1. Introduction
Nanoparticles (NPs) can act as core materials, providing a useful platform for surface functionalisation, for applications in biomedicine, catalysis, and electronic devices [
1]. The use of composite NPs in analytical chemistry has also been specifically reviewed [
2].
Since the early 2000s, much effort has been made to combine the intrinsic properties of biomolecules with the unusual and size-dependent material properties of NPs [
3]. When integrated with biomolecules, NPs with metallic, metal oxide (MO), semiconductor, or silica cores have been used in a wide range of applications in biology and medicine [
4,
5]. Moreover, Au NPs are emerging as therapeutics in their own right, with the capability of modulating cellular processes based on their surface functionality [
6].
A recent review summarised the synthesis and biofunctionalisation of inorganic metal, semiconductor and magnetic nanoparticles for applications in many areas of biomedicine, from diagnostics to the treatment of diseases [
7]. The rapid detection of Hendra virus using magnetic particles and quantum dots has also been reported recently [
8].
A special issue on theranostic nanomedicine [
9] listed twenty-eight accounts of various inorganic NPs, lipid aggregates and synthetic polymer systems, detailing the physicochemical properties of these biocompatible NP platforms. Unfortunately, however, the very active research on new nanomaterials that are potentially useful in medicine has not been counterbalanced by an adequate knowledge of their pharmacokinetics and toxicity [
10].
In this study, we describe how to engineer transition metal oxide NPs of the three ferromagnetic elements, Fe, Co and Ni (i.e., Fe3O4, Co3O4, and NiO) with heparin, with the aim of merging the properties of the individual components.
The natural occurring anticoagulant heparin is a heterogeneous, polydisperse, highly sulphated glycosaminoglycan composed of 1→4 linked disaccharide repeating units, which consist of an α-
d-glucosamine (A) and a hexuronic acid, α-
l-iduronic (I) or β-
d-glucuronic (G) acid unit, with
O-sulphate groups at different positions on the disaccharide unit [
11]. The most frequently occurring repeating disaccharide sequence is α-
l-iduronic acid-2-
O-sulphate-α-
D-glucosamine-
N,6-disulphate (I2S-ANS,6S), which represents the highly sulphated segment of heparin, whereas under-sulphated sequences contain ANAc (α-
d-glucosamine-
N-acetyl) and non-sulphated, I and G. Heparin and low molecular weight heparin have both been recognised as anti-angiogenic agents and vectors that are able to bind proteins that are involved and over-expressed in the tumour microenvironment [
12,
13]. Some chemically modified heparin species have also been reported to exert a beneficial effect in cancer patients, regardless of the mode of action [
14].
Iron oxide (Fe
3O
4) and Fe-based alloy NPs, such as iron-cobalt (FeCo), provide a unique nanoplatform for theranostic applications because of their biocompatibility, their responses to external magnetic fields, and their sizes, which are comparable to those of functional biomolecules [
15]. Furthermore, because of the ease of fabrication and their approved clinical usage, Fe
3O
4 NPs with controlled sizes and controlled surface chemistry have been studied extensively for Magnetic Resonance Imaging (MRI) and Magnetic Fluid Hyperthermia (MFH) applications [
16,
17]. Superparamagnetic Iron Oxide NPs (SPION) were recently introduced for MRI, drug delivery, drug-targeting, and hyperthermia through intra- or extra-venous injection [
18–
20]. Surface engineering of iron oxide NPs has been widely studied for targeted cancer therapy [
21].
As reported by Klostergaard [
22], based on the advanced stage of preclinical human tumour xenograft studies with Fe-based magnetically responsive nanoparticles, there is great confidence that the broader requirements for moving forward to clinical applicability will be met. Therefore, our interest focuses on Fe
3O
4.
The same paper also investigated Co
3O
4 and NiO and discussed their toxicity and the risk of their use. Although we are conscious of the toxicity aspects of administering NPs [
23–
25], the many applications of Co
3O
4 and NiO nanohybrids prompted us to prepare and characterise metal oxide-heparin nanohybrids. The presence of cobalt and nickel within biofunctional magnetic NPs for the manipulation of proteins and cells supported our challenge [
26].
On the nanoscale, Co
3O
4 exhibits magnetic, optical, field emission and electrochemical properties that are attractive in device applications [
27–
29]. Cobalt ferrite (mixed cobalt iron oxide) NP formation was studied in the presence of a synthetic polypeptide, and coating this material with histidine induced cellular biocompatibility [
30,
31]. NiO can form part of electrochemical devices, in which Ni ions are also used as an agent for chelating biomolecules [
32,
33]. A nonenzymatic glucose sensor based on a nickel(II)oxide/ordered mesoporous carbon-modified glassy carbon electrode has also been recently reported [
34].
Concerning polysaccharides, dextran-coated iron oxide NPs have been proposed as a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy [
35].
In addition, Chertok
et al. [
36] argued clearly how and why iron oxide NPs could be promising drug delivery vehicles for MRI-monitored magnetic targeting of brain tumours. Currently, we focus on a heparin skeleton with the aim of taking advantage of the multiple roles that heparin can play. In particular, the heparin coating could specifically direct iron oxide NPs to a tumour mass microenvironment, where their paramagnetic properties can be simultaneously exploited for MRI and hyperthermia, and could exert non-conventional drug activity inside the tumour cells because of the ability of the NPs to cross the cell membrane.
2. Results and Discussion
Complex nanostructures of inorganic elements coated by polymers have been covalently linked to heparin through spacers [
37,
38]. Covalent linkages between nanomaterials and biomolecules often lead to a reduction in the biological activity of the biomolecules once attached to the nanoparticle’s surface [
39,
40]. Thus, non-covalent bioconjugation techniques, based on hydrogen bonding, electrostatic interactions and antibody–antigen interactions, are often advantageous for the biofunctionalisation of nanomaterials [
41,
42].
In this work, the creation of MO@heparin NPs was achieved via the electrostatic attachment of heparin to the surface of NPs. Because heparin was not manipulated, its structure in MO@heparin NPs was reasonably preserved. Another example of non-covalent heparin bioconjugation on poly-
l-lysine-coated iron oxide NPs, [
43] has been published, to which we refer in detail in the discussion.
MO@heparin NPs were prepared, as detailed in the experimental section, using a one-pot coating reaction at room temperature; by this method, the formation of any by-products was avoided, and the excess of heparin as a starting material could be easily recovered and recycled. The washing procedure with diethyl ether removed water and residual non-linked heparin and provided the final MO@heparin NPs which were characterised using a colorimetric assay, FT-IR spectroscopy, DLS and zeta potential measurements, TEM and 1H NMR spectroscopy.
The NP weight increase observed after coating (
Table 1) indicates the order of magnitude of the heparin:metal ratio.
Gravimetric data can sometimes be underestimated because of the difficulty of recovering and handling the wet material. Therefore, the toluidine blue (TB) colorimetric method, used to assay the heparin content in immobilised heparin preparations, was tested on heparin-coated NPs [
44]. This method is based on the metachromatic effect induced by heparin sulphate groups on TB in the 400 to 650 nm wavelength range. All of the coated NPs were effective at inducing the metachromatic effect. The same experiment performed with the addition of bare NPs did not influence the TB absorbance.
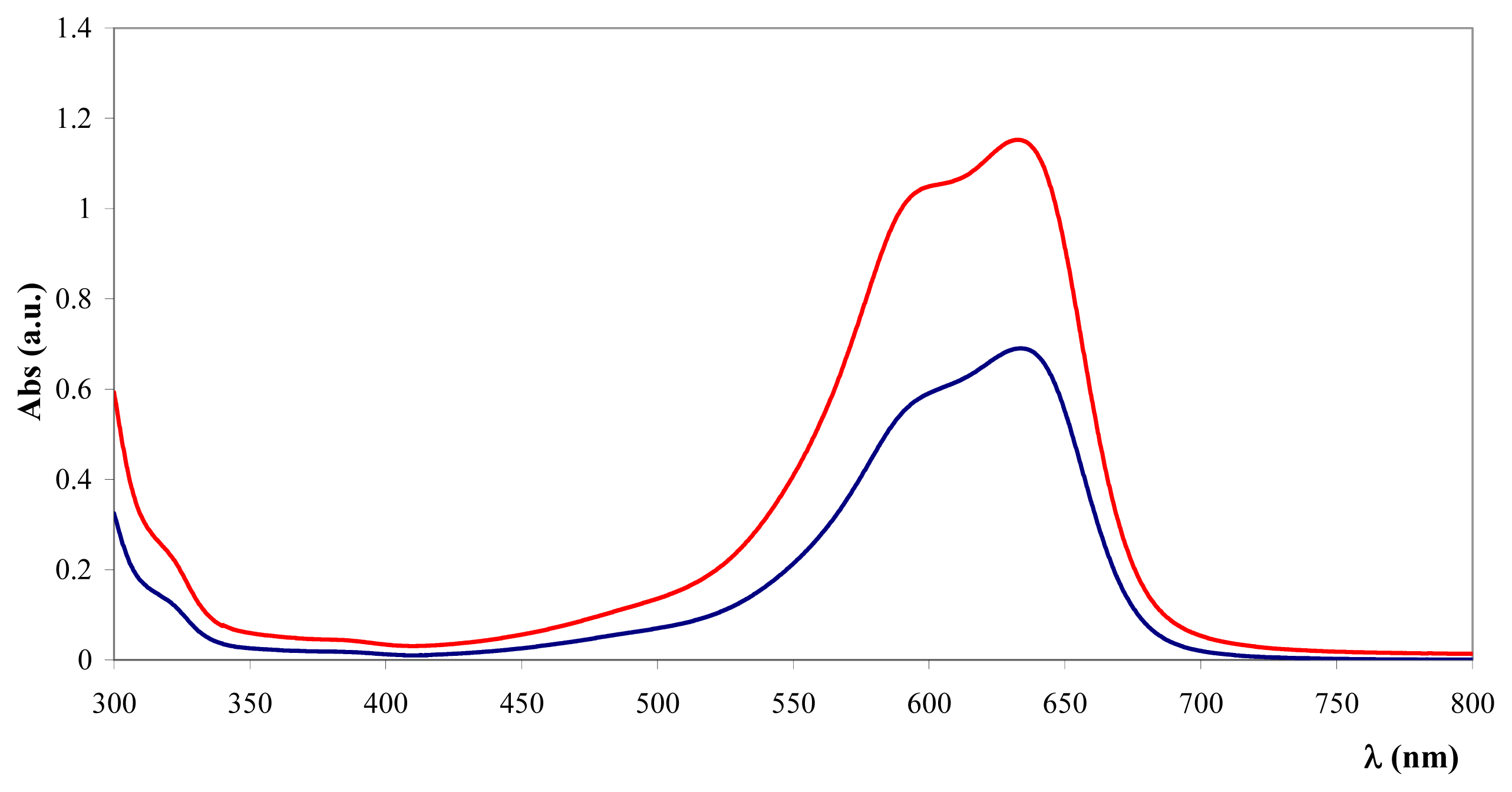
Figure 1 reveals the spectral characteristics of TB supernatant depletion upon addition of Fe
3O
4@heparin NP.
Based on a standard curve reported in the experimental section, the amount of coating for 100 mg of NPs was measured (
Table 1).
Co
3O
4@heparin and NiO@heparin NPs were repeatedly analysed by TB assay. The data range reported in
Table 1 fit well with the gravimetric data. The Fe
3O
4@heparin NP colorimetric data range appears a little narrower and the gravimetric datum seems underestimated. Fe
3O
4@heparin NP was prepared several times and their TB assay appears quite reproducible.
By averaging the gravimetric and colorimetric data in
Table 1, the amount of immobilised heparin can be reasonably evaluated to be in the range of 10%–20% (
w:w) when heparin is in large excess. Decreasing the amount of heparin during the synthesis, as for Fe
3O
4@heparin NPs (
Table 2), results in a lower functionalisation of Fe
3O
4.
