Colorectal Carcinogenesis: A Cellular Response to Sustained Risk Environment

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Current Models of Colorectal Cancer
3. Diet and the Normal Colonic Environment
3.1. Fermentation of NSP and RS
3.2. Protein Fermentation and Ammonia Production
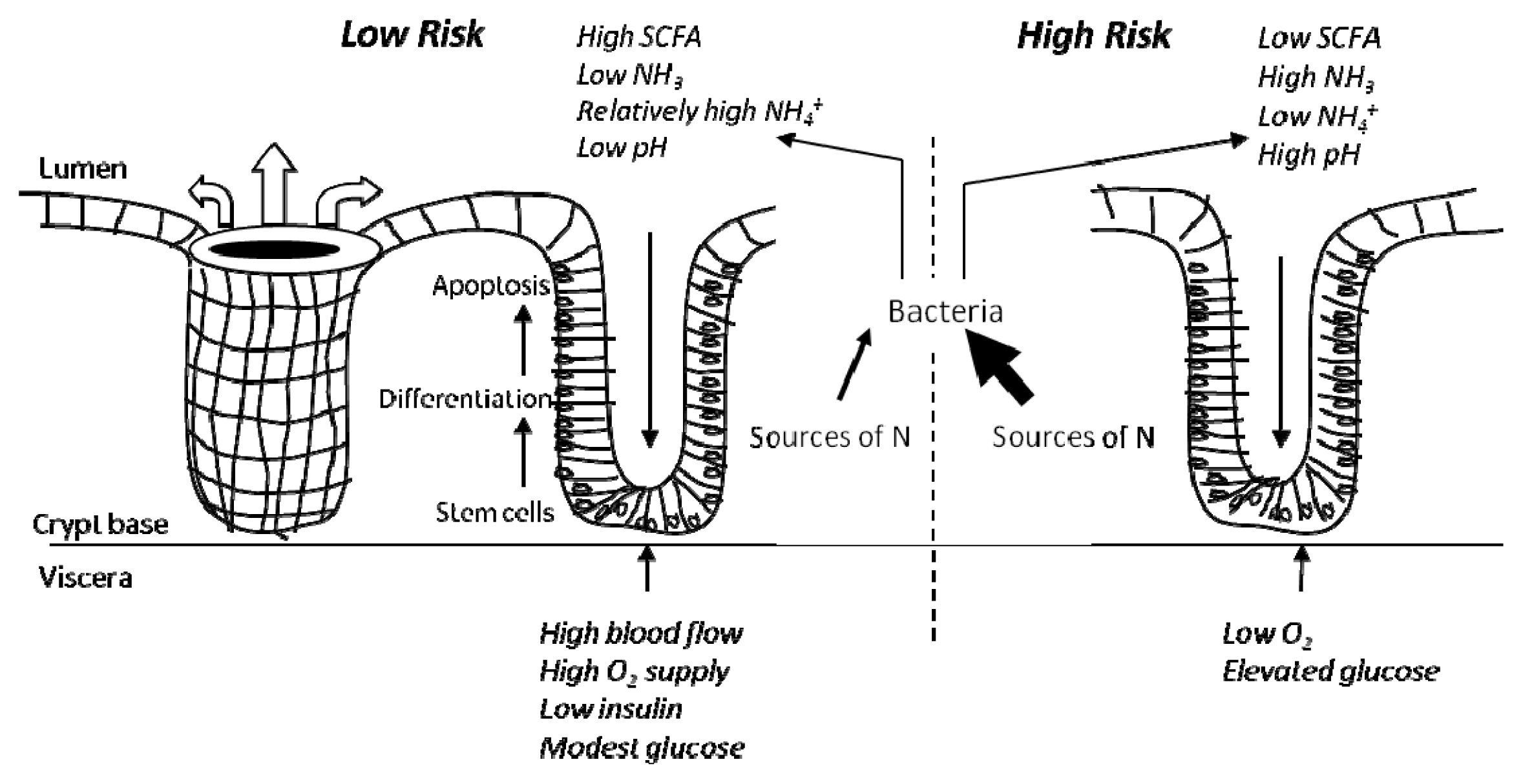
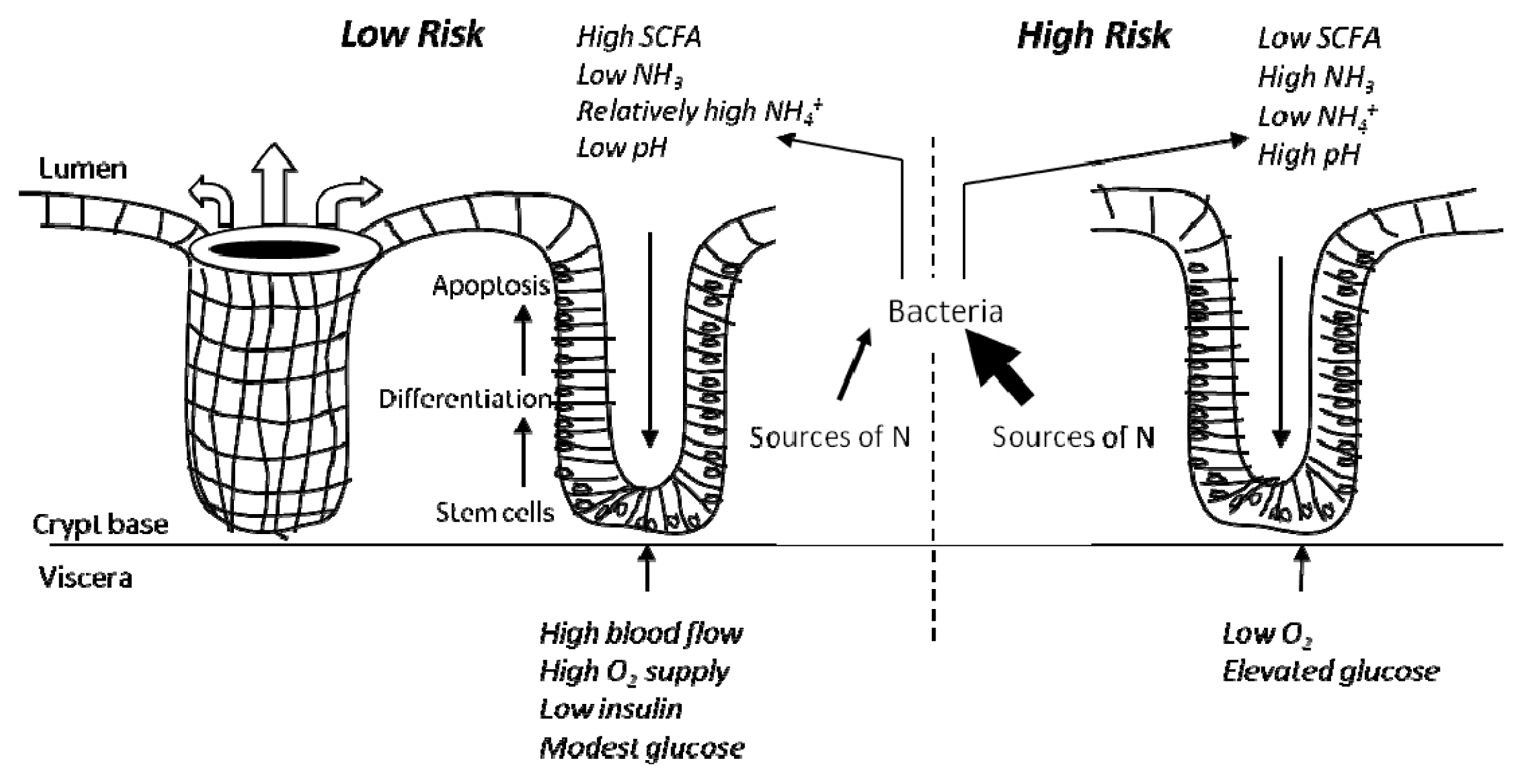
4. Colonic Environment for Risk of Colorectal Cancer and Related Problems
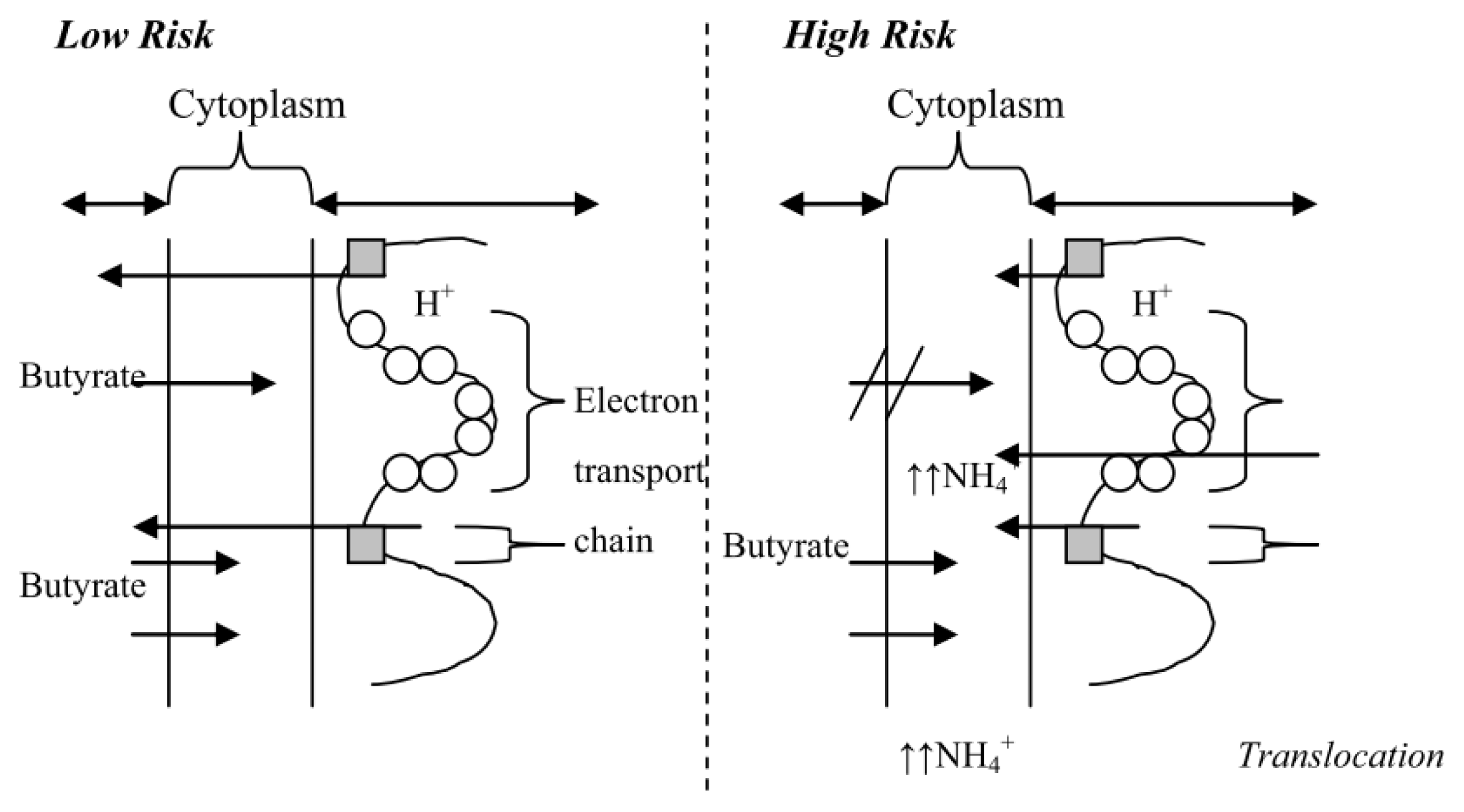
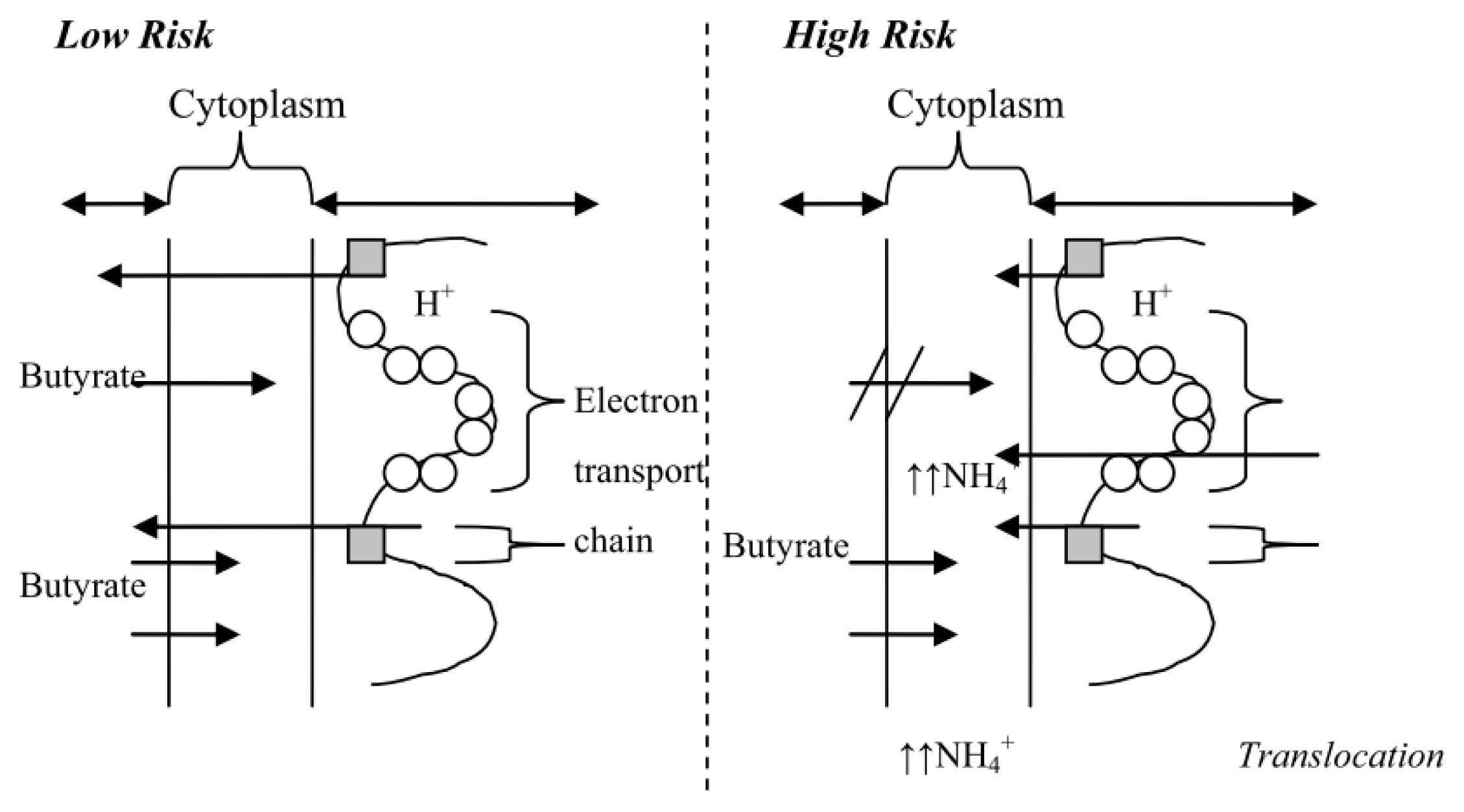
4.1. The High Risk Environment
4.2. Influence of Diet and in vivo Studies
4.3. In vitro Data to Support a “High Risk” Environment
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2011, 127, 2893–2917. [Google Scholar]
- Cancer in Australia: An Overview, 2010. Available online: http://www.aihw.gov.au/publication-detail/?id=6442472459 (on accessed 18 June 2013).
- Ferlay, J.; Parkin, D.M.; Steliarova-Foucher, E. Estimates of cancer incidence and mortality in europe in 2008. Eur. J. Cancer 2010, 46, 765–781. [Google Scholar]
- Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective; World Cancer Research Fund and American Institute for Cancer Research: Washington, WA, USA, 2007.
- Lutz, W.K.; Schlatter, J. Chemical carcinogens and overnutrition in diet-related cancer. Carcinogenesis 1992, 13, 2211–2216. [Google Scholar]
- National Registry of Diseases Office, Singapore Cancer Registry Report No.7. In Trends in Cancer Incidence in Singapore 1968–2007; Health Promotion Board: Singapore, Singapore, 2008.
- Burn, J.; Mathers, J.; Bishop, D.T. Genetics, inheritance and strategies for prevention in populations at high risk of colorectal cancer (crc). Recent Results Cancer Res 2013, 191, 157–183. [Google Scholar]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med 2003, 348, 919–932. [Google Scholar]
- Weitz, J.; Koch, M.; Debus, J.; Hohler, T.; Galle, P.R.; Buchler, M.W. Colorectal cancer. Lancet 2005, 365, 153–165. [Google Scholar]
- Bingham, S.A.; Day, N.E.; Luben, R.; Ferrari, P.; Slimani, N.; Norat, T.; Clavel-Chapelon, F.; Kesse, E.; Nieters, A.; Boeing, H.; et al. Dietary fibre in food and protection against colorectal cancer in the european prospective investigation into cancer and nutrition (epic): An observational study. Lancet 2003, 361, 1496–1501. [Google Scholar]
