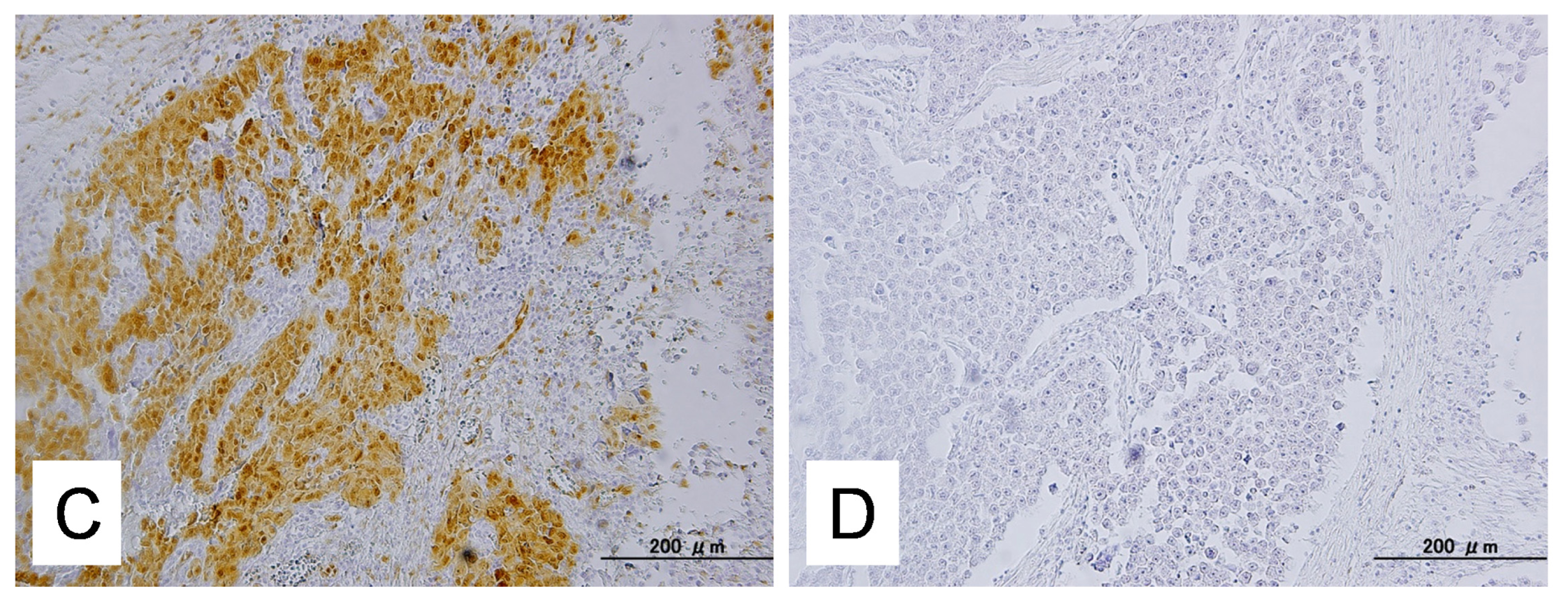
Expression of active phosphorylated ERK1/2 (p-ERK1/2) was assessed by immunohistochemistry and western blot analysis. The antibody used in this study was a rabbit polyclonal antibody that reacted with phosphorylated but not unphosphorylated ERK1/2 (Cell Signaling Technology, Danvers, MA, USA). Immunohistochemistry was carried out on tissue microarrays at a dilution of 1:1000 followed by detection with the En Vision+ System using the peroxidase method (DAKO, Carpinteria, CA, USA). The percentage of positive cells was estimated by randomly counting ~500 tumor cells from 3 different high-power fields (40×) within one specimen. A positive reaction was defined as a discrete localization of the brown chromagen in the nucleus or cytoplasm. Cases in which more than 5% of the tumor cells showed detectable immunoreactivity were scored as positive.
4.5. Western Blot Analysis
Cell lysates were prepared by dissolving cell pellets in Laemmli sample buffer (Bio-Rad) supplemented with 5% β-mercaptoethanol (Sigma). Western blot analysis was performed on type II ovarian carcinoma cell lines, including OVCAR3, SKOV3, A2780, MDAH2774, KF28, and OVK18. Similar amounts of total protein from each lysate were loaded and separated on 10% Tris-Glycine-SDS polyacrylamide gels (Novex, San Diego, CA, USA) and electroblotted to Millipore Immobilon-P polyvinylidene difluoride membranes. The membranes were probed with p-ERK1/2 antibodies (pTEpY, 1:1000; Santa Cruz Biotechnology, CA, USA) followed by peroxidase-conjugated anti-mouse immunoglobulin (1:10,000). The same membrane was probed with an antibody that reacted with total ERK1/2 (1:5000; Santa Cruz Biotechnology, CA, USA) as a loading control. Western blots were developed by chemiluminescence (Pierce, Rockford, IL, USA).
