Comparison of Membrane Targeting Strategies for the Accumulation of the Human Immunodeficiency Virus p24 Protein in Transgenic Tobacco

{kind=link}
{kind=link}
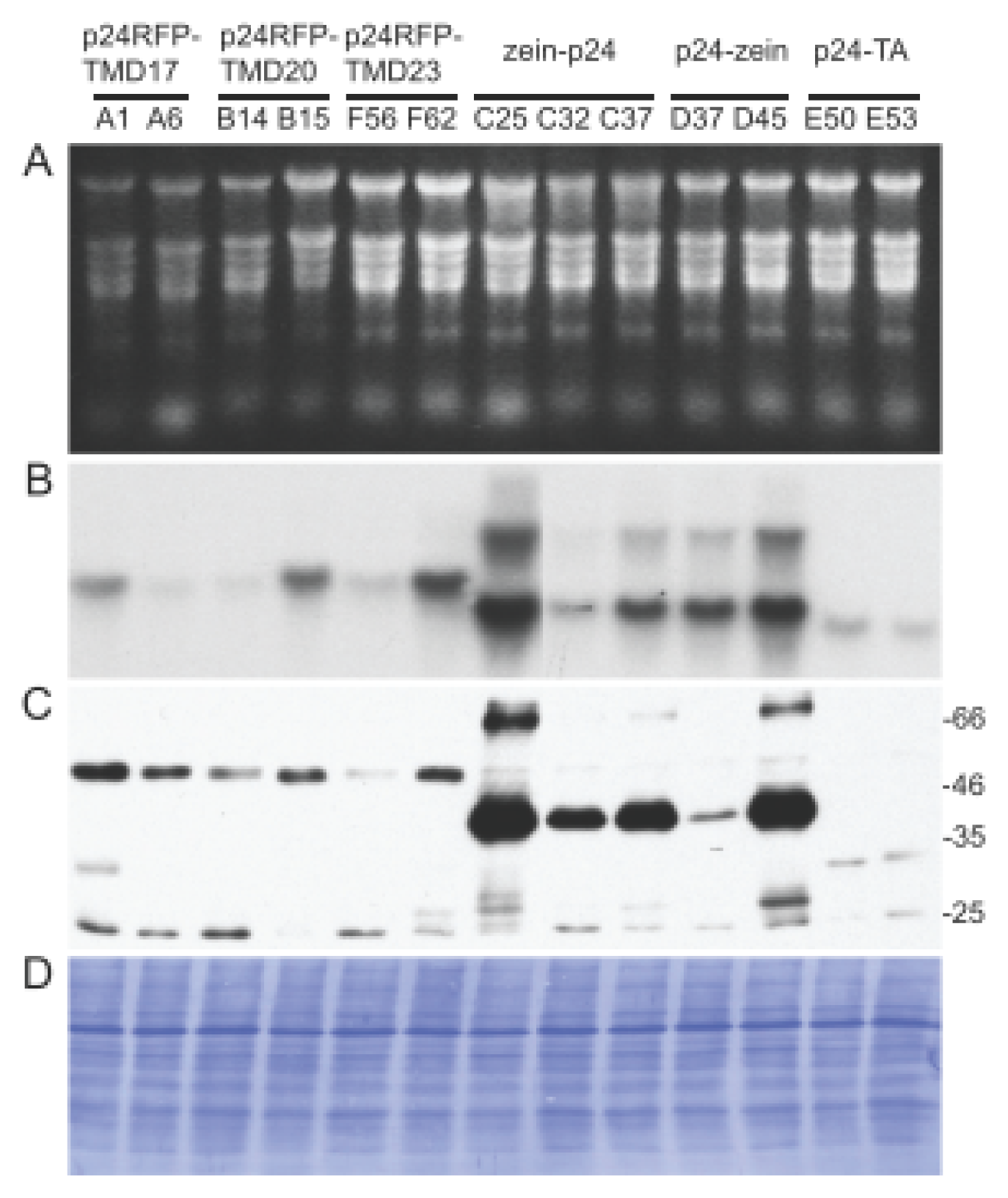{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
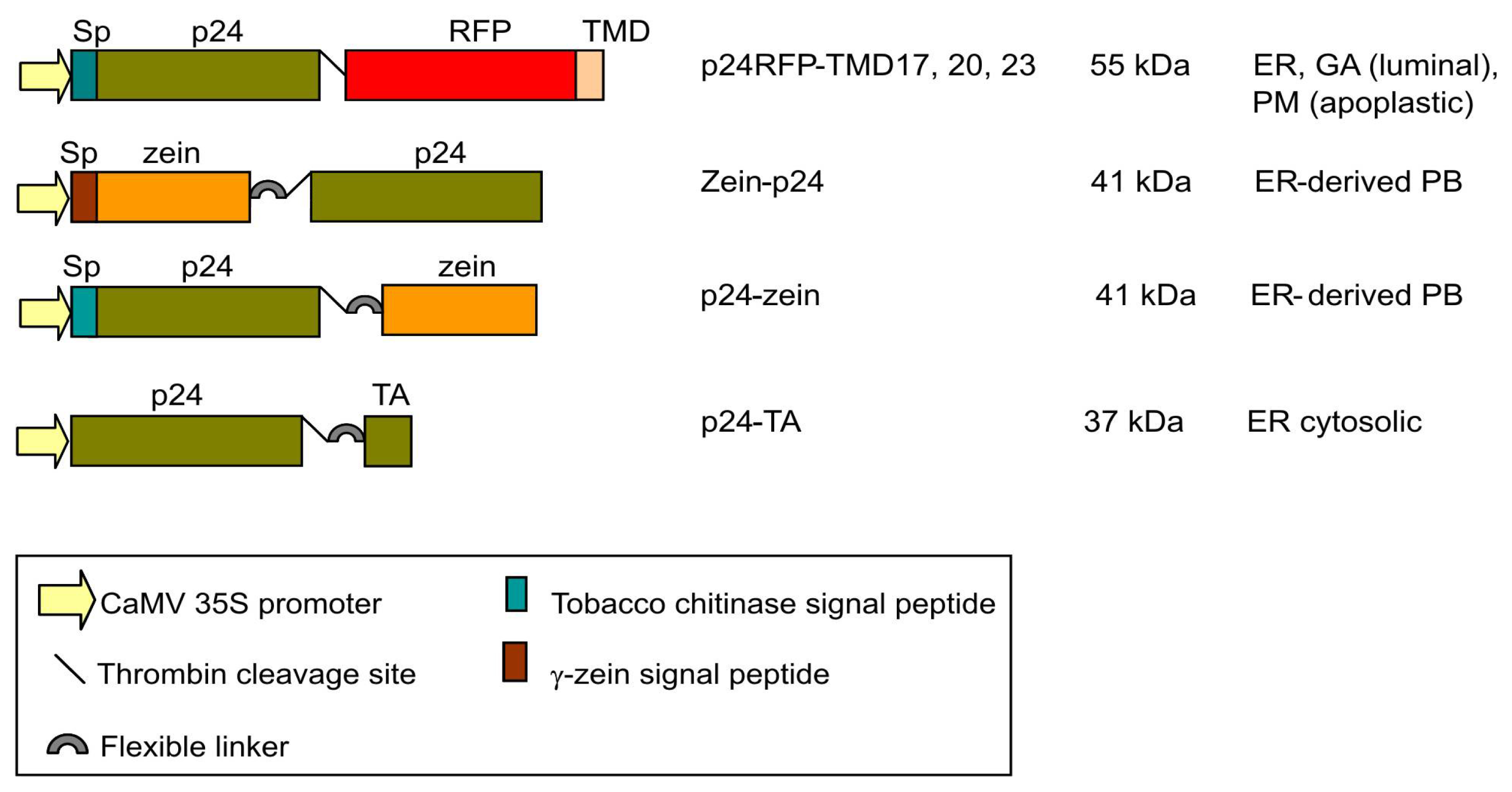
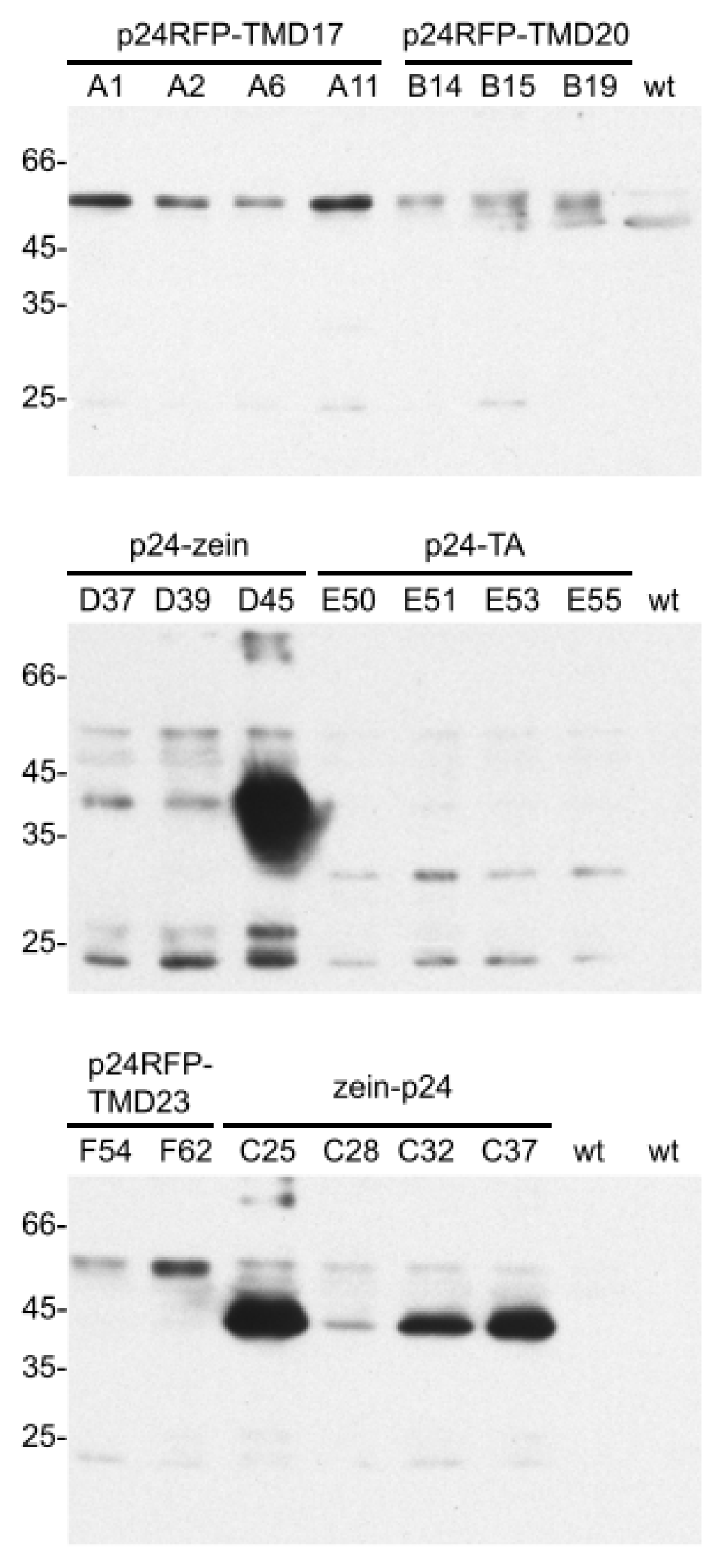
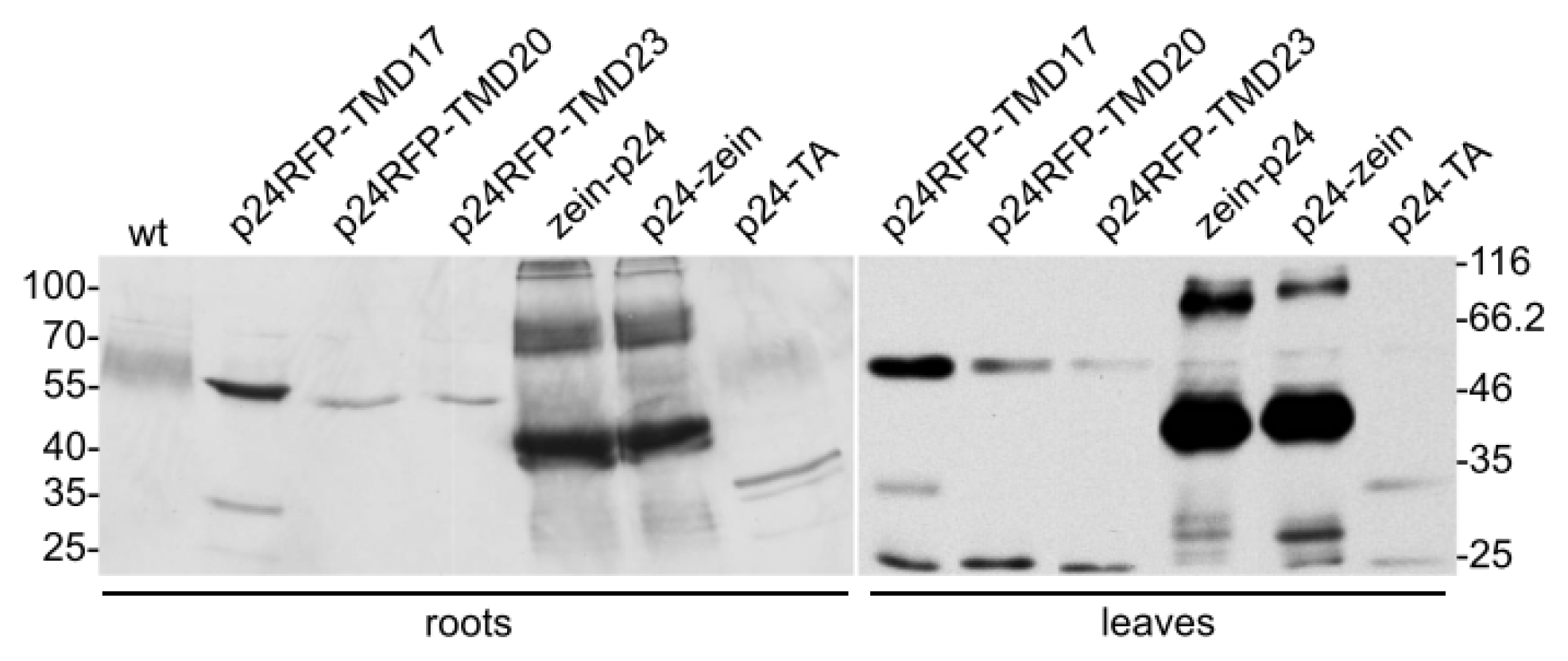
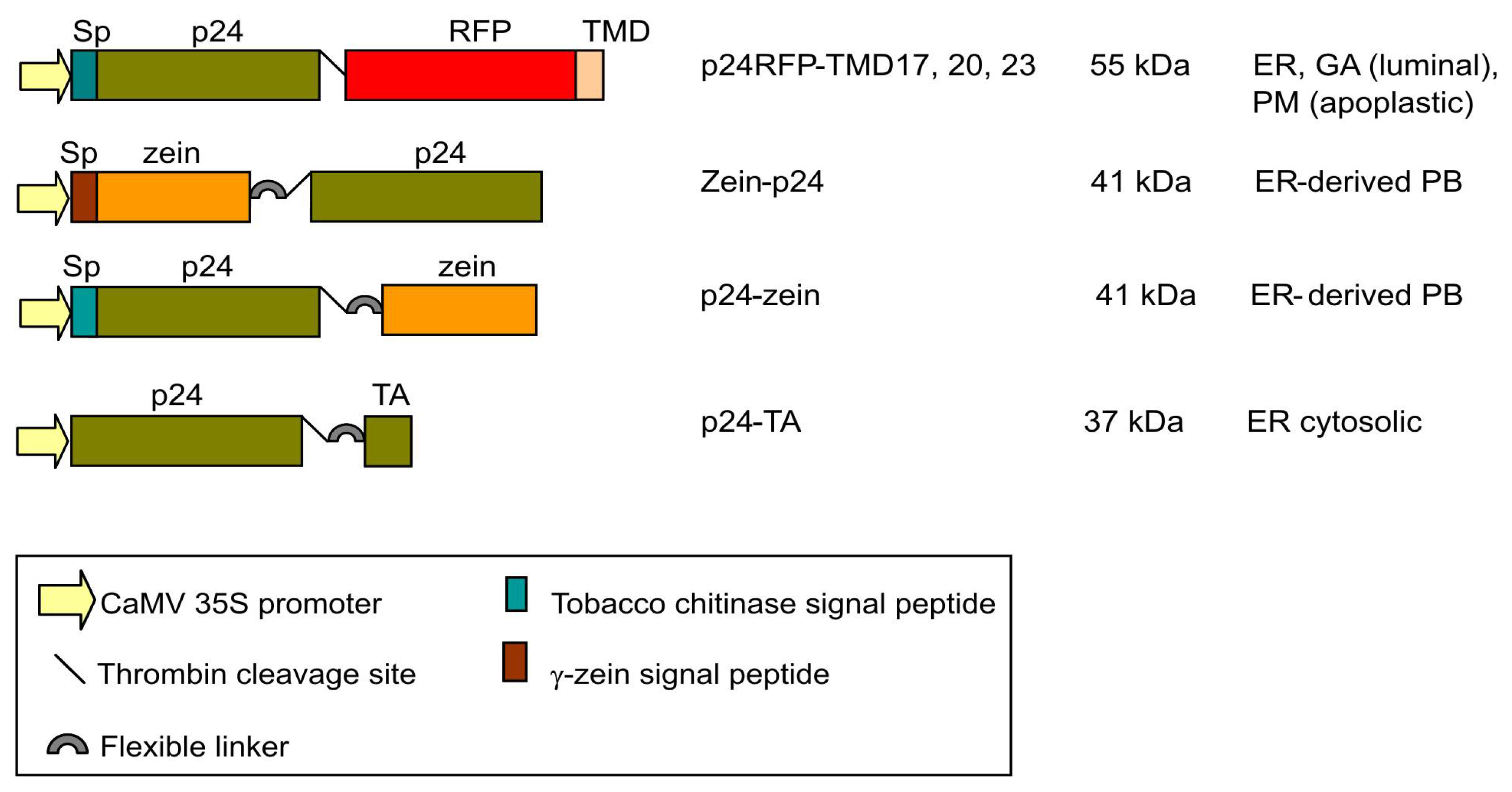
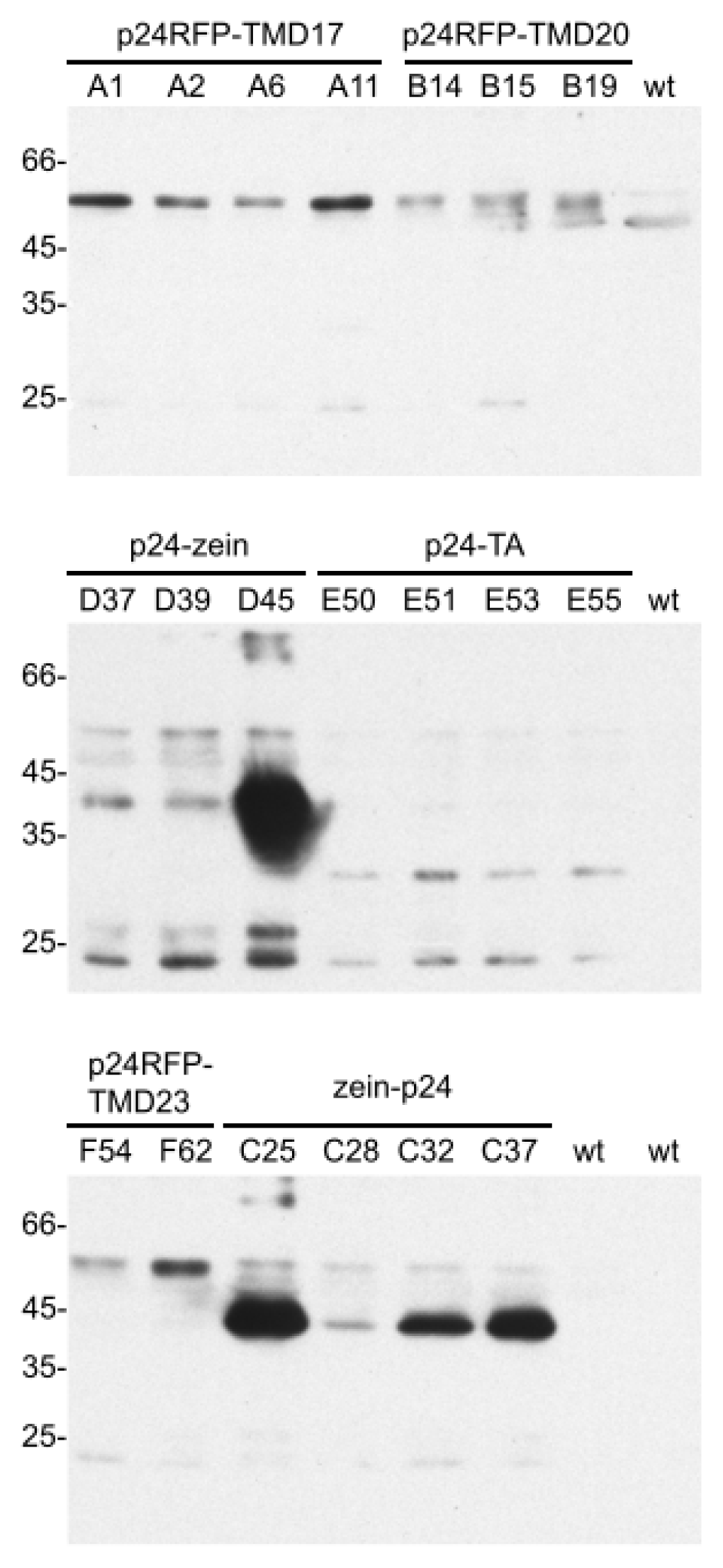
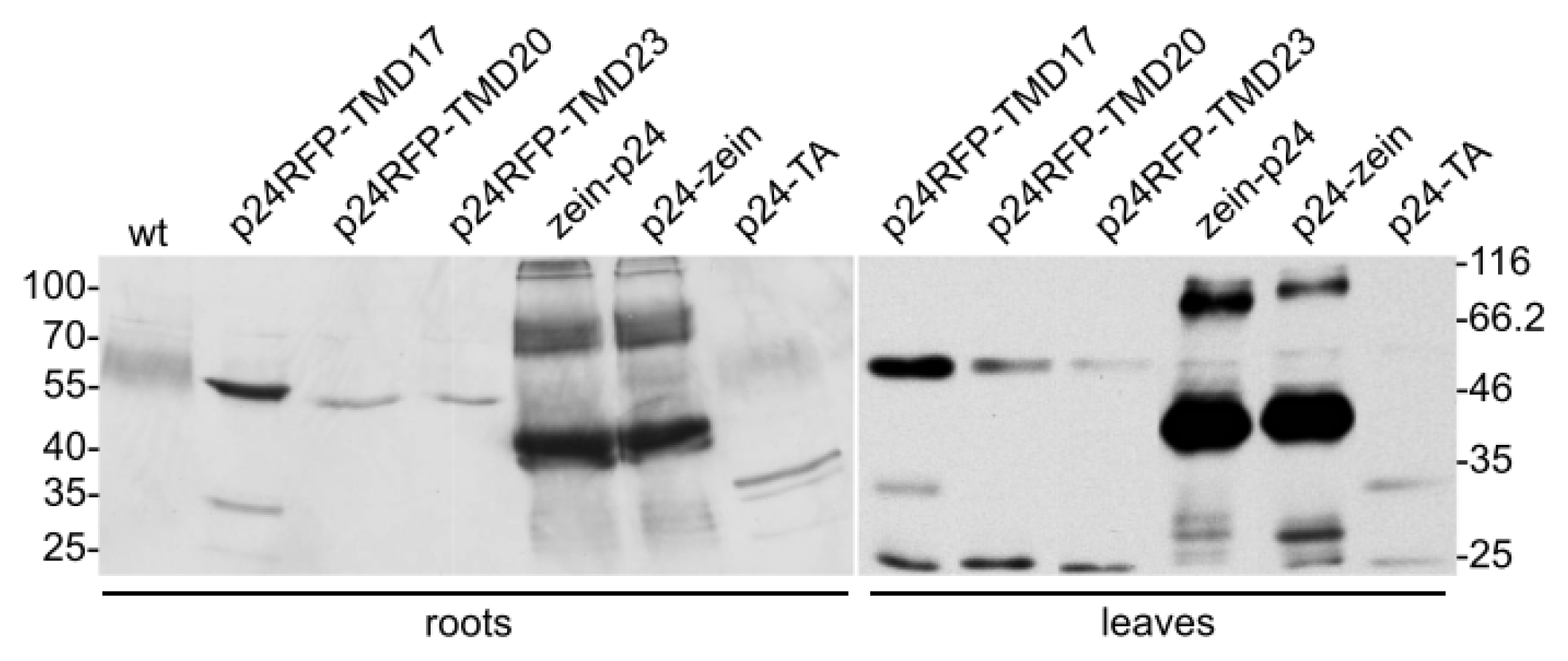
2.1. Accumulation of p24 Engineered into either Type I or TA Integral Membrane Protein, or Fused to the N-Terminal Domain of γ-Zein