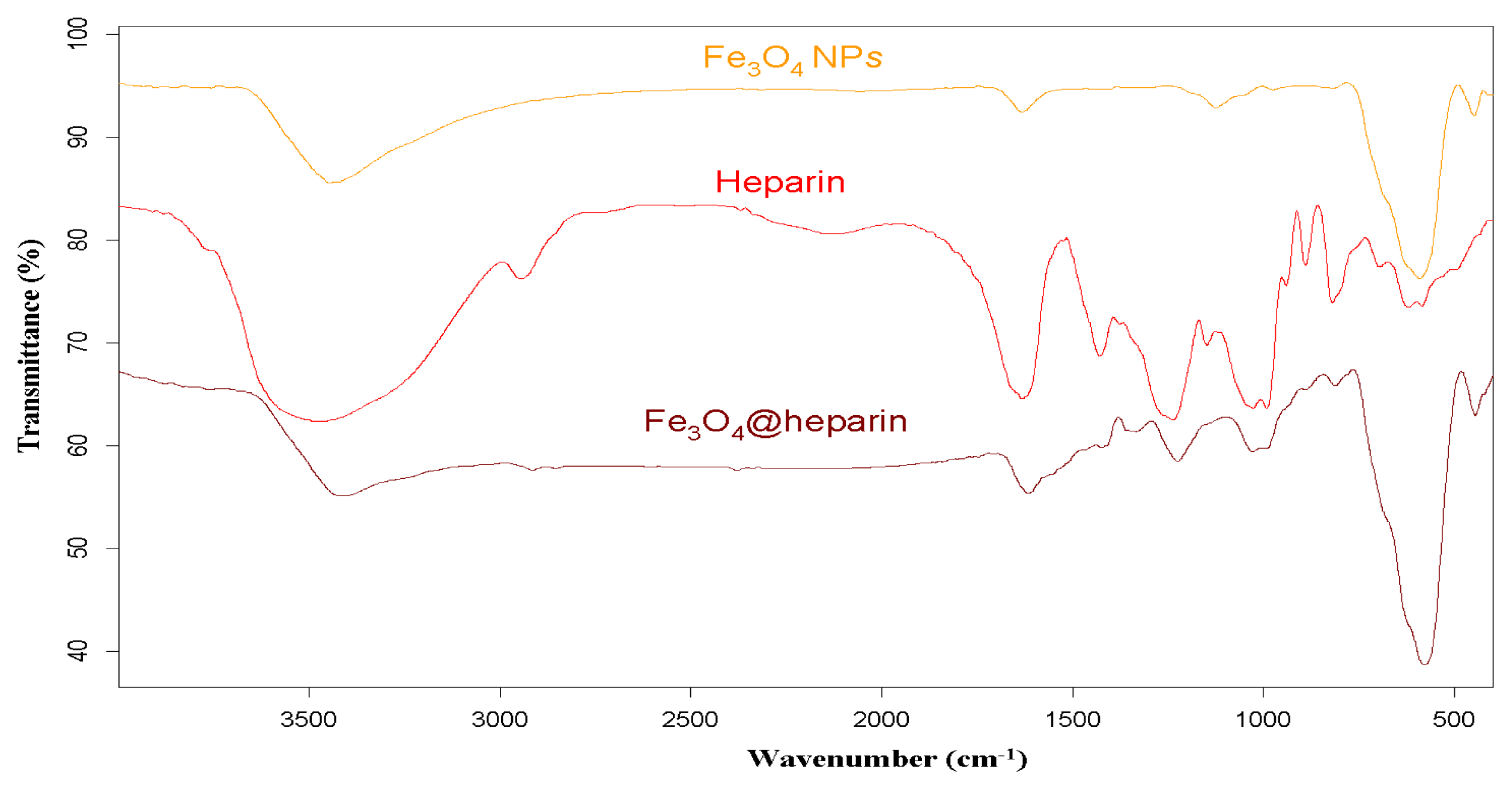
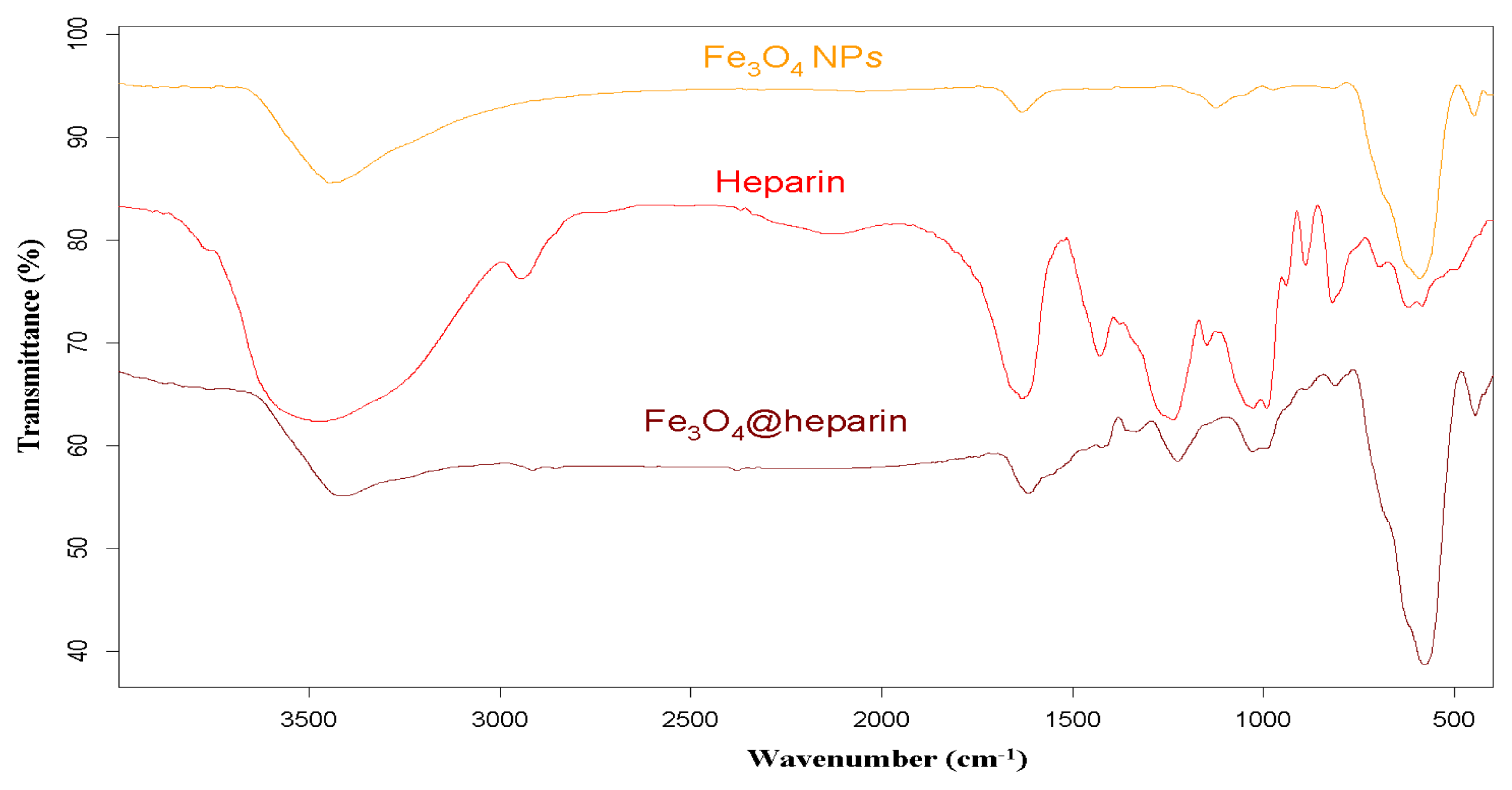
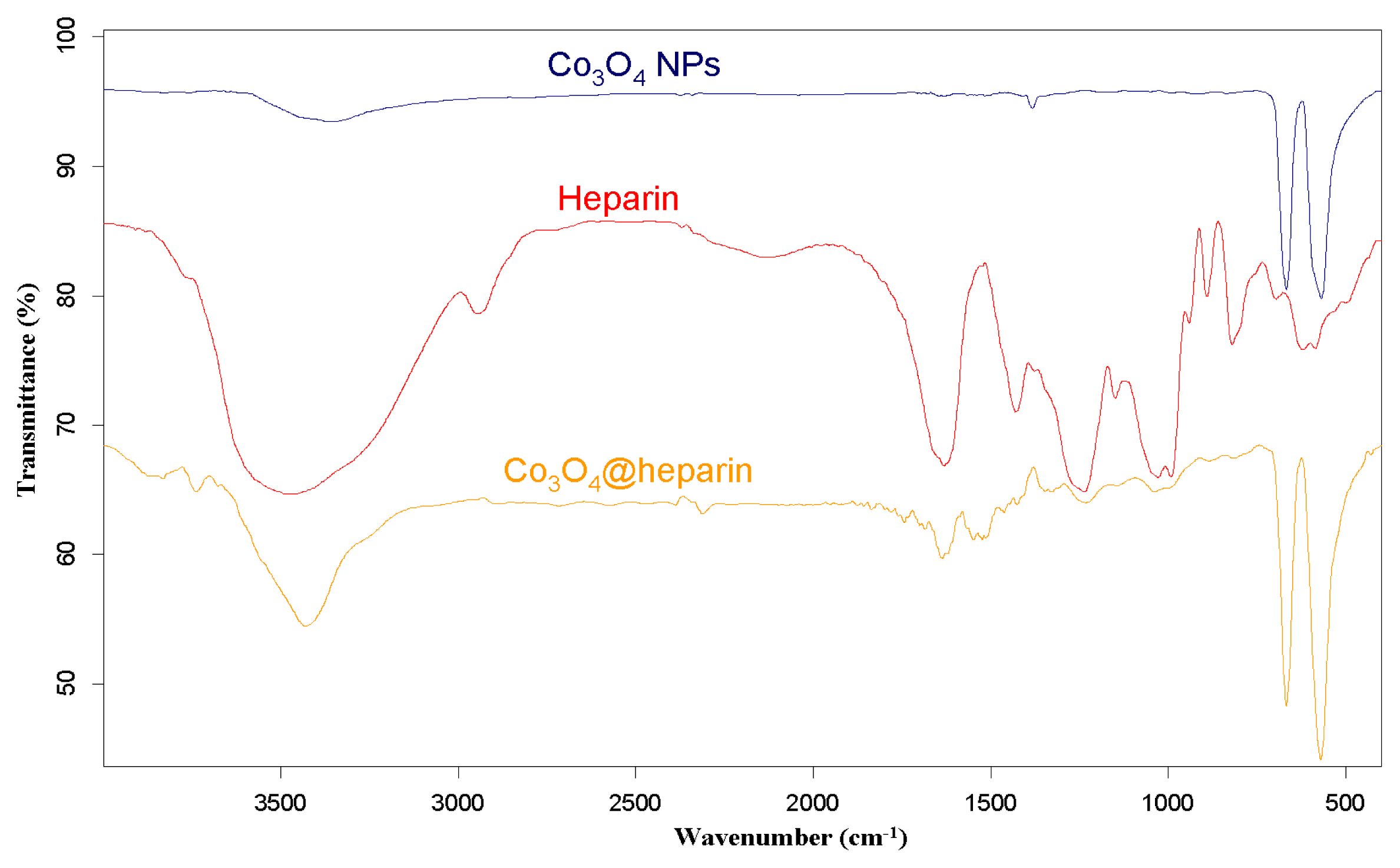
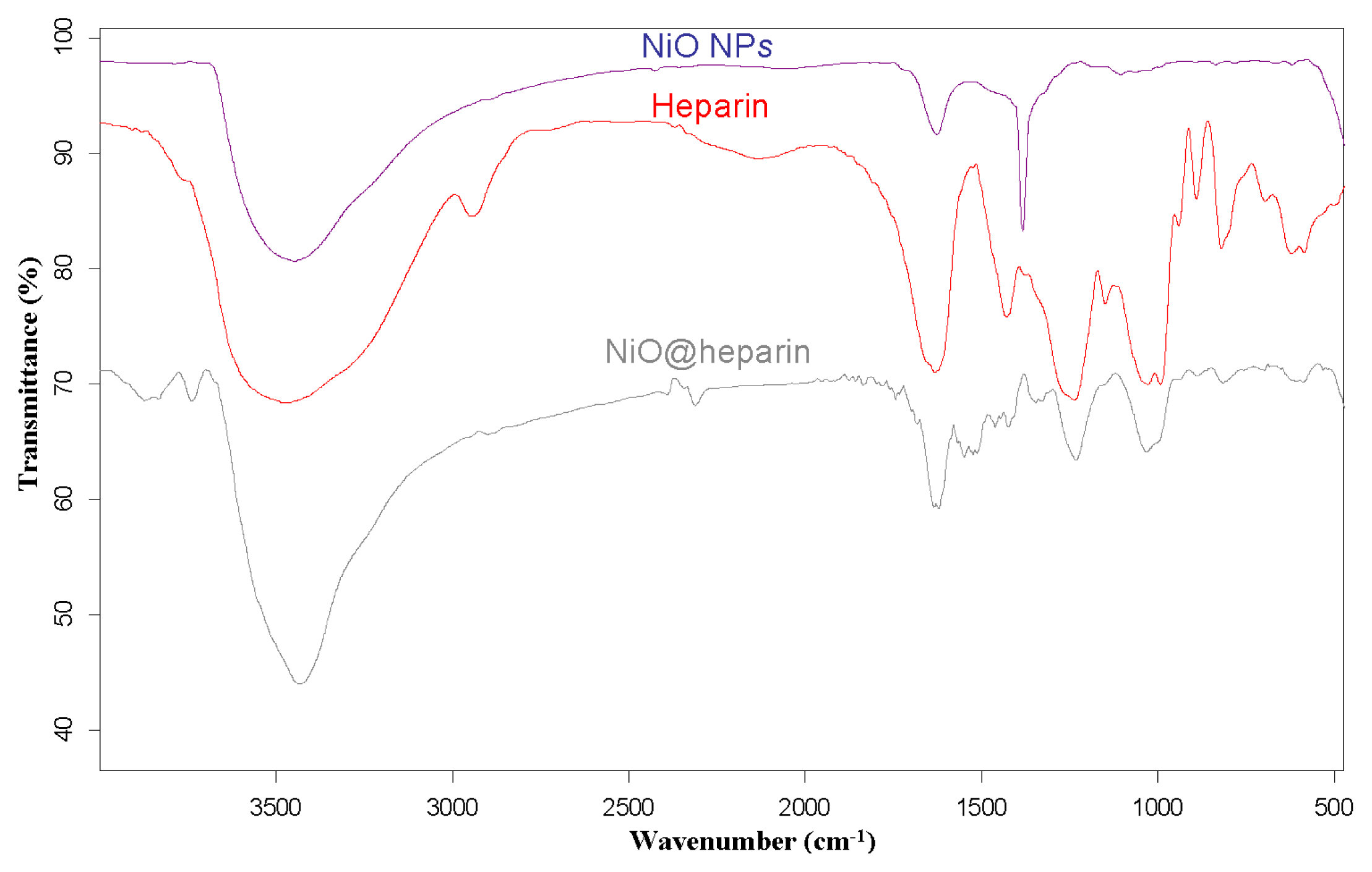
For each MO@heparin NP preparation, the presence of heparin was detected by FT-IR spectra. FT-IR spectra of NPs before and after modification with heparin are presented in
Figures 2–
4, along with the reference spectra for heparin.