- Benamouzig, R.; Uzzan, B.; Deyra, J.; Martin, A.; Girard, B.; Little, J.; Chaussade, S. Prevention by daily soluble aspirin of colorectal adenoma recurrence: 4-year results of the apacc randomised trial. Gut 2012, 61, 255–261. [Google Scholar]
- Hughes, L.A.; Simons, C.C.; van den Brandt, P.A.; Goldbohm, R.A.; van Engeland, M.; Weijenberg, M.P. Body size and colorectal cancer risk after 16.3 years of follow-up: An analysis from the netherlands cohort study. Am. J. Epidemiol 2011, 174, 1127–1139. [Google Scholar]
- Ruder, E.H.; Laiyemo, A.O.; Graubard, B.I.; Hollenbeck, A.R.; Schatzkin, A.; Cross, A.J. Non-steroidal anti-inflammatory drugs and colorectal cancer risk in a large, prospective cohort. Am. J. Gastroenterol 2011, 106, 1340–1350. [Google Scholar]
- Alexander, D.D.; Weed, D.L.; Cushing, C.A.; Lowe, K.A. Meta-analysis of prospective studies of red meat consumption and colorectal cancer. Eur. J. Cancer Prev 2011, 20, 293–307. [Google Scholar]
- Anderson, J.C.; Moezardalan, K.; Messina, C.R.; Latreille, M.; Shaw, R.D. Smoking and the association of advanced colorectal neoplasia in an asymptomatic average risk population: Analysis of exposure and anatomical location in men and women. Dig. Dis. Sci 2011, 56, 3616–3623. [Google Scholar]
- Belobrajdic, D.P.; Bird, A.R.; Conlon, M.A.; Williams, B.A.; Kang, S.; McSweeney, C.S.; Zhang, D.; Bryden, W.L.; Gidley, M.J.; Topping, D.L. An arabinoxylan-rich fraction from wheat enhances caecal fermentation and protects colonocyte DNA against diet-induced damage in pigs. Br. J. Nutr 2011, 107, 1–9. [Google Scholar]
- Le Leu, R.K.; Brown, I.L.; Hu, Y.; Morita, T.; Esterman, A.; Young, G.P. Effect of dietary resistant starch and protein on colonic fermentation and intestinal tumourigenesis in rats. Carcinogenesis 2007, 28, 240–245. [Google Scholar]
- Toden, S.; Bird, A.R.; Topping, D.L.; Conlon, M.A. Differential effects of dietary whey, casein and soya on colonic DNA damage and large bowel scfa in rats fed diets low and high in resistant starch. Br. J. Nutr 2007, 97, 535–543. [Google Scholar]
- O’Callaghan, N.J.; Toden, S.; Bird, A.R.; Topping, D.L.; Fenech, M.; Conlon, M.A. Colonocyte telomere shortening is greater with dietary red meat than white meat and is attenuated by resistant starch. Clin. Nutr 2012, 31, 60–64. [Google Scholar]
- Kenar, L.; Karayilanoglu, T.; Aydin, A.; Serdar, M.; Kose, S.; Erbil, M.K. Protective effects of diets supplemented with omega-3 polyunsaturated fatty acids and calcium against colorectal tumor formation. Dig. Dis. Sci 2008, 53, 2177–2182. [Google Scholar]