2.2. In Vivo Protein Stability
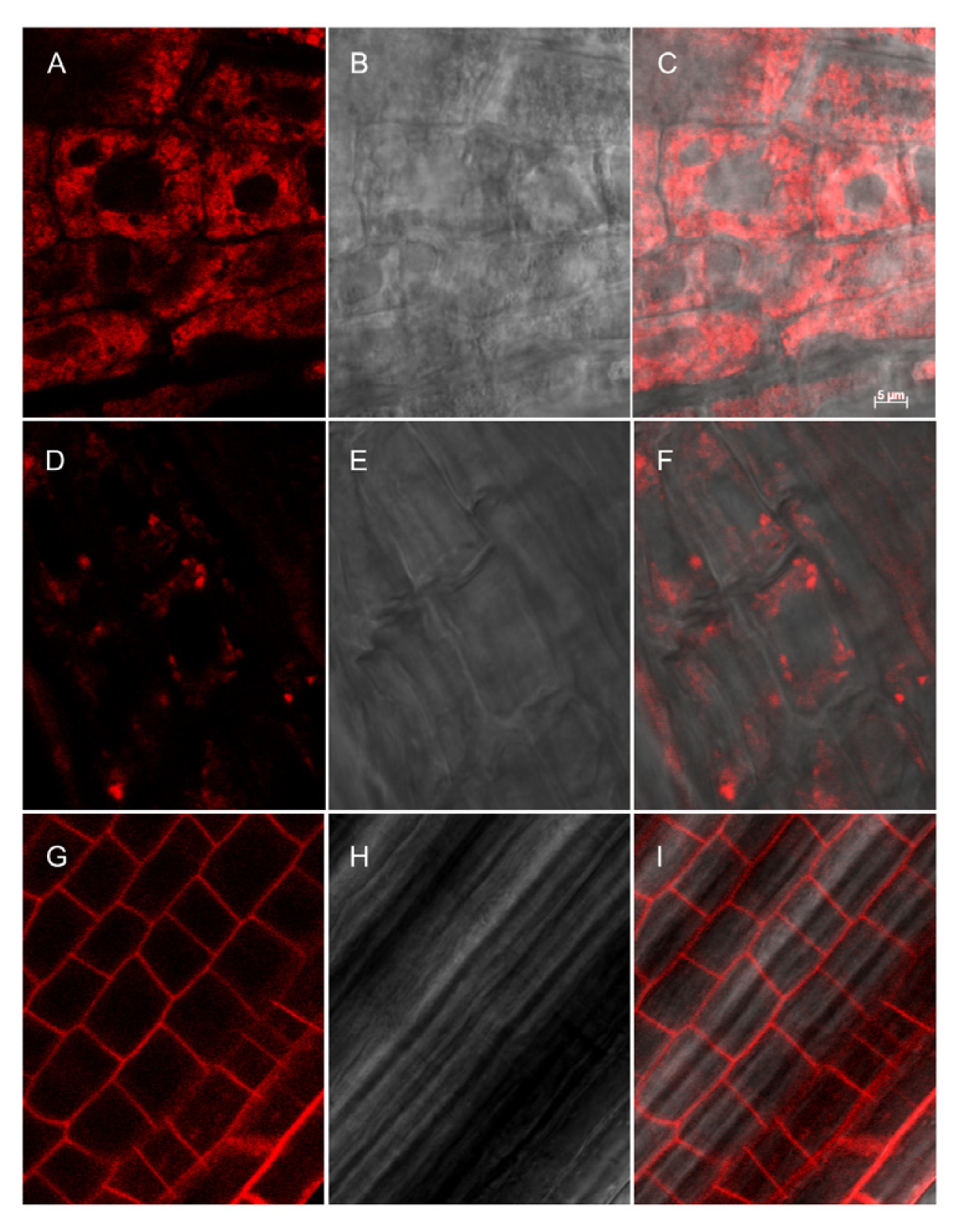
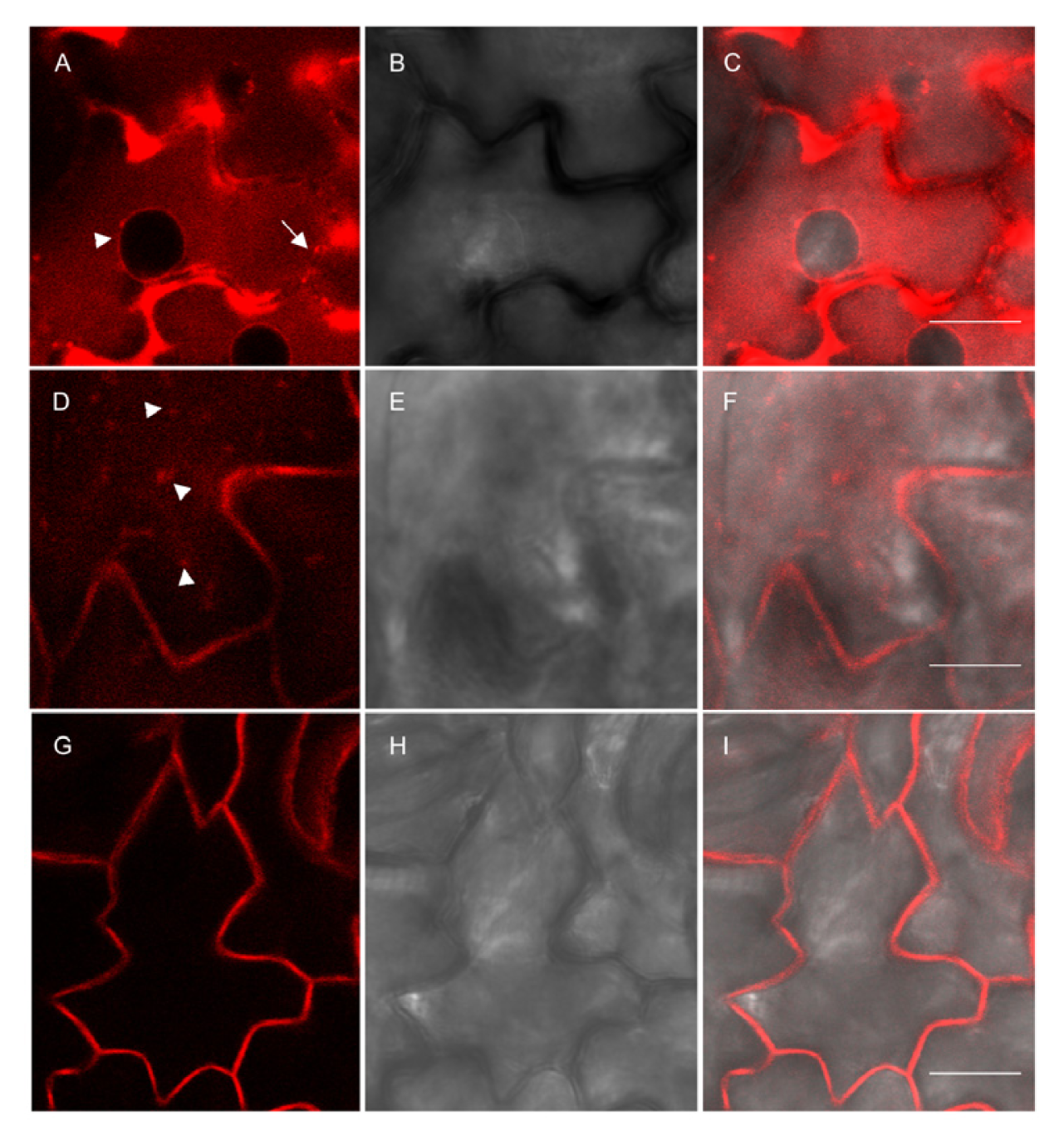
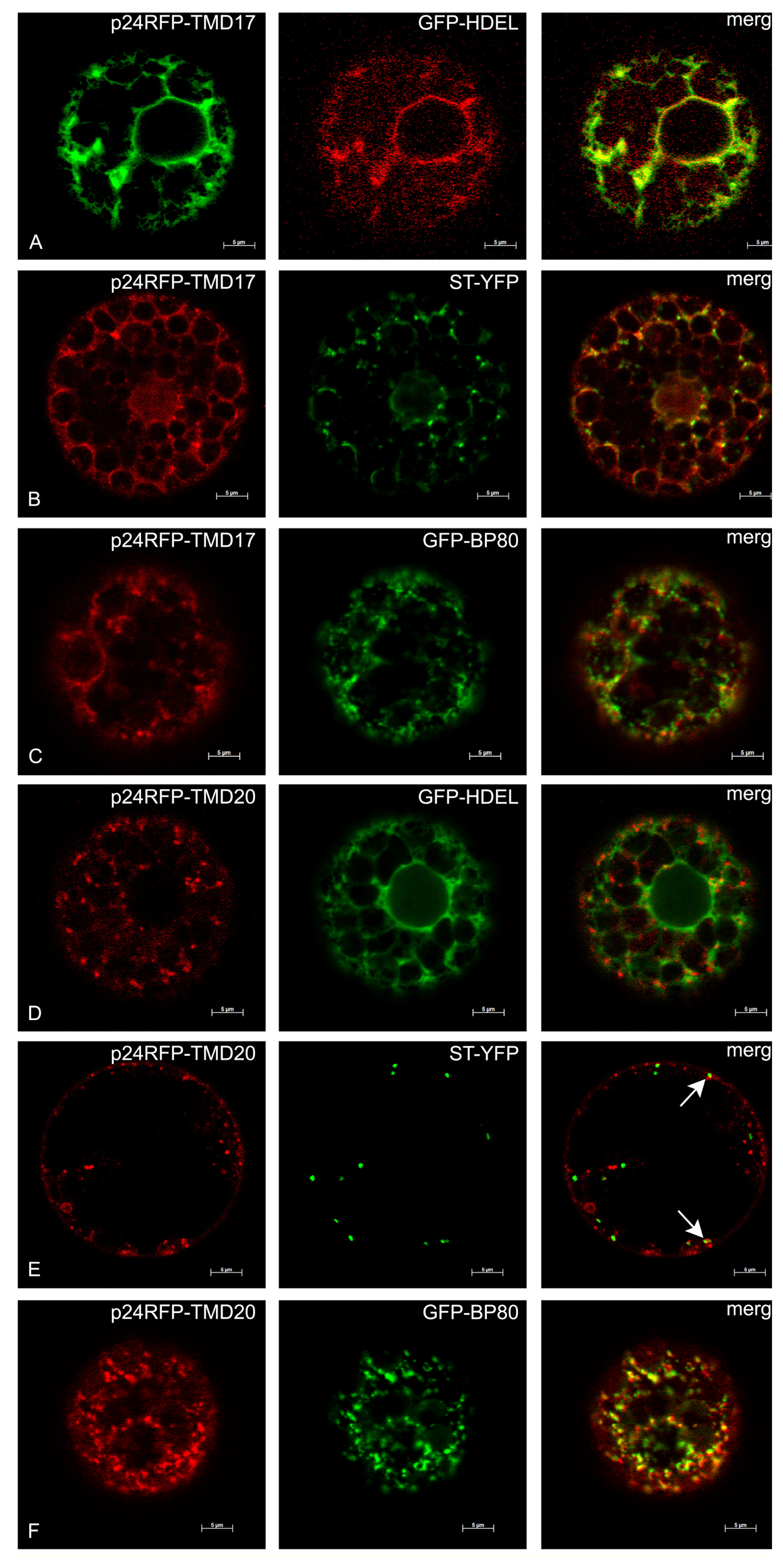
2.3. Subcellular Localization
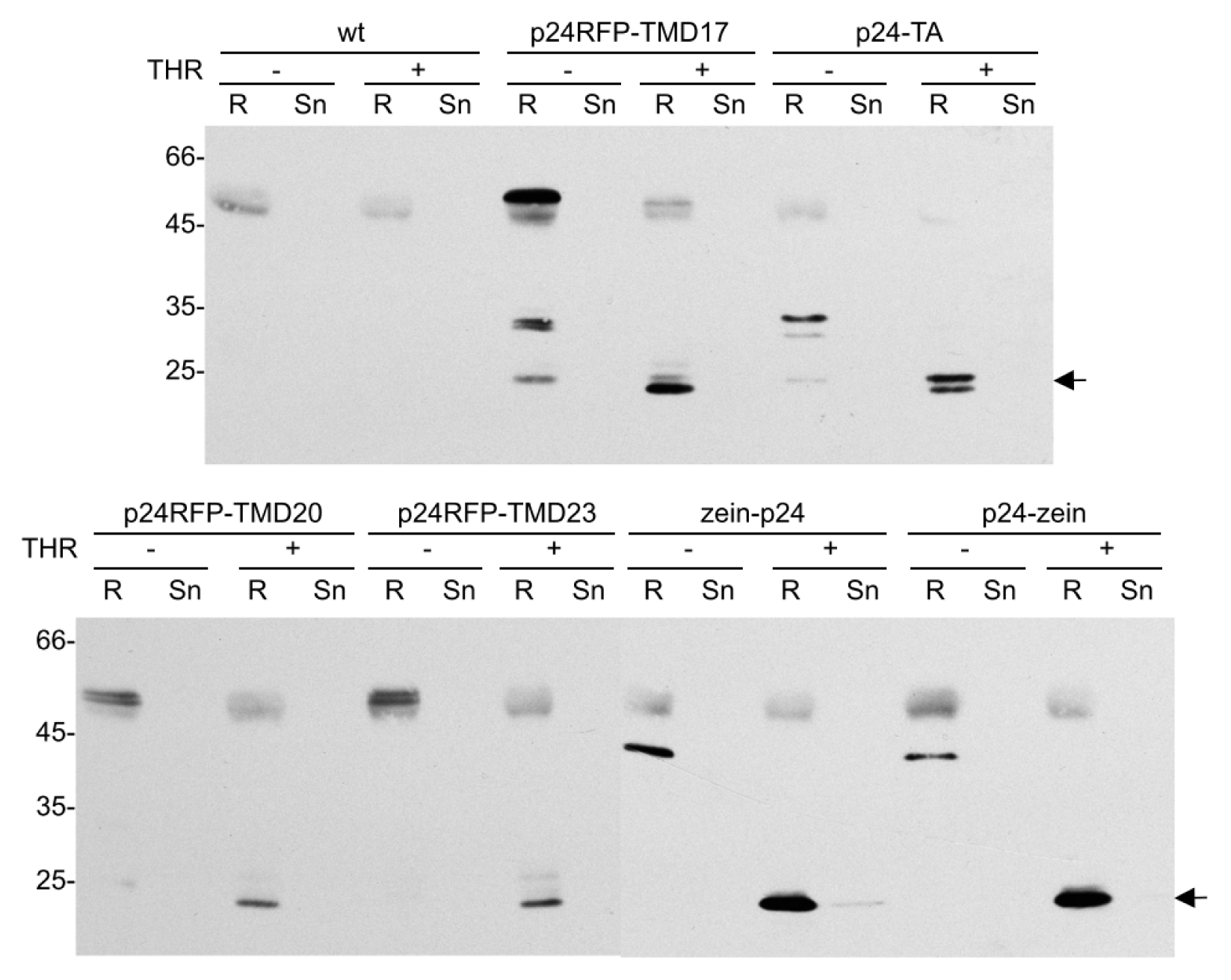
2.4. In Vitro Release of p24 upon Thrombin Cleavage
3. Discussion
3.1. TMD Length and Subcellular Localization
3.2. Attachment to the Cytosolic Face of the ER
3.3. Fusion to the Zein Domain
3.4. Comparison with Other Strategies
4. Experimental Section