In the NP spectra, two bands characteristic of adsorbed water (1631 cm
−1) and carbonate stretching (1384 cm
−1) are observed, resulting from the exposure of the samples to air [
45,
46]. A broad OH stretching band of hydrogen-bonded water is observed at 3438 cm
−1. The two broad bands at 1631 cm
−1 from moisture and at 1125 cm
−1 exhibited by Fe
3O
4 are quite similar to those observed for the unmodified iron oxide sample by Khurshid [
43]. In the range 900–400 cm
−1, the IR bands of solids are usually assigned to the vibration of ions in the crystal lattice [
47]. The spectra of pure solids consisted of two bands in the range 592–499 cm
−1 for Fe
3O
4 and in the range 667–568 cm
−1 for Co
3O
4, whereas one band appeared at approximately 438 cm
−1 for NiO.
In the FT-IR spectra of the MO@heparins NPs, the main bands of heparin are clearly distinguishable. After coating the particles with heparin, the sulphate bands of heparin at 1240–1260 cm
−1 and 1019 cm
−1 were clearly visible, confirming the presence of heparin on MO@heparin NPs. Some bands in the range 1550–1400 cm
−1 can be assigned to CO
2− stretching. In MO@heparin NPs, the sulphonamide –NHSO
3− stretching bands at 891 cm
−1 are of lower intensity than in heparin, in perfect agreement with the spectra of heparin-coated Fe
3O
4 described by Khurshid [
43].
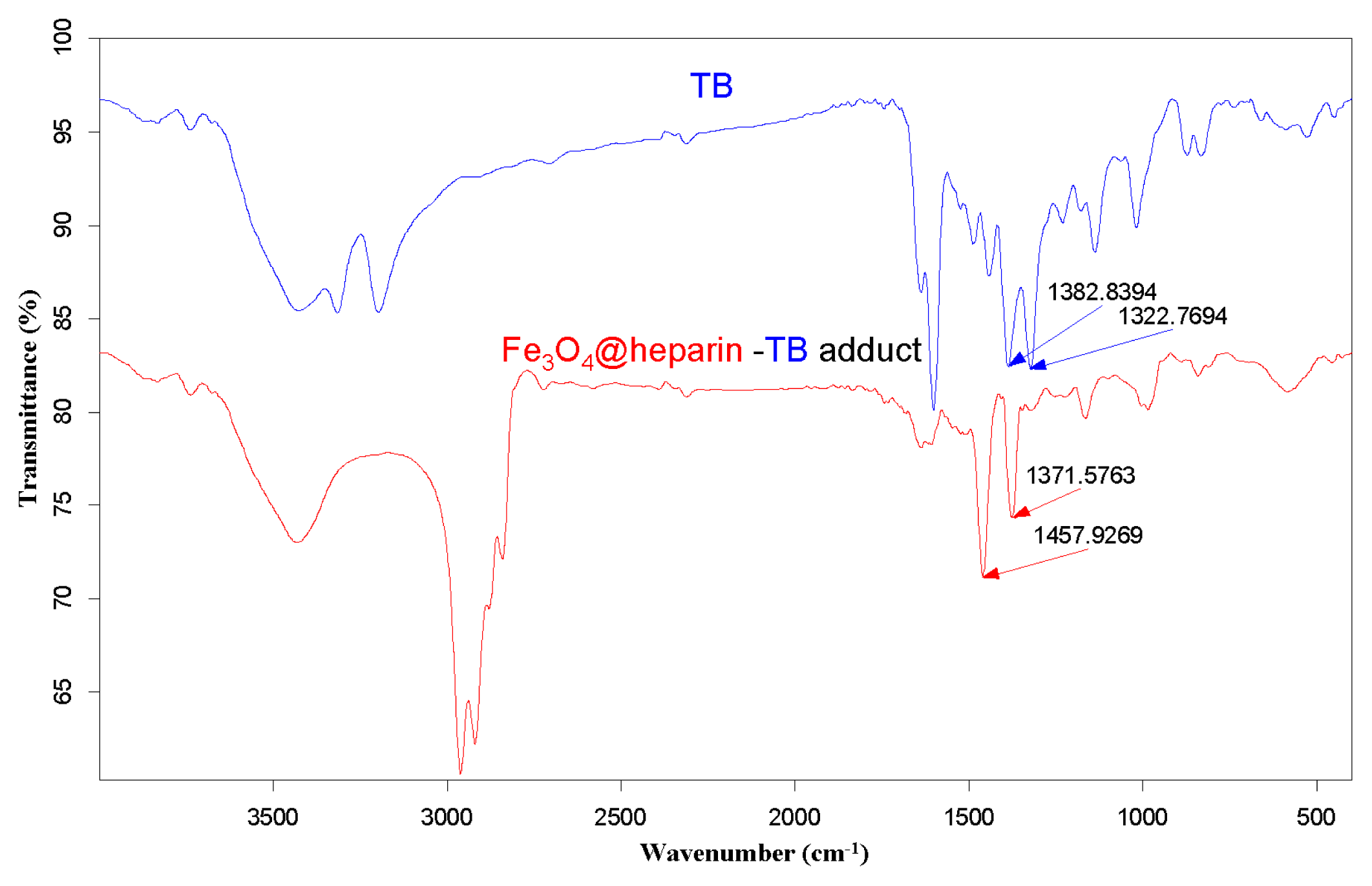
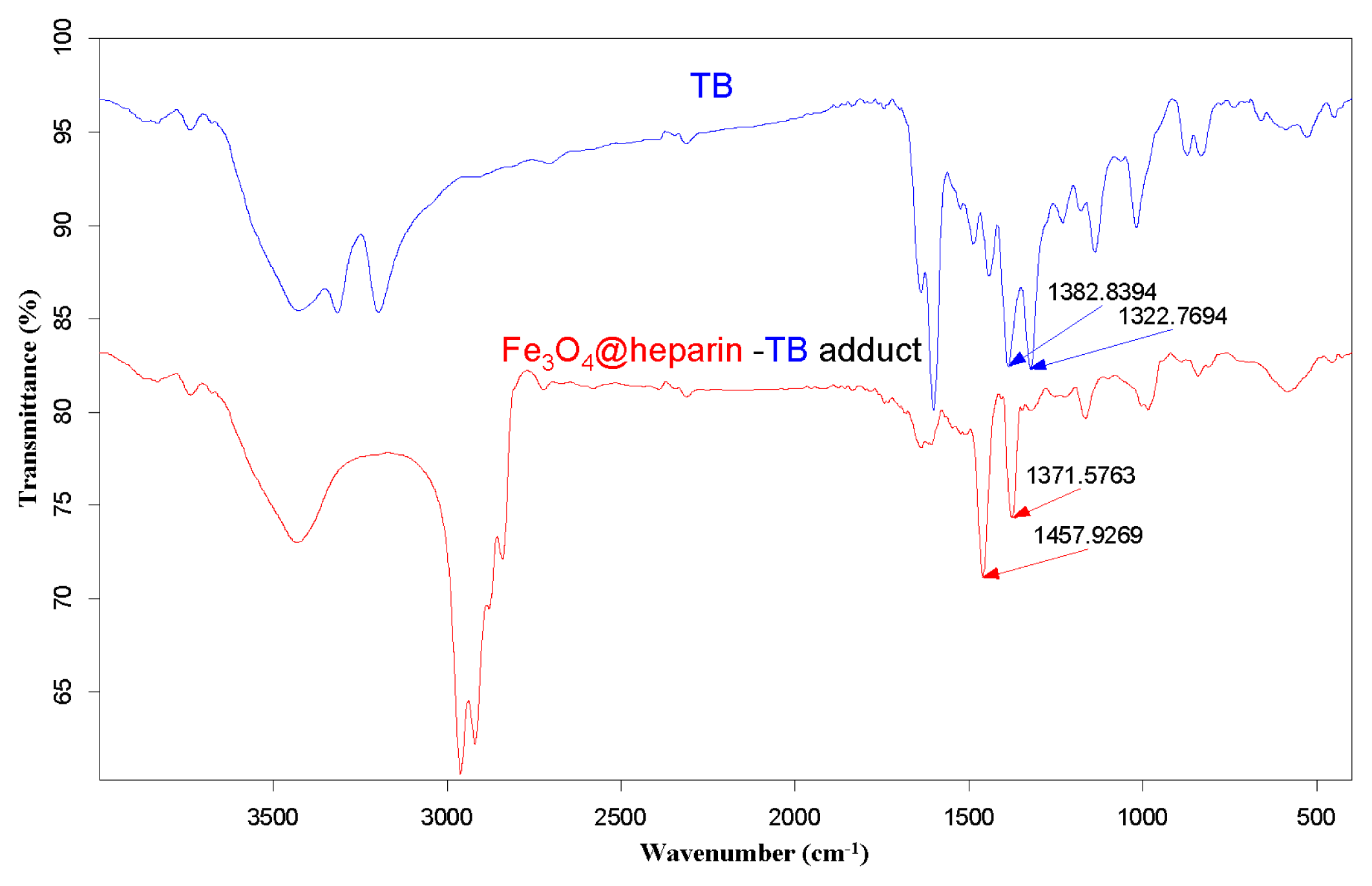
The purple centrifuged MO@heparin-TB adducts were dried under vacuum and were also investigated using IR. The FT-IR spectra in
Figure 5 demonstrate that the stretching frequencies of the TB functional group -N
+(CH
3)
2 shifted from 1382.84 cm
−1 and 1322.77 cm
−1 (free TB) to 1457–1460 cm
−1 and 1371–1377 cm
−1 (TB interacting with heparin). These shifts are consistent with the structure of the MO@heparin-TB adducts, in which heparin’s negative charges are associated with TB’s positive quaternary amino group -N
+(CH
3)
2.