- Sarotra, P.; Kansal, S.; Sandhir, R.; Agnihotri, N. Chemopreventive effect of different ratios of fish oil and corn oil on prognostic markers, DNA damage and cell cycle in colon carcinogenesis. Eur. J. Cancer Prev 2012, 21, 147–154. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet 1993, 9, 138–141. [Google Scholar]
- Young, G.P.; Hu, Y.; Le Leu, R.K.; Nyskohus, L. Dietary fibre and colorectal cancer: A model for environment—Gene interactions. Mol. Nutr. Food Res 2005, 49, 571–584. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar]
- Radtke, F.; Clevers, H. Self-renewal and cancer of the gut: Two sides of a coin. Science 2005, 307, 1904–1909. [Google Scholar]
- Burkitt, D.P. Epidemiology of cancer of the colon and rectum. Cancer 1971, 28, 3–13. [Google Scholar]
- Goldbohm, R.A.; van den Brandt, P.A.; van’t Veer, P.; Brants, H.A.; Dorant, E.; Sturmans, F.; Hermus, R.J. A prospective cohort study on the relation between meat consumption and the risk of colon cancer. Cancer Res 1994, 54, 718–723. [Google Scholar]
- Willett, W.C.; Stampfer, M.J.; Colditz, G.A.; Rosner, B.A.; Speizer, F.E. Relation of meat, fat, and fiber intake to the risk of colon cancer in a prospective study among women. N. Engl. J. Med 1990, 323, 1664–1672. [Google Scholar]
- Freudenheim, J.L.; Graham, S.; Marshall, J.R.; Haughey, B.P.; Wilkinson, G. A case-control study of diet and rectal cancer in western new york. Am. J. Epidemiol 1990, 131, 612–624. [Google Scholar]
- Potter, J.D.; McMichael, A.J. Diet and cancer of the colon and rectum: A case-control study. J. Natl. Cancer Inst 1986, 76, 557–569. [Google Scholar]
- Burkitt, D.P.; Walker, A.R.; Painter, N.S. Dietary fiber and disease. JAMA 1974, 229, 1068–1074. [Google Scholar]
- Cummings, J.H. Fermentation in the human large intestine: Evidence and implications for health. Lancet 1983, 1, 1206–1209. [Google Scholar]
- Giovannucci, E.; Stampfer, M.J.; Colditz, G.; Rimm, E.B.; Willett, W.C. Relationship of diet to risk of colorectal adenoma in men. J. Natl. Cancer Inst 1992, 84, 91–98. [Google Scholar]
- Heilbrun, L.K.; Nomura, A.; Hankin, J.H.; Stemmermann, G.N. Diet and colorectal cancer with special reference to fiber intake. Int. J. Cancer 1989, 44, 1–6. [Google Scholar]