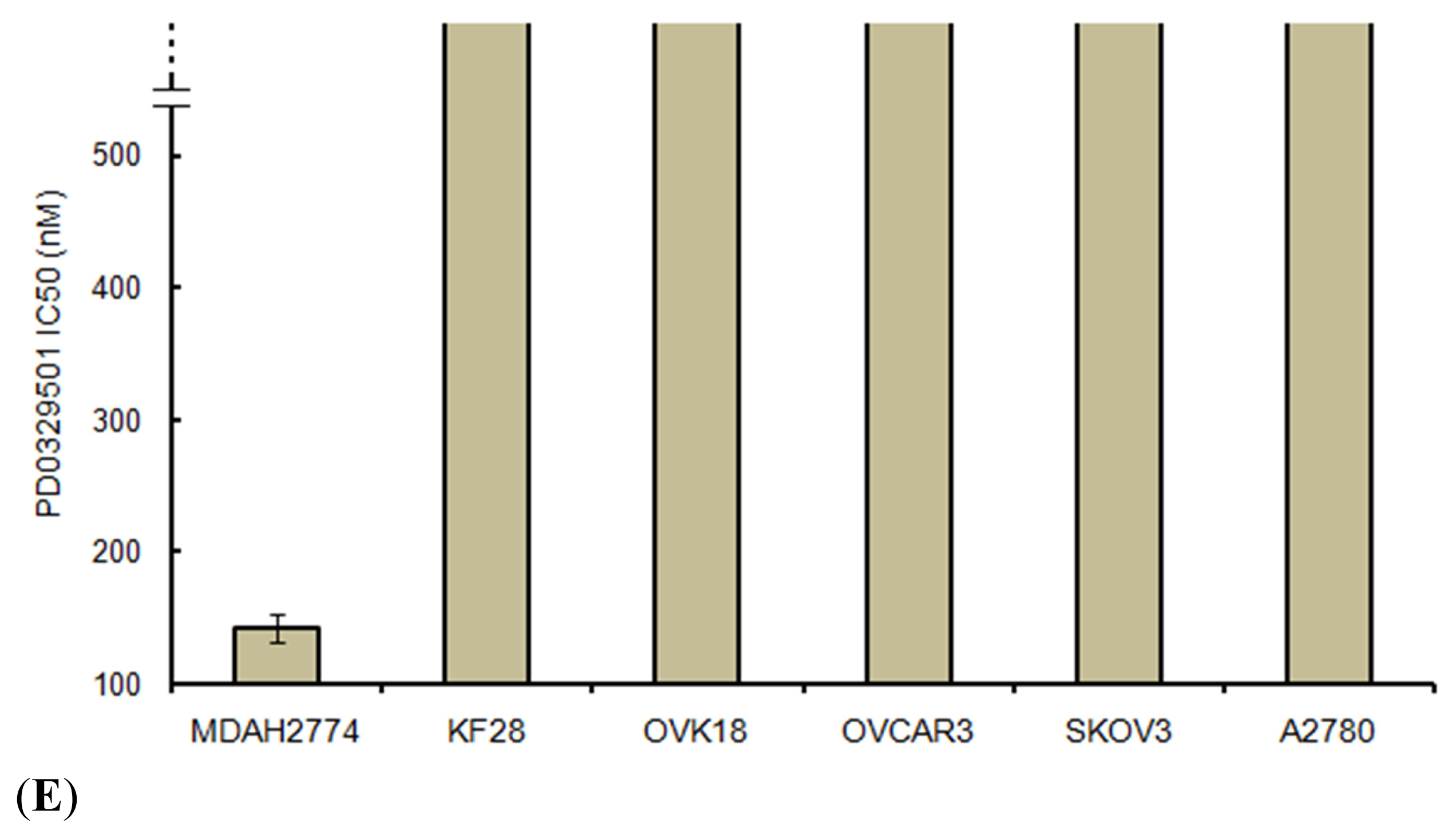
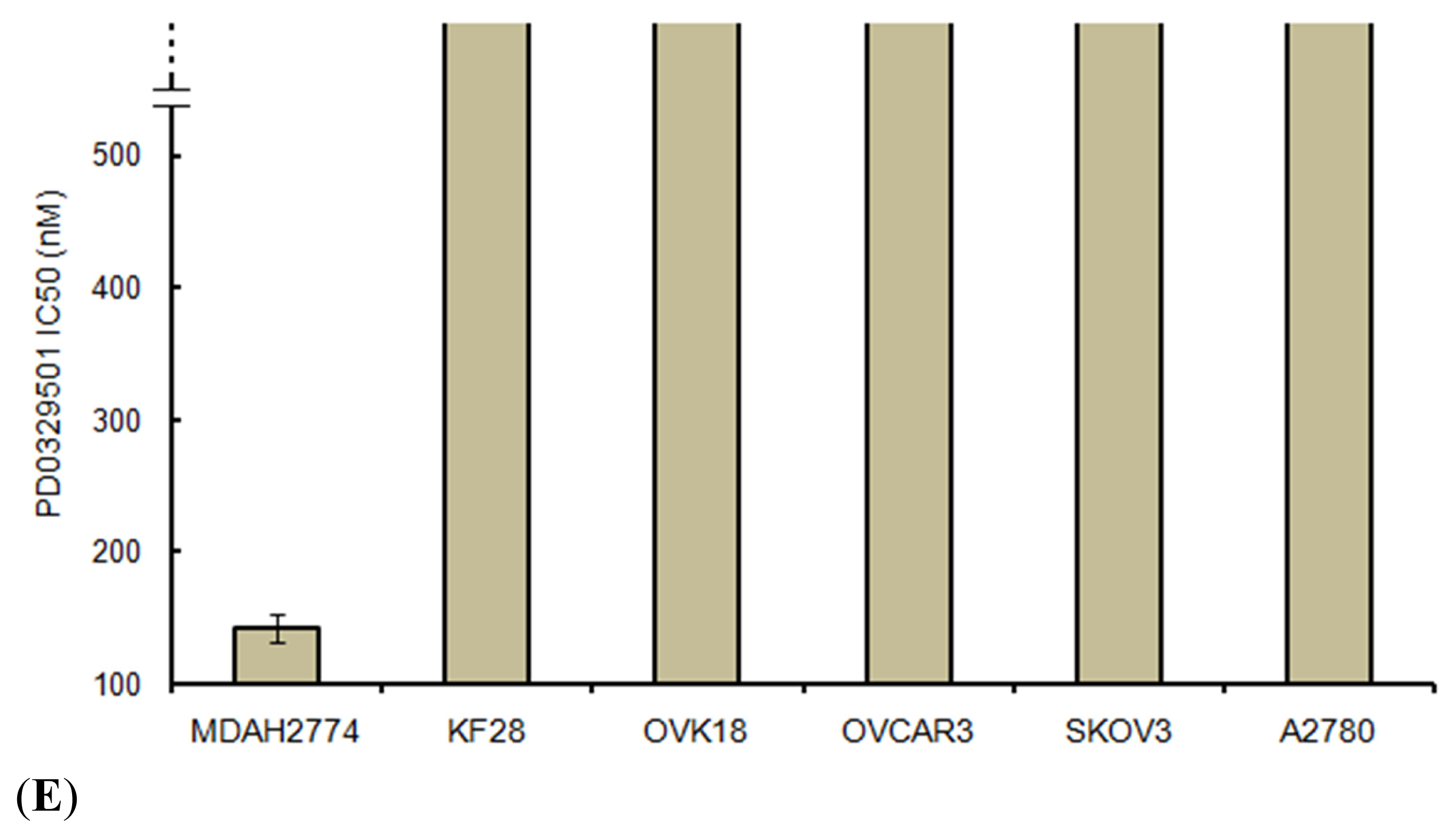
4.6. Cell Growth Assays
For cell growth assays, cells were plated at the same density (5 × 10
3 cells per well) in 96-well plates. A methyl thiazoyl tetrazorium (MTT) cell growth assay was performed [
24] 96 h after treating the cells with PD0325901 (Selleck Chemicals, Houston, TX, USA) at 1–200 nM or with dimethyl sulfoxide (DMSO) as a control. The IC
50 was determined on the basis of dose-response curves by a cytotoxicity assay. The data were expressed as the mean ± SD of triplicates.
4.7. Statistical Methods for Clinical Correlations
Overall survival was calculated from date of diagnosis to date of death or last follow-up. Progression-free survival was defined as the time from the first day of surgery or chemotherapy until the first of either death from any cause or disease progression (based on an increase in the CA 125 levels and/or on the findings of imaging studies). Survival data were plotted as Kaplan-Meier curves, and statistical significance was determined by the Log-rank test. Multivariate prognostic analysis was performed using a Cox proportional hazards model. Data were censored when patients were lost to follow-up. The chi-square test or Fischer’s exact test was used for comparisons of categorical data.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}