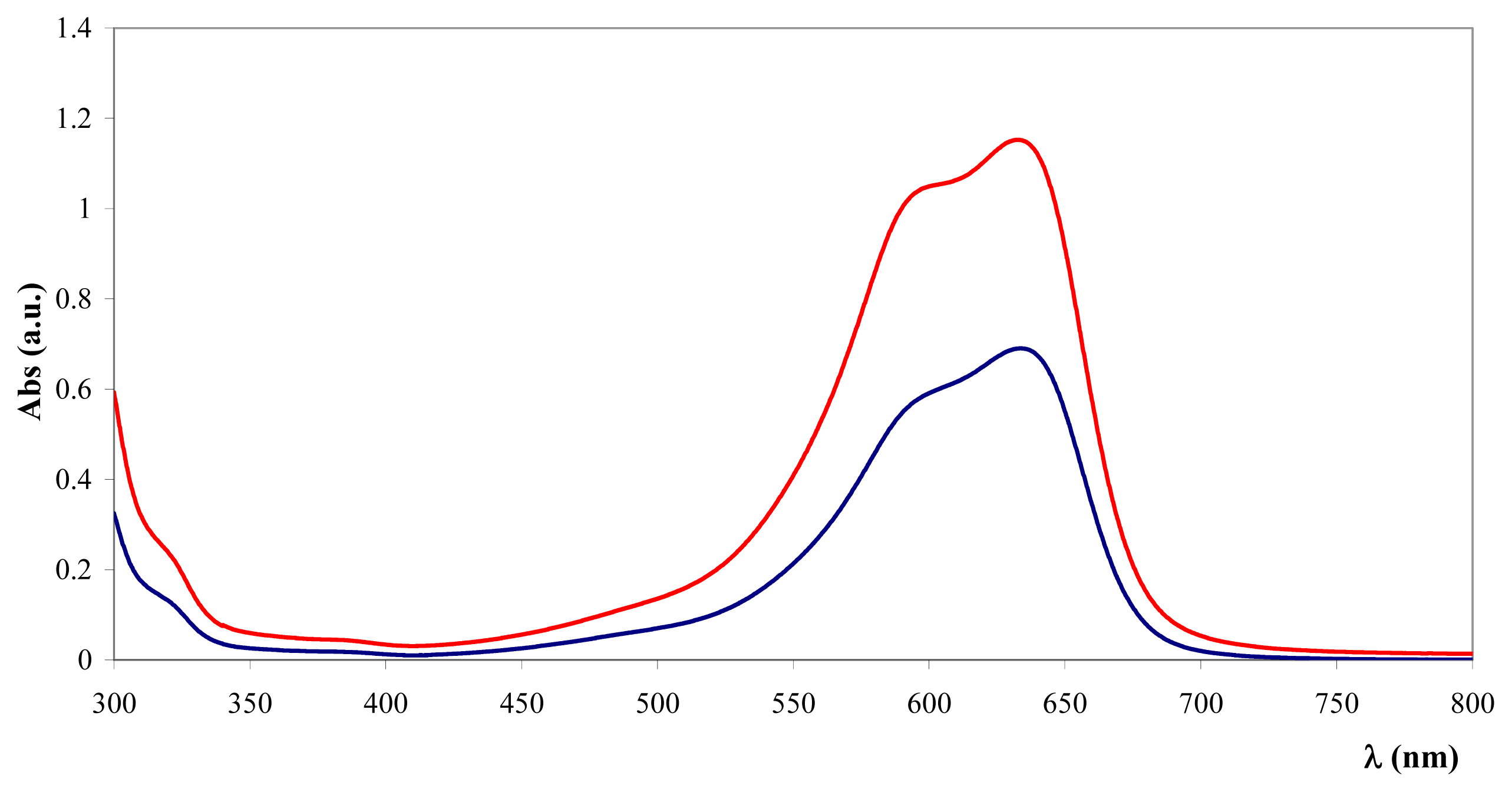
Figure 6 presents a comparison between TB and the Fe
3O
4@heparin-TB adduct. The bare NPs that were added to the TB solution were black in color when recovered and did not yield significant IR spectra.
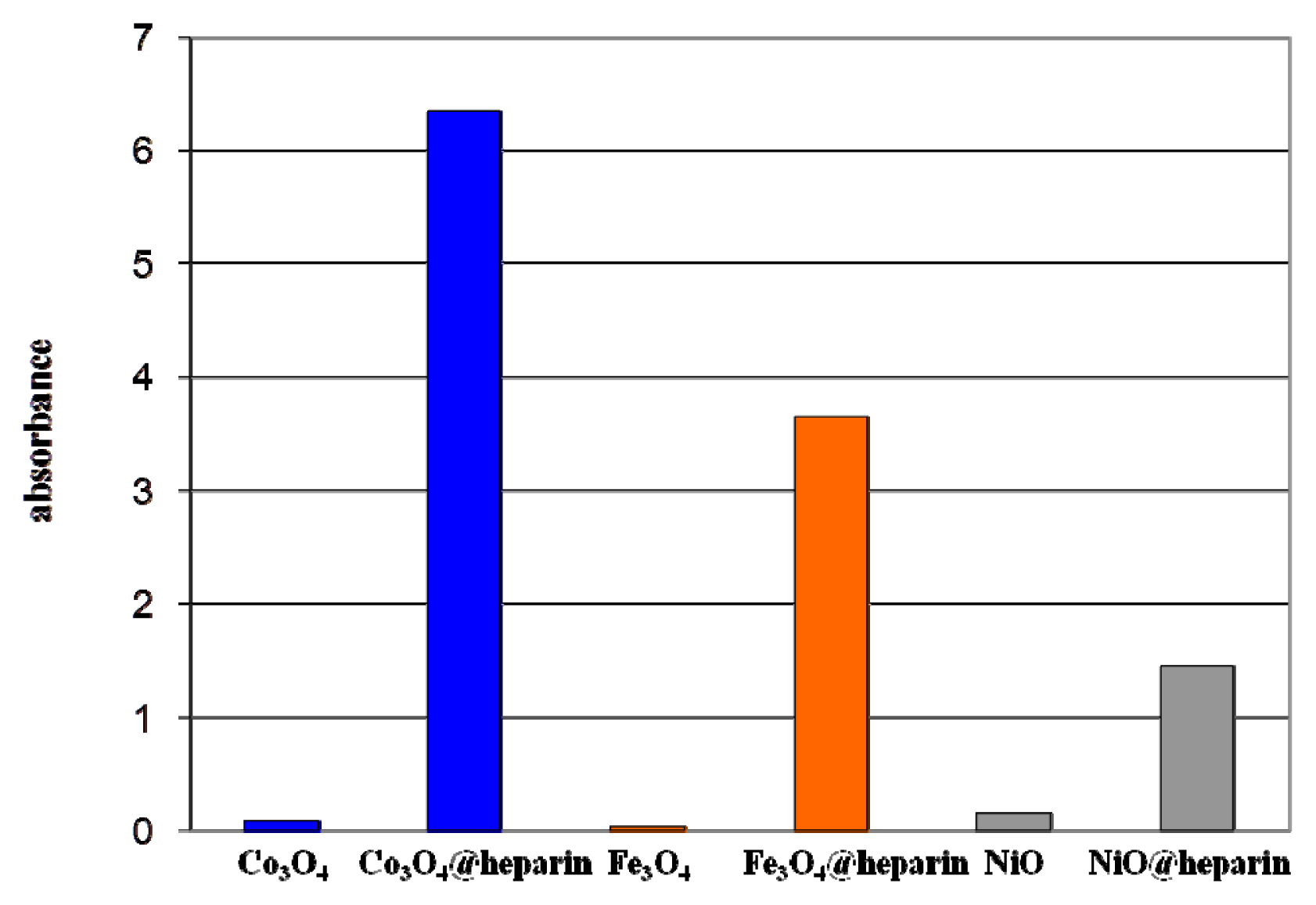
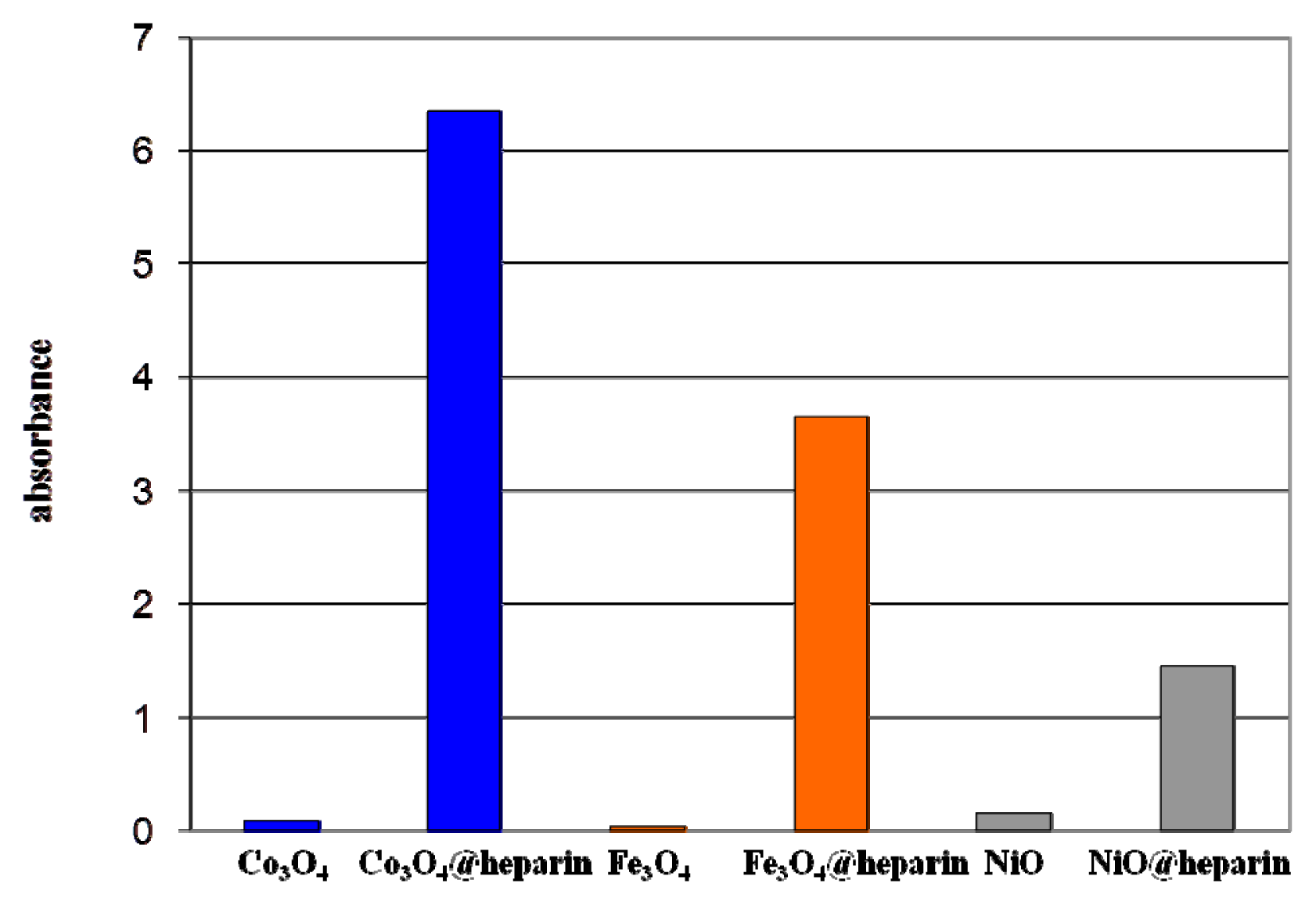
Figure 6 reports light absorption at 600 nm for the coated and bare NPs suspended in distilled water (1 mg/mL). The coated NP suspensions reach higher absorbance values (NiO@heparin < Fe
3O
4@heparin < Co
3O
4@heparin). This effect is permanent, as the coated NP suspensions remained unaltered.
Dynamic light scattering (DLS) experiments produced further information about the supernatant of the MO@heparin NPs water suspensions. For samples described in
Table 1, more than 50% of NiO@heparin NP preparation and Co
3O
4@heparin NP preparation was solubilised, whereas only approximately 20% of the Fe
3O
4@heparin NP preparation was solubilised.