- Park, Y.; Hunter, D.J.; Spiegelman, D.; Bergkvist, L.; Berrino, F.; van den Brandt, P.A.; Buring, J.E.; Colditz, G.A.; Freudenheim, J.L.; Fuchs, C.S.; et al. Dietary fiber intake and risk of colorectal cancer: A pooled analysis of prospective cohort studies. JAMA 2005, 294, 2849–2857. [Google Scholar]
- Topping, D. The contribution of resistant starch to health benefits of dietary fibre. J. Japan Assoc. Diet. Fiber Res 2012, 16, s11–s19. [Google Scholar]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar]
- Robles Alonso, V.; Guarner, F. Linking the gut microbiota to human health. Br. J. Nutr 2013, 109, S21–S26. [Google Scholar]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar]
- Szelenyi-Galatai, M.; Febel, H.; Szegedi, B.; Zsolnai-Harczi, I.; Huszar, S. Evaluation of the digestibility of nutrients in pigs by ileal cannulation technique. Acta Vet. Hung 1996, 44, 411–432. [Google Scholar]
- Topping, D.L.; Segal, I.; Regina, A.; Bajka, B.H.; Toden, S.; Clarke, J.M.; Morell, M.K.; Bird, A.R. Resistant Starch and Human Health. In Dietary Fibre: New Frontiers in Food and Health; van der Kamp, J.W., Jones, J., McCleary, B., Topping, D., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2010; pp. 311–321. [Google Scholar]
- Segal, I. Physiological small bowel malabsorption of carbohydrates protects against large bowel diseases in africans. J. Gastroenterol. Hepatol 2002, 17, 249–252. [Google Scholar]
- Scheppach, W. Effects of short chain fatty acids on gut morphology and function. Gut 1994, 35, S35–S38. [Google Scholar]
- McNeil, N.I.; Cummings, J.H.; James, W.P. Short chain fatty acid absorption by the human large intestine. Gut 1978, 19, 819–822. [Google Scholar]
- Al-Lahham, S.H.; Peppelenbosch, M.P.; Roelofsen, H.; Vonk, R.J.; Venema, K. Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochim. Biophys. Acta 2010, 1801, 1175–1183. [Google Scholar]
- O’Keefe, S.J.; Chung, D.; Mahmoud, N.; Sepulveda, A.R.; Manafe, M.; Arch, J.; Adada, H.; van der Merwe, T. Why do african Americans get more colon cancer than native Africans? J. Nutr 2007, 137, 175S–182S. [Google Scholar]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev 2001, 81, 1031–1064. [Google Scholar]
- Yajima, T. Contractile effect of short-chain fatty acids on the isolated colon of the rat. J. Physiol 1985, 368, 667–678. [Google Scholar]