4.1. Plasmid Construction and Protein Engineering
4.2. Production and Growth of Transgenic Plants
4.3. Protein Extraction, Western Blot Analysis and Subcellular Fractionation
4.4. Total RNA Isolation and Northern Blot
4.5. Pulse-Chase and Immunoprecipitation
4.6. Confocal Microscopy
4.7. Transient Expression in Protoplasts
4.8. Thrombin Digestion
5. Conclusions
Acknowledgments
Conflict of Interests
References
- Thomas, D.R.; Penney, C.A.; Majumder, A.; Walmsley, A.M. Evolution of plant-made pharmaceuticals. Int. J. Mol. Sci 2011, 12, 3220–3236. [Google Scholar]
- Egelkrout, E.; Rajan, V.; Howard, J.A. Overproduction of recombinant proteins in plants. Plant Sci 2012, 184, 83–101. [Google Scholar]
- Doran, P.M. Foreign protein degradation and instability in plants and plant tissue cultures. Trends Biotechnol 2006, 24, 426–432. [Google Scholar]
- Streatfield, S.J. Approaches to achieve high-level heterologous protein production in plants. Plant Biotechnol. J 2007, 5, 2–15. [Google Scholar]
- Vitale, A.; Pedrazzini, E. Recombinant pharmaceuticals from plants: The plant endomembrane system as bioreactor. Mol. Interv 2005, 5, 216–225. [Google Scholar]