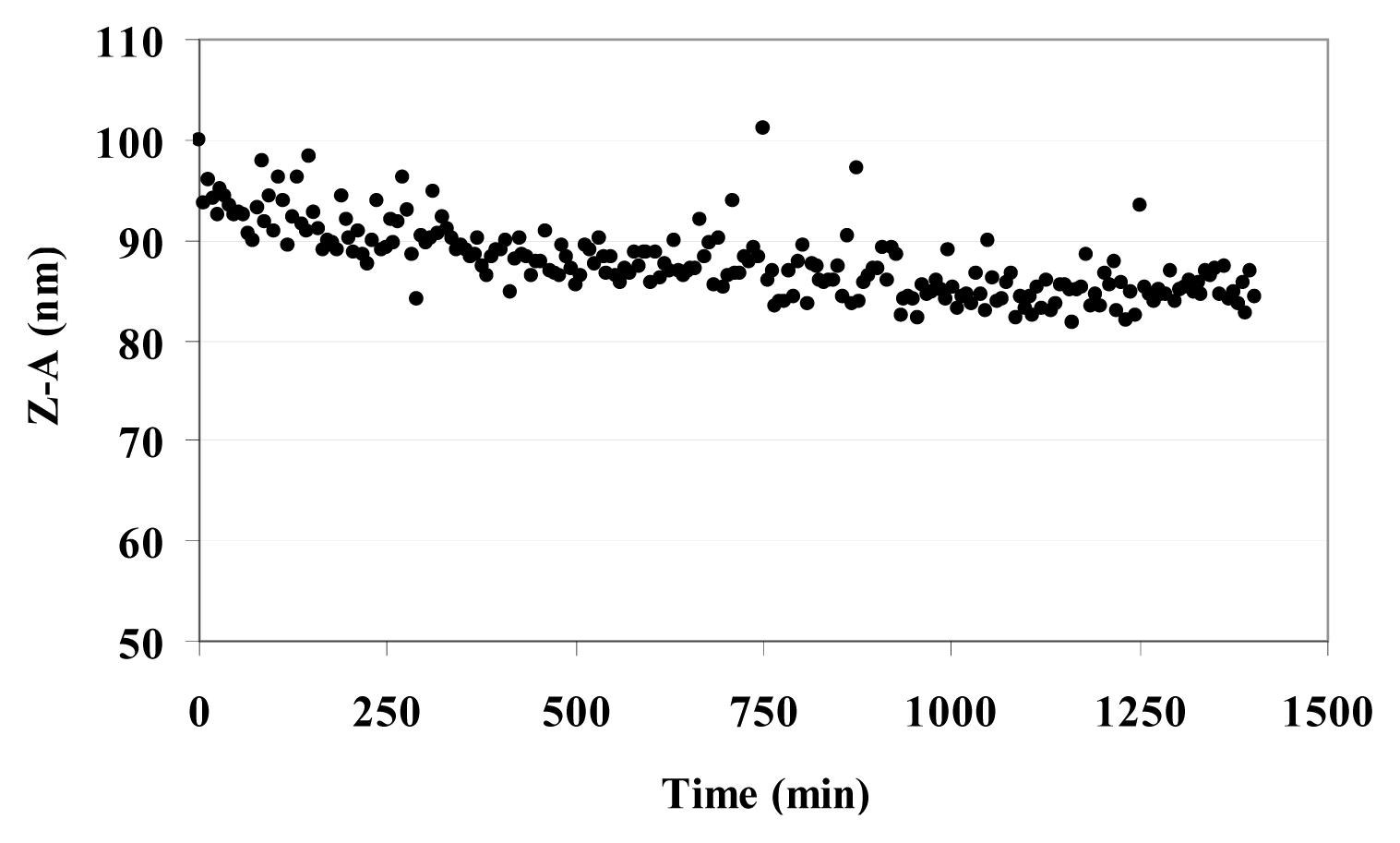
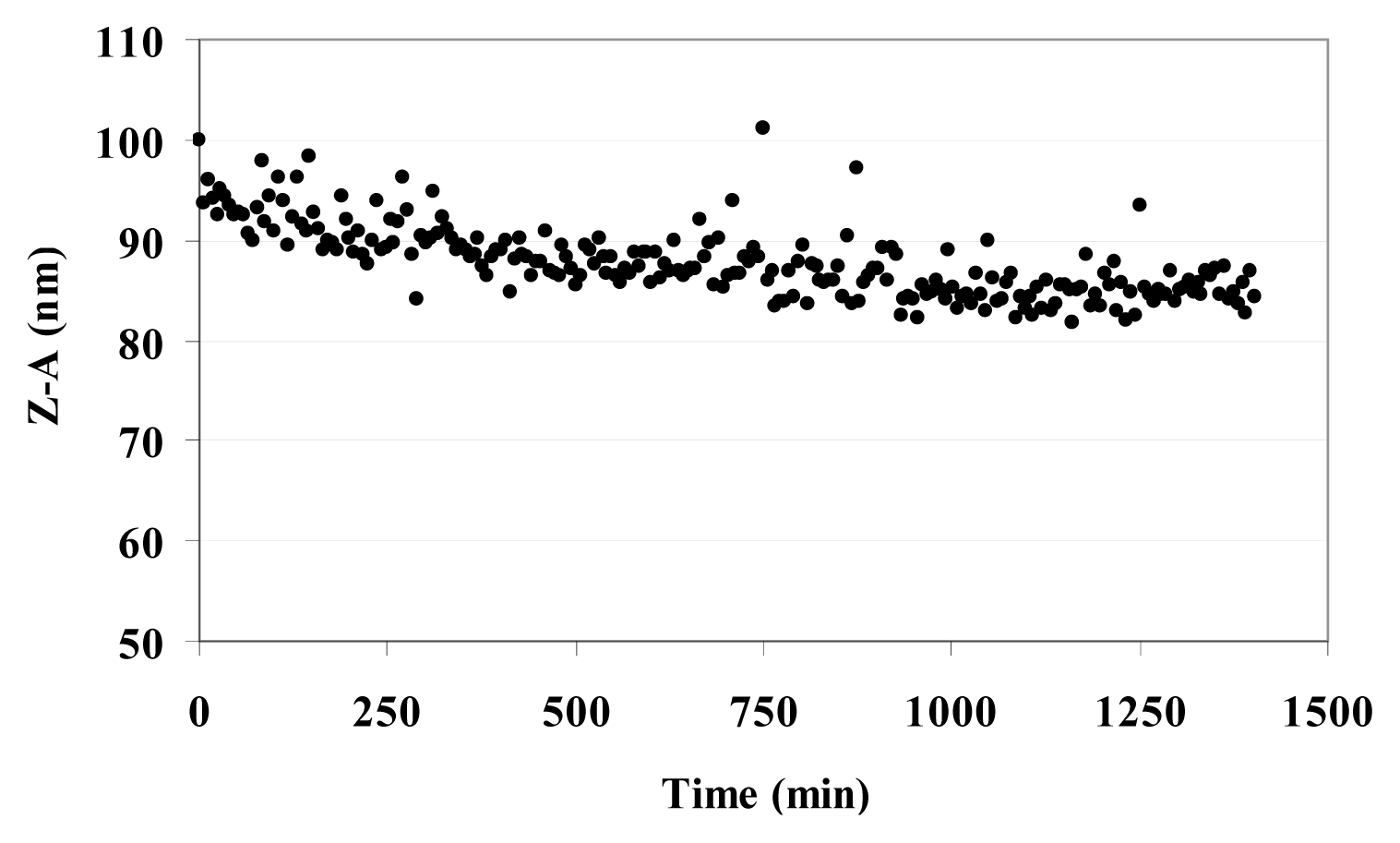
Table 3 reports the Z-Average (Z-A) value as the mean value of the hydrodynamic radius of suspended MO@heparin NPs and the PolyDispersity Index (PDI) which is a measure of the width of the NP size distribution. The Z-A for all three MO@heparin NPs appear to be quite similar, with PDI values <0.3; in contrast, DLS experiments on bare NPs showed a dispersion of larger NPs that rapidly aggregate, yielding a high PDI (data not shown). The lower aggregation of MO@heparin NPs is a result of the presence of the heparin coating. The behaviour of a suspension of Fe
3O
4@heparin NP in water was followed for 24 h. The Z-A tended to a constant value in 12 h (
Figure 7).
The ζ measurements of bare NPs and of coated NPs described in
Table 1 were performed at a concentration of 25 μg/mL in water and recorded at 25 °C, see
Table 4. The heparin coating resulted in MO@heparin NPs having a highly negative surface charge, consistent with the colorimetric assays in the presence of TB. The same trend has been observed for non-covalent bioconjugation of heparin to poly-
l-lysine-coated iron oxide NPs [
43].
The size and the surface charge of Fe
3O
4@heparin NP were also measured in the presence of free heparin (
Table 5). The addition of different amounts of heparin to a solution of Fe
3O
4@heparin in water did not result in any change in its Z-A and PDI values (as determined by DLS measurements). Fe
3O
4@heparin NP maintained the highly negative surface charge as shown by ζ measurements. Before running the analyses, all the samples were left for 1 h to reach equilibrium. This delay could explain the difference of Z-A between 92 and 118 and of the PDI between 0.17 and 0.24 for the starting sample, see
Table 3.
Fe
3O
4@heparin NPs were also prepared in PBS and in the presence of the surfactant Tween 20
®, as detailed in the experimental section. The heparin coatings are quite the same as for the Fe
3O
4@heparin NP synthesised in water (see
Table 6). The PDI values are, however, much higher than those measured for the Fe
3O
4@heparin NP synthesised in water (see
Table 3). The Z-A calculation makes sense only for the Fe
3O
4@heparin NP prepared in the presence of Tween 20
®. ζ values put in evidence a negative surface charge for all the samples. During the pre-treatment for DLS analysis, a large portion of the Fe
3O
4@heparin NP prepared in PBS was solubilised in water. The coating in PBS seems interesting as it could make coated NPs soluble in water.