- Boren, J.; Lee, W.N.; Bassilian, S.; Centelles, J.J.; Lim, S.; Ahmed, S.; Boros, L.G.; Cascante, M. The stable isotope-based dynamic metabolic profile of butyrate-induced ht29 cell differentiation. J. Biol. Chem 2003, 278, 28395–28402. [Google Scholar]
- Krishnan, S.; Ramakrishna, B.S.; Binder, H.J. Stimulation of sodium chloride absorption from secreting rat colon by short-chain fatty acids. Dig. Dis. Sci 1999, 44, 1924–1930. [Google Scholar]
- Cats, A.; Devries, E.; Mulder, N.; Kleibeuker, J. Regional differences of physiological functions and cancer susceptibility in the human large intestine. Int. J. Oncol 1996, 9, 1055–1069. [Google Scholar]
- Lin, H.C.; Visek, W.J. Colon mucosal cell damage by ammonia in rats. J. Nutr 1991, 121, 887–893. [Google Scholar]
- Visek, W.J. Diet and cell growth modulation by ammonia. Am. J. Clin. Nutr 1978, 31, S216–S220. [Google Scholar]
- Wrong, O.M.; Vince, A.J.; Waterlow, J.C. The contribution of endogenous urea to faecal ammonia in man, determined by 15n labelling of plasma urea. Clin. Sci. (Lond) 1985, 68, 193–199. [Google Scholar]
- Vince, A.; Dawson, A.M.; Park, N.; O’Grady, F. Ammonia production by intestinal bacteria. Gut 1973, 14, 171–177. [Google Scholar]
- Birkett, A.; Muir, J.; Phillips, J.; Jones, G.; O’Dea, K. Resistant starch lowers fecal concentrations of ammonia and phenols in humans. Am. J. Clin. Nutr 1996, 63, 766–772. [Google Scholar]
- Dykhuizen, R.S.; Fraser, A.; McKenzie, H.; Golden, M.; Leifert, C.; Benjamin, N. Helicobacter pylori is killed by nitrite under acidic conditions. Gut 1998, 42, 334–337. [Google Scholar]
- Shimizu, T.; Marusawa, H.; Endo, Y.; Chiba, T. Inflammation-mediated genomic instability: Roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci 2012, 103, 1201–1206. [Google Scholar]
- Izzotti, A.; Durando, P.; Ansaldi, F.; Gianiorio, F.; Pulliero, A. Interaction between helicobacter pylori, diet, and genetic polymorphisms as related to non-cancer diseases. Mutat. Res 2009, 667, 142–157. [Google Scholar]
- Fukushi, K.; Ito, H.; Kimura, K.; Yokota, K.; Saito, K.; Chayama, K.; Takeda, S.; Wakida, S. Determination of ammonium in river water and sewage samples by capillary zone electrophoresis with direct uv detection. J. Chromatogr. A 2006, 1106, 61–66. [Google Scholar]
- Karpinets, T.V.; Foy, B.D. Tumorigenesis: The adaptation of mammalian cells to sustained stress environment by epigenetic alterations and succeeding matched mutations. Carcinogenesis 2005, 26, 1323–1334. [Google Scholar]