- Khan, I.; Twyman, R.M.; Arcalis, E.; Stöger, E. Using storage organelles for the accumulation and encapsulation of recombinant proteins. Biotechnol. J 2012, 7, 1099–1108. [Google Scholar]
- Wandelt, C.I.; Khan, M.R.; Craig, S.; Schroeder, H.E.; Spencer, D.; Higgins, T.J. Vicilin with carboxy-terminal KDEL is retained in the endoplasmic reticulum and accumulates to high levels in the leaves of transgenic plants. Plant J 1992, 2, 181–192. [Google Scholar]
- De Virgilio, M.; de Marchis, F.; Bellucci, M.; Mainieri, D.; Rossi, M.; Benvenuto, E.; Arcioni, S.; Vitale, A. The human immunodeficiency virus antigen Nef forms protein bodies in leaves of transgenic tobacco when fused to zeolin. J. Exp. Bot 2008, 10, 2815–2829. [Google Scholar]
- Obregon, P.; Chargelegue, D.; Drake, P.M.W.; Prada, A.; Nuttall, J.; Frigerio, L.; Ma, J.K.-C. HIV-1 p24-immunoglobulin fusion molecule: A new strategy for plant-based protein production. Plant Biotechnol. J 2006, 4, 195–207. [Google Scholar]
- Scheller, J.; Leps, M.; Conrad, U. Forcing single-chain variable fragment production in tobacco seeds by fusion to elastin-like polypeptides. Plant Biotech. J 2006, 4, 243–249. [Google Scholar]