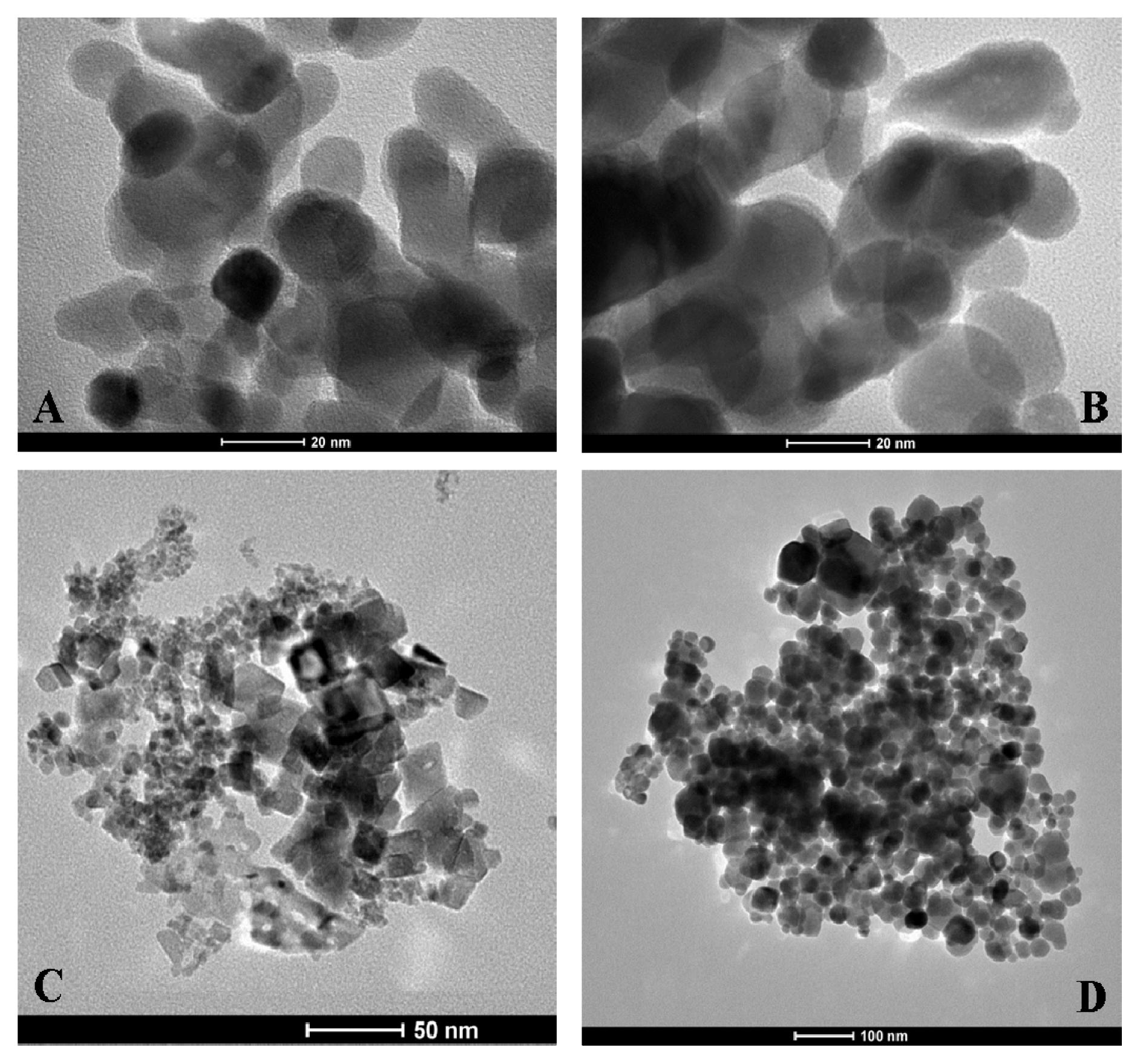
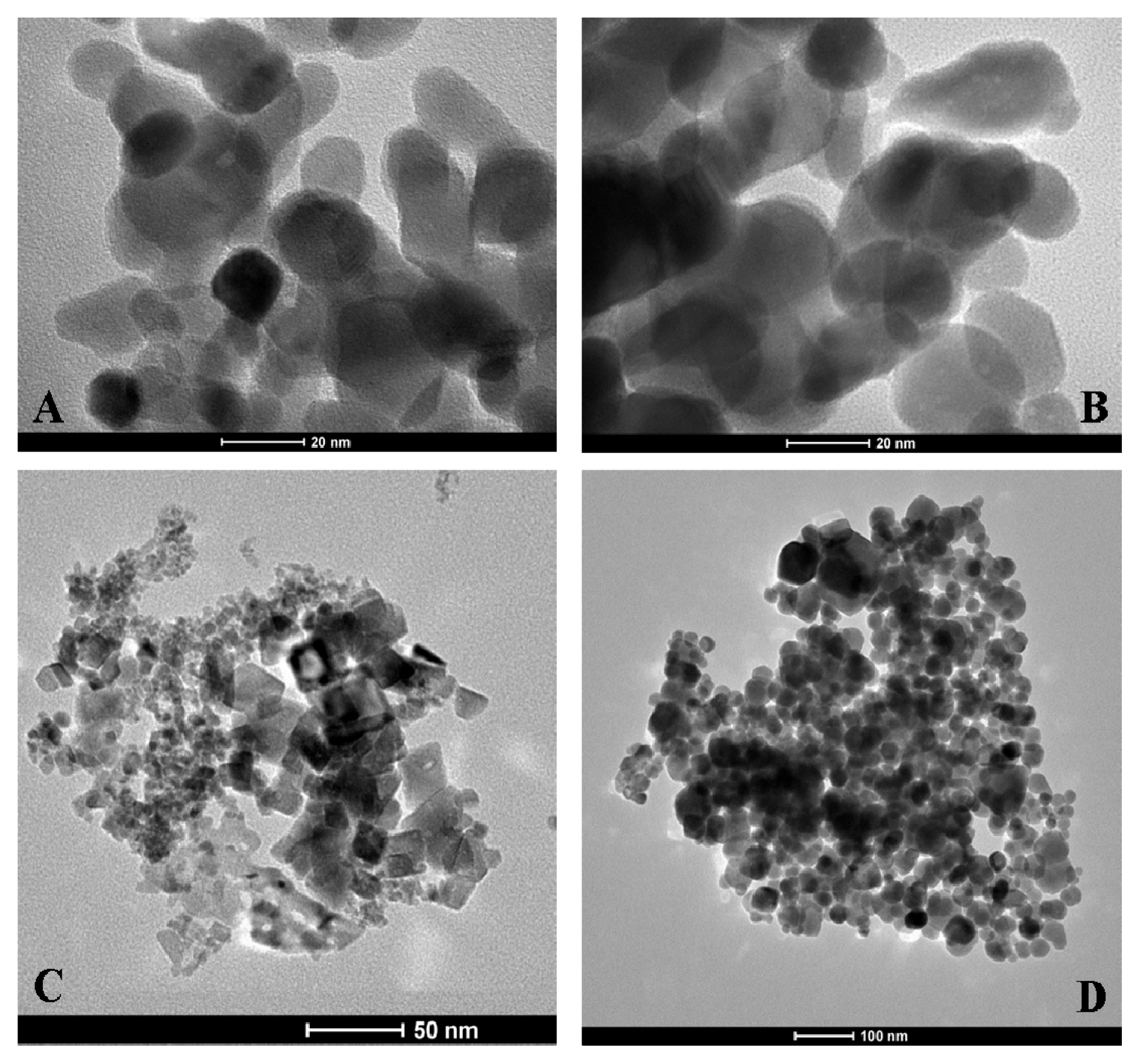
Transmission electron microscopy (TEM) images of coated and uncoated NPs are presented in
Figure 8. As expected, electron microscopy does not reveal any apparent differences between the coated and uncoated NPs (
Figure 8A,B). This finding is possibly due to insufficient contrast of the heparin molecules with respect to the electron-dense metal NPs. The shape and dimensions of the NiO and Fe
3O
4 NPs can be observed in
Figure 8C,D, respectively. The Co
3O
4 NP morphology appears quite different from the NiO and Fe
3O
4 NPs morphology; while the former shows globular shapes, the latter show much more crystalline shapes.
The formation of MO@heparin adducts has been confirmed by NMR spectroscopy. MO@heparin NPs described in
Table 1 were analysed using
1H High-Resolution Magic Angle Spinning (HR MAS) NMR (
Figure 9), which combines the typical advantages of solid- and liquid-state NMR techniques, and, when possible, by solution NMR spectrometry (
Figure 10). With the mass ratio of MO@heparin components being in favour of the inorganic part, the solid-state
13C CP-MAS and MAS techniques were not of sufficient sensitivity to detect the polysaccharide.
1H HR-MAS NMR spectra should, in principle, permit the detection of the carbohydrate connected to the particle surface [
48].
In our case, this detection was successful only for Co
3O
4@heparin NP. The lack of heparin detection for Fe
3O
4@heparin and NiO@heparin NPs can be explained by the drastic reduction of its mobility due to a very strong connection to the metal oxide surface. Starting from the assumption that the heparin NPs coating occurred in a heterogeneous phase, TEM results could be used to argue about HR-MAS NMR results.