- Lewis, S.J.; Heaton, K.W. Increasing butyrate concentration in the distal colon by accelerating intestinal transit. Gut 1997, 41, 245–251. [Google Scholar]
- Silviera, M.L.; Smith, B.P.; Powell, J.; Sapienza, C. Epigenetic differences in normal colon mucosa of cancer patients suggest altered dietary metabolic pathways. Cancer Prev. Res. (Phila) 2012, 5, 374–384. [Google Scholar]
- Visek, W.J. Effects of urea hydrolysis on cell life-span and metabolism. Feder. Proc 1972, 31, 1178–1193. [Google Scholar]
- Karpinets, T.V.; Foy, B.D. Model of the developing tumorigenic phenotype in mammalian cells and the roles of sustained stress and replicative senescence. J. Theor. Biol 2004, 227, 253–264. [Google Scholar]
- Taylor, R.W.; Barron, M.J.; Borthwick, G.M.; Gospel, A.; Chinnery, P.F.; Samuels, D.C.; Taylor, G.A.; Plusa, S.M.; Needham, S.J.; Greaves, L.C.; et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest 2003, 112, 1351–1360. [Google Scholar]
- Kim, Y.H.; Park, J.W.; Lee, J.Y.; Kwon, T.K. Sodium butyrate sensitizes trail-mediated apoptosis by induction of transcription from the dr5 gene promoter through sp1 sites in colon cancer cells. Carcinogenesis 2004, 25, 1813–1820. [Google Scholar]
- Pajak, B.; Gajkowska, B.; Orzechowski, A. Sodium butyrate sensitizes human colon adenocarcinoma colo 205 cells to both intrinsic and tnf-alpha-dependent extrinsic apoptosis. Apoptosis 2009, 14, 203–217. [Google Scholar]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar]
- Wang, L.; Luo, H.S.; Xia, H. Sodium butyrate induces human colon carcinoma ht-29 cell apoptosis through a mitochondrial pathway. J. Int. Med. Res 2009, 37, 803–811. [Google Scholar]
- Kurtovic, J.; Liu, Q.; Duan, Z.P.; Ha, D.K.; Bengmark, S.; Riordan, S.M. Reply. Hepatology (Baltimore, Md. ) 2005, 41, 219. [Google Scholar]
- Lin, H.C.; Visek, W.J. Large intestinal ph and ammonia in rats: Dietary fat and protein interactions. J. Nutr 1991, 121, 832–843. [Google Scholar]
- Xiao, R.; Badger, T.M.; Simmen, F.A. Dietary exposure to soy or whey proteins alters colonic global gene expression profiles during rat colon tumorigenesis. Mol. Cancer 2005, 4, 1. [Google Scholar]
- Morita, T.; Kasaoka, S.; Ohhashi, A.; Ikai, M.; Numasaki, Y.; Kiriyama, S. Resistant proteins alter cecal short-chain fatty acid profiles in rats fed high amylose cornstarch. J. Nutr 1998, 128, 1156–1164. [Google Scholar]
- Duncan, S.H.; Belenguer, A.; Holtrop, G.; Johnstone, A.M.; Flint, H.J.; Lobley, G.E. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol 2007, 73, 1073–1078. [Google Scholar]
- Lewin, M.H.; Bailey, N.; Bandaletova, T.; Bowman, R.; Cross, A.J.; Pollock, J.; Shuker, D.E.; Bingham, S.A. Red meat enhances the colonic formation of the DNA adduct o6-carboxymethyl guanine: Implications for colorectal cancer risk. Cancer Res 2006, 66, 1859–1865. [Google Scholar]
- Tan, S.L.; Gerber, J.P.; Cosgrove, L.J.; Lockett, T.J.; Clarke, J.M.; Williams, D.B.; Head, R.J. Is the tissue persistence of o(6)-methyl-2′-deoxyguanosine an indicator of tumour formation in the gastrointestinal tract? Mutat. Res 2011, 721, 119–126. [Google Scholar]
- Fenech, M.; El-Sohemy, A.; Cahill, L.; Ferguson, L.R.; French, T.A.; Tai, E.S.; Milner, J.; Koh, W.P.; Xie, L.; Zucker, M.; et al. Nutrigenetics and nutrigenomics: Viewpoints on the current status and applications in nutrition research and practice. J. Nutrigenet. Nutrigenomics 2011, 4, 69–89. [Google Scholar]
- Fung, K.Y.; Lewanowitsch, T.; Henderson, S.T.; Priebe, I.; Hoffmann, P.; McColl, S.R.; Lockett, T.; Head, R.; Cosgrove, L.J. Proteomic analysis of butyrate effects and loss of butyrate sensitivity in ht29 colorectal cancer cells. J. Proteome Res 2009, 8, 1220–1227. [Google Scholar]
- Lambert, D.W.; Wood, I.S.; Ellis, A.; Shirazi-Beechey, S.P. Molecular changes in the expression of human colonic nutrient transporters during the transition from normality to malignancy. Br. J. Cancer 2002, 86, 1262–1269. [Google Scholar]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar]
- Kerr, C.A.; Dunne, R.; Hines, B.M.; Zucker, M.; Cosgrove, L.; Ruszkiewicz, A.; Lockett, T.; Head, R. Measuring the combinatorial expression of solute transporters and metalloproteinases transcripts in colorectal cancer. BMC Res. Notes 2009, 2, 164. [Google Scholar]
- Lopez de Silanes, I.; Olmo, N.; Turnay, J.; Gonzalez de Buitrago, G.; Perez-Ramos, P.; Guzman-Aranguez, A.; Garcia-Diez, M.; Lecona, E.; Gorospe, M.; Lizarbe, M.A. Acquisition of resistance to butyrate enhances survival after stress and induces malignancy of human colon carcinoma cells. Cancer Res 2004, 64, 4593–4600. [Google Scholar]
- Chiaro, C.; Lazarova, D.L.; Bordonaro, M. Tcf3 and cell cycle factors contribute to butyrate resistance in colorectal cancer cells. Biochem. Biophys. Res. Commun 2012, 428, 121–126. [Google Scholar]