Figure 8 seems to suggest a relationship between the Fe
3O
4/NiO NPs morphology and the lack of heparin detection. The crystalline Fe
3O
4/NiO NPs could reduce the mobility of the coated highly charged heparin by forming ordered and rigid Fe
3O
4@heparin and NiO@heparin NPs. The interaction between the apparently amorphous Co
3O
4 NP and heparin could allow the heparin coating to maintain a certain grade of mobility.
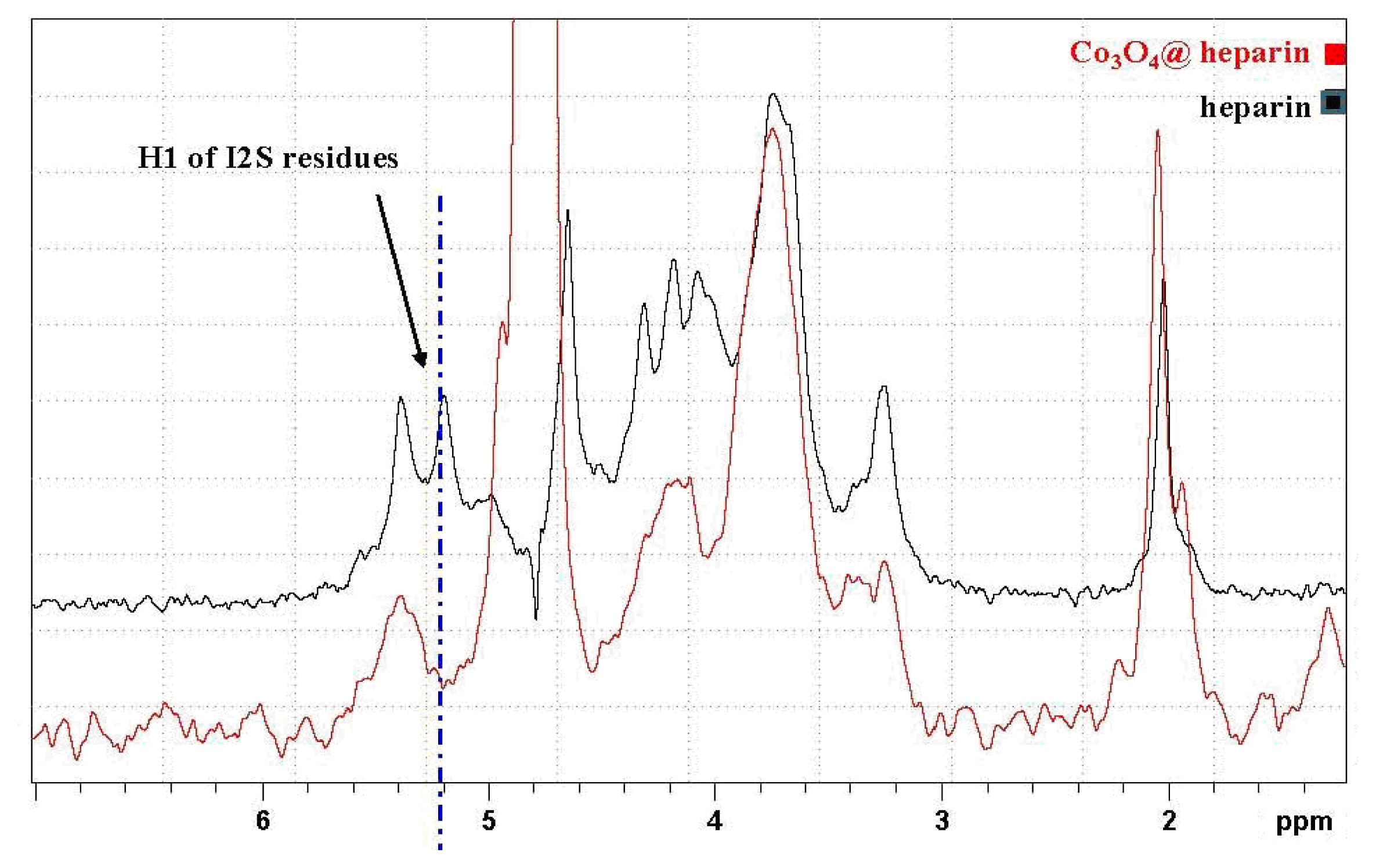
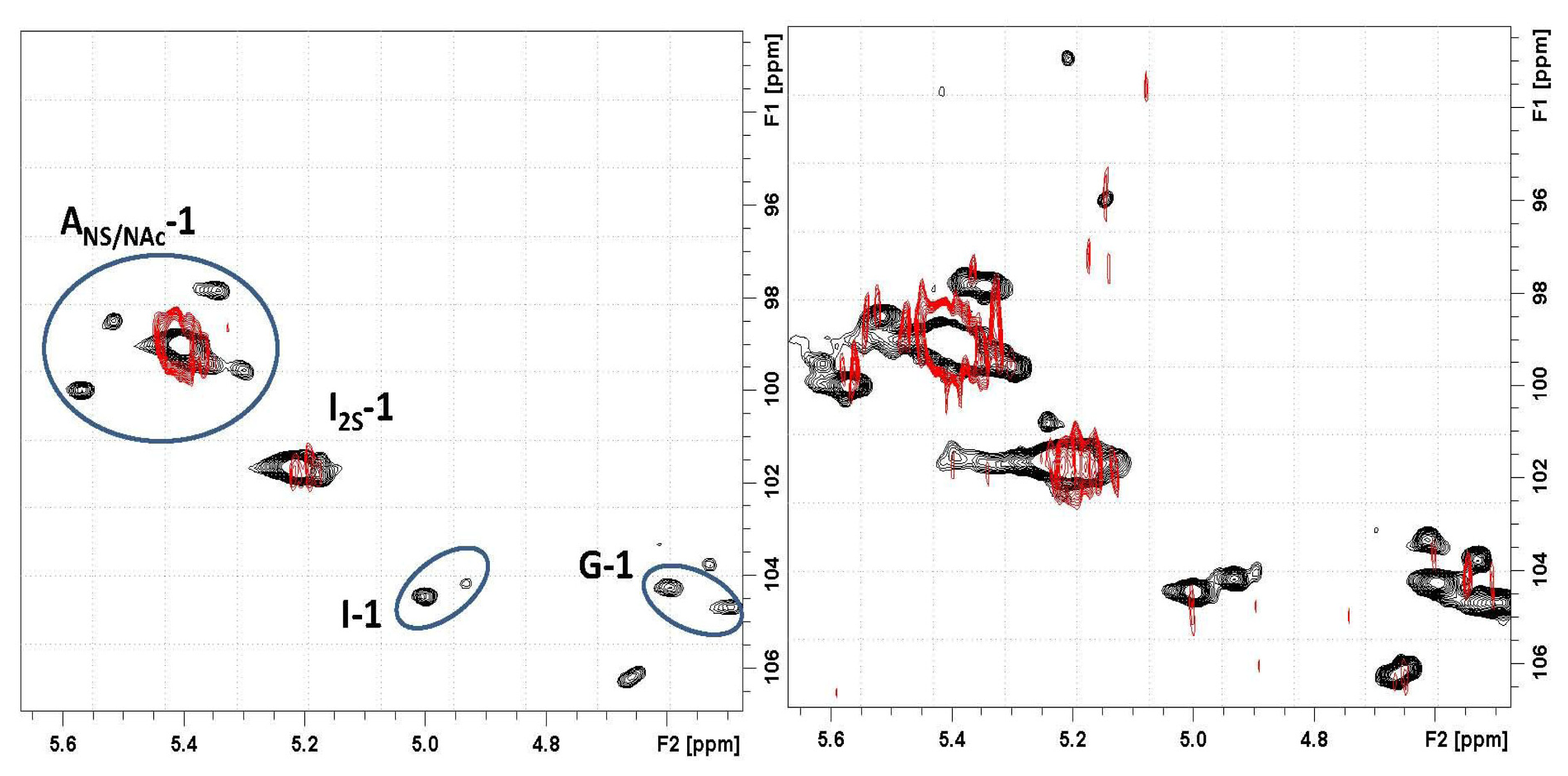
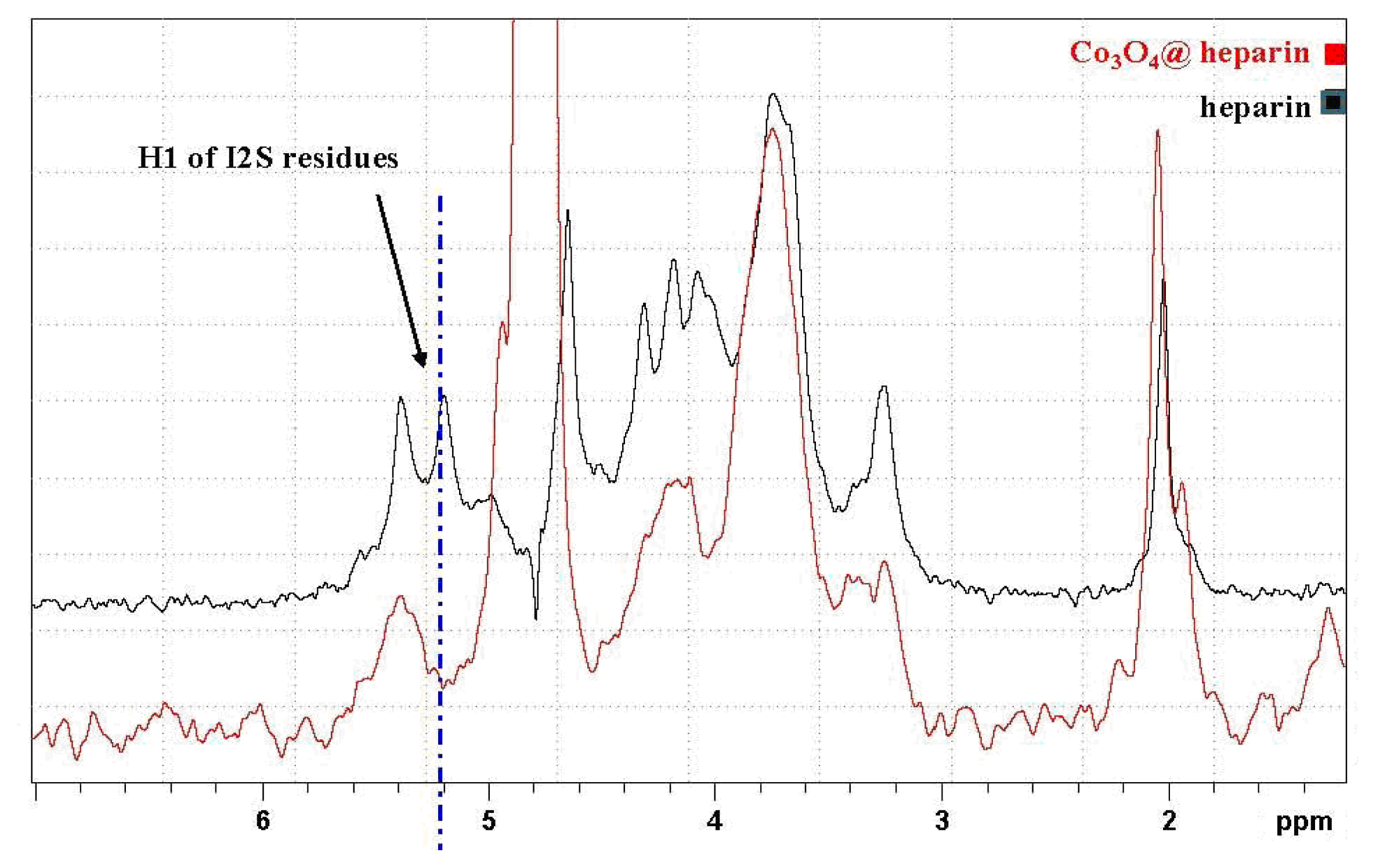
In
Figure 9, the HR-MAS spectrum of Co
3O
4@heparin NP suspended in D
2O is compared with that of heparin alone. The selective disappearance of the anomeric signal (H1) of 2-
O-sulphate iduronic acid (I2S) residues is evident and indicates their interaction with the surface of Co
3O
4.
To investigate this interaction in more detail, we used conventional NMR techniques to examine the supernatant of the Co
3O
4@heparin centrifugation isolating step, which contains some residual Co
3O
4@heparin NP and an excess of heparin (see the monodimensional 1D spectra presented in
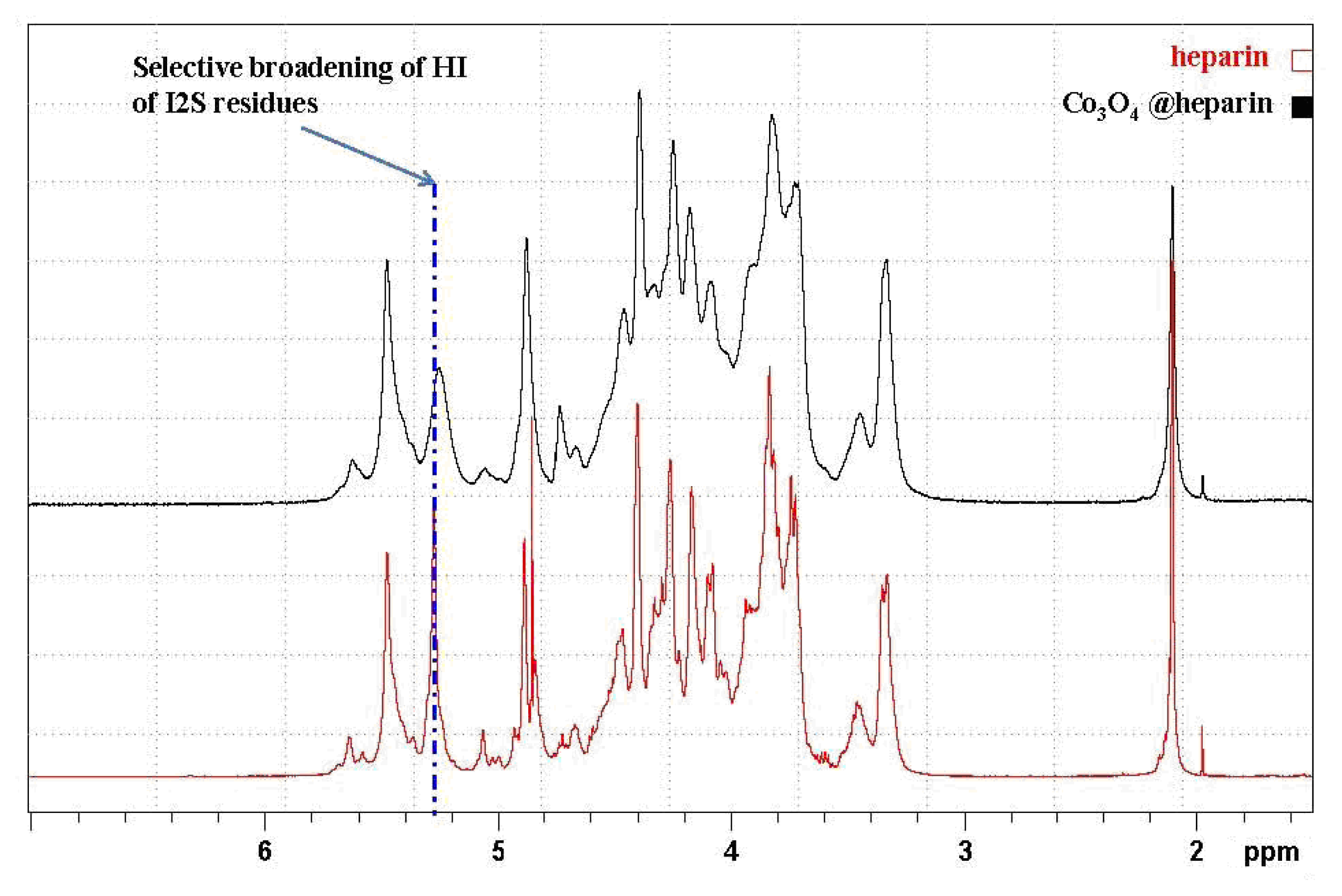
Figure 10). The same selective broadening of the uronic acid was observed upon introduction of paramagnetic species [
49,
50].
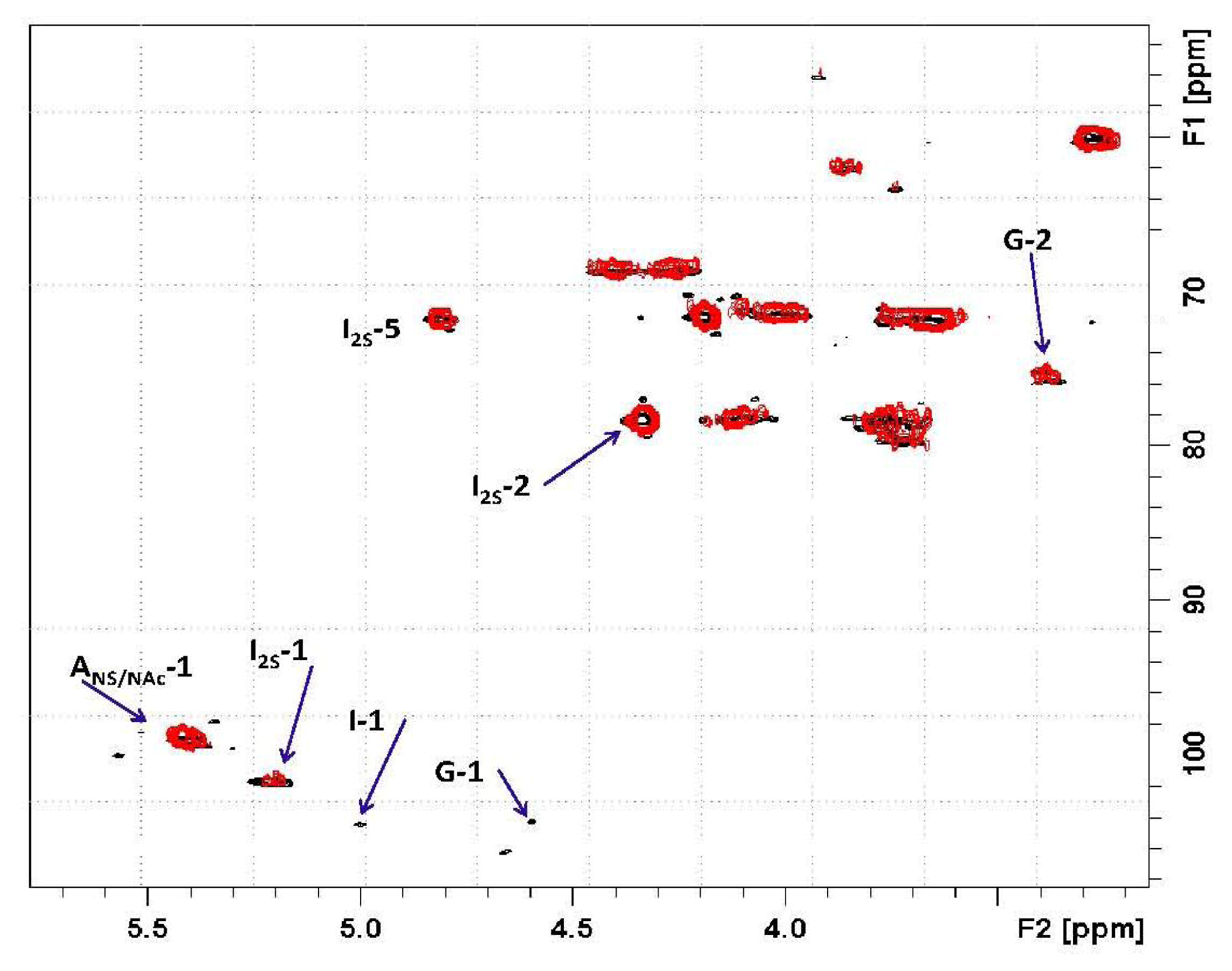
Based on the 2D heteronuclear correlation NMR spectrum of the Co
3O
4@heparin supernatant, the previous HR-MAS result was confirmed and additional details were observed (see
Figures 11 and
12). Indeed, not only the I2S residues but also minor non-sulphated iduronic and glucuronic residues are involved in the interaction, indicating that all of the uronic acid residues are responsible for the electrostatic interaction.
Moreover, the signal of the proton linked to the carbon bearing the sulphate residue of I2S is unaffected, supporting the hypothesis that the site of heparin- Co3O4 NP interaction is the carboxyl group.
The influence of substitution pattern and cation binding on conformation of heparin in solution has been studied by NMR [
51]. The I2S anomeric signal collapses because of metal ion interaction; a similar effect occurred for H5, the closest proton to the carboxyl moiety. For Co
3O
4@heparin NP, this phenomenon did not occur, as the unaffected I2S H5 signal remains evident in both the 1D and the 2D hetero-correlation spectra.
Whereas the FT-IR technique helped to visualise the MO@heparin NPs and the TB test quantified the heparin coating, the NMR technique investigations offered an original method of examining MO@heparin NPs. According to NMR technique results, we can argue that the core in Fe3O4@heparin and NiO@heparin NPs seems to immobilize the heparin shell, while the core in Co3O4@heparin NP has specific interactions with the heparin shell that anyway maintains its mobility.
3. Experimental Section
3.1. Synthesis of MO@heparin NPs (Core@Shell: MO@heparin)
Fe3O4 (CAS Number 1317-61-9), Co3O4 (CAS Number 1308-06-1), and NiO (CAS Number 1313-99-1) NPs (particle size <50 nm by TEM measurements, as described by supplier) were purchased from Sigma-Aldrich (St. Louis, USA) and used as received. Heparin, in the form of sodium salt, was provided by LDO Company (Trino Vercellese, Italy). In a general synthesis experiment, a suspension of NPs in distilled water (4 mg/mL) obtained by ultra-sonication for 5 min (Sonica 5300MH-Soltec, Milan, Italy) was transferred into a solution of heparin (40 mg/mL, pH 7 adjusted with 0.01 N NaOH). The mixture was stirred overnight (130–150 rpm, 20–25 °C, Julabo SW22). Co3O4@heparin and NiO@heparin NPs were separated by centrifugation (1 h, 6300g, Hettich Zentrifugen-Rotina 35 F, Tuttlingen, Germany), whereas Fe3O4@heparin NP was separated using a magnet (Ni–Cu–Ni Nickel plated, magnetisation N45). MO@heparin NPs were exhaustively washed with diethyl ether and then dried in air and in a conventional oven at 50 °C for 1 h.
Following the same reaction conditions and work up in water, Fe3O4@heparin NPs were prepared in a phosphate buffered saline (PBS) solution (0.137 M NaCl, 3 mM KCl, 0.01 M Na2HPO4, and 2 mM KH2PO4), in water with 1% w/w Tween 20® (Sigma Aldrich, St. Louis, USA) as the surfactant and in PBS with 1% w/w Tween 20®. The pH value after the dissolution of heparin in PBS was 7.2.
Fe3O4@heparin NPs were also prepared in water by changing the heparin/Fe3O4 ratio. Six milliliter of a 40 mg/mL solution of Fe3O4 was added, after 5 min of ultra-sonication, to different volumes of a 40 mg/mL heparin solution at pH 7.
3.2. IR Characterisation of MO@heparin NPs
Heparin, the final MO@heparin NPs and the starting NPs were mixed with infrared grade KBr in a convenient proportion. The solid phase Fourier transform infrared (FT-IR) spectra were collected on a Bruker IFS 25.
3.3. Standard Curve for the Toluidine Blue (TB) Assay
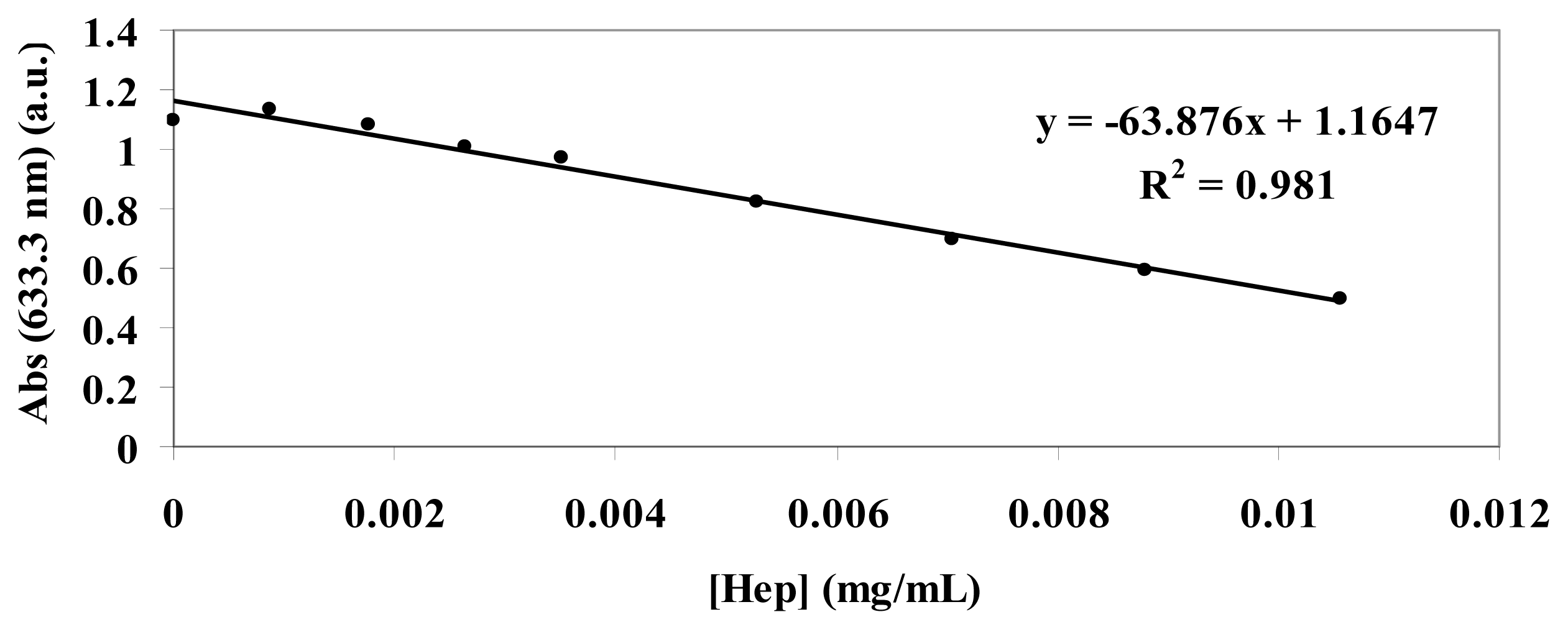
The standard curve of TB absorbance at 633 nm was constructed at increasing concentrations of heparin (range 0.01 mg/mL:0.0009 mg/mL) by mixing 1 mL of TB (0.04 mg/mL), 1 mL of water and 1 mL of different concentration heparin solutions (
Figure 13). A second standard curve was obtained after extraction of each heparin:TB mixture with hexane. The two curves overlapped.
3.4. TB Assay Heparin Content in MO@heparin NPs and IR Characterisation of MO@heparin-TB Adducts
Suspensions obtained by mixing 1 mL of MO@heparin NPs or of bare NPs (0.05 mg/mL), 1 mL of TB (0.04 mg/mL) and 1 mL of water were stirred (30 min, 130–150 rpm, 25 °C, Julabo SW22) and centrifuged (20 min, 5000 rpm, 20 °C, Hettich Zentrifugen-Rotina 35 F). The supernatant TB blue solutions were UV analysed from 300 to 800 nm, and the absorbance at 633 nm was examined. The purple MO@heparin-TB adducts and the black bare NPs were dried under vacuum and investigated using IR, following the same procedure detailed for the MO@heparin NPs.
3.5. NMR Characterisation of Co3O4@heparin
The 1H High-Resolution Magic Angle Spinning (HR MAS) NMR spectra were recorded using a Bruker Avance 300 WB spectrometer (Karlsruhe, Germany) equipped with a 4-mm MAS probe. The samples were placed in a zirconium rotor, 4 mm in diameter and 21 mm high. Wet (D2O) sample analysis was run at a spin rate of 2500 Hz. The measurements were performed using a cpmg1ld standard sequence for water signal presaturation, under the following experimental parameters: D1 3 s, AQ 270 ms, P90 4 μs, NS 1024, T2 loop 16 ms, using tetramethylsilane as the reference.
Two-dimensional heteronuclear single quantum coherence (2D-HSQC) spectra were recorded at 25 °C on a Bruker Avance 500 spectrometer (Karlsruhe, Germany) equipped with a 5-mm TCI inverse probe under the following experimental conditions: carbon decoupling during acquisition, 320 increments of 32–64 scans. The polarisation transfer delay (D = 1/[2 × 1JC–H]) was established using 1JC–H coupling values of 150 Hz. The matrix size 1 K × 512 was zero filled to 4 K × 2 K by applying a squared cosine function before Fourier transformation.
3.6. Dynamic Light Scattering (DLS) and Zeta Potential (ζ) of MO@heparin NPs and of Bare NPs
A 0.025 mg/mL solution of MO and MO@heparin NPs in water was prepared by immersing the solution in an ultrasonic bath for 1–5 s followed by centrifugation (Minispin, 1 min, 5000 rpm). The supernatant was immediately analysed in a folded capillary cell using DLS and Zetasizer Nano ZS, ZEN3600 from Malvern Instruments, at 25 °C and a scattering angle of 173°. The kinetic behaviour of Fe3O4@heparin in water was analysed based on DLS measurements at 37 °C using a quartz cuvette of 1 cm optic length Zetasizer Software 6.20 was used to analyse the field autocorrelation function in terms of the distribution of relaxation rates. The zeta potential size was automatically calculated from the electrophoretic mobility based on the Henry equation, UE = 2ɛζf(ka)/3η, where ζ is the zeta potential, UE is the measured electrophoretic velocity, η is the viscosity, ɛ is the dielectric constant and f(ka) is Henry’s function (adopted value: 1.5, referring to the Smoluchowski approximation). The Z-Average values obtained by DLS technique were calculated by the software as the average of ten measurements of the same suspension, each one obtained registering ten scans. The ζ values were calculated by the software as the average of six measurements of the same suspension.
The DLS and ζ measurements were also registered at increasing concentrations of free heparin by mixing 5 mL of Fe3O4@heparin (0.05 mg/mL) with different volumes of heparin (0.05 mg/mL) and adding water to reach a total volume of 10 mL. After 1 h., the solutions were centrifuged (Minispin, 5000 rpm, 1 min), and the supernatant was analysed at 25°C.
3.7. Transmission Electron Microscope (TEM) Characterisation of MO@heparin NPs and of Bare NPs
Five microliter drops of the different NP suspensions were deposited on 400 mesh copper grids coated with a formvar-carbon film (Società Italiana Chimici, Roma, Italy). After a few minutes, the excess liquid was removed using blotting paper. The grids were allowed to air dry at room temperature in a clean area. The samples were analysed using a Tecnai T12 TEM (FEI, Eindhoven, The Netherlands) operating at 120 kV.

 ,
, 
 ) and after (
) and after (
 ) the addition of Fe3O4@heparin NP. The comparison highlights the depletion of TB in the supernatant upon the addition of Fe3O4@heparin NP.
) the addition of Fe3O4@heparin NP. The comparison highlights the depletion of TB in the supernatant upon the addition of Fe3O4@heparin NP.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}