- Goncalves, P.; Gregorio, I.; Catarino, T.A.; Martel, F. The effect of oxidative stress upon the intestinal epithelial uptake of butyrate. Eur. J. Pharmacol 2012, 699, 88–100. [Google Scholar]
- Goncalves, P.; Gregorio, I.; Martel, F. The short-chain fatty acid butyrate is a substrate of breast cancer resistance protein. Am. J. Physiol. Cell. Physiol 2011, 301, C984–C994. [Google Scholar]
- Sun, B.; Liu, R.; Xiao, Z.D.; Zhu, X. C-met protects breast cancer cells from apoptosis induced by sodium butyrate. PLoS One 2012, 7, e30143. [Google Scholar]
- Mathers, J.C.; Movahedi, M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of resistant starch on cancer risk in carriers of hereditary colorectal cancer: An analysis from the capp2 randomised controlled trial. Lancet Oncol 2012, 13, 1242–1249. [Google Scholar]
- Bajka, B.H.; Clarke, J.M.; Cobiac, L.; Topping, D.L. Butyrylated starch protects colonocyte DNA against dietary protein-induced damage in rats. Carcinogenesis 2008, 29, 2169–2174. [Google Scholar]
- Segal, I. Rarity of colorectal adenomas in the african black population. Eur. J. Cancer Prev 1998, 7, 387–391. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fung, K.Y.C.; Ooi, C.C.; Zucker, M.H.; Lockett, T.; Williams, D.B.; Cosgrove, L.J.; Topping, D.L. Colorectal Carcinogenesis: A Cellular Response to Sustained Risk Environment. Int. J. Mol. Sci. 2013, 14, 13525-13541. https://doi.org/10.3390/ijms140713525
Fung KYC, Ooi CC, Zucker MH, Lockett T, Williams DB, Cosgrove LJ, Topping DL. Colorectal Carcinogenesis: A Cellular Response to Sustained Risk Environment. International Journal of Molecular Sciences. 2013; 14(7):13525-13541. https://doi.org/10.3390/ijms140713525
Chicago/Turabian StyleFung, Kim Y. C., Cheng Cheng Ooi, Michelle H. Zucker, Trevor Lockett, Desmond B. Williams, Leah J. Cosgrove, and David L. Topping. 2013. "Colorectal Carcinogenesis: A Cellular Response to Sustained Risk Environment" International Journal of Molecular Sciences 14, no. 7: 13525-13541. https://doi.org/10.3390/ijms140713525
APA StyleFung, K. Y. C., Ooi, C. C., Zucker, M. H., Lockett, T., Williams, D. B., Cosgrove, L. J., & Topping, D. L. (2013). Colorectal Carcinogenesis: A Cellular Response to Sustained Risk Environment. International Journal of Molecular Sciences, 14(7), 13525-13541. https://doi.org/10.3390/ijms140713525