- Floss, D.M.; Sack, M.; Stadlmann, J.; Rademacher, T.; Scheller, J.; Stöger, E.; Fischer, R.; Conrad, U. Biochemical and functional characterization of anti-HIV antibody-ELP fusion proteins from transgenic plants. Plant Biotechnol. J 2008, 6, 379–391. [Google Scholar]
- Floss, D.M.; Schallau, K.; Rose-John, S.; Conrad, U.; Scheller, J. Elastin-like polypeptides revolutionize recombinant protein expression and their biomedical application. Trends Biotechnol 2010, 28, 37–45. [Google Scholar]
- Conley, A.J.; Joensuu, J.J.; Menassa, R.; Brandle, J.E. Induction of protein body formation in plant leaves by elastin- like polypeptide fusions. BMC Biol 2009, 7, 48. [Google Scholar]
- Capuano, F.; Beaudoin, F.; Napier, J.A.; Shewry, P.R. Properties and exploitation of oleosins. Biotechnol. Adv. 2007, 25, 203–206. [Google Scholar]
- Parmenter, D.L.; Boothe, J.G.; van Rooijen, G.J.; Yeung, E.C.; Moloney, M.M. Production of biologically active hirudin in plant seeds using oleosin partitioning. Plant Mol. Biol 1995, 29, 1167–1180. [Google Scholar]
- Nykiforuk, C.L.; Boothe, J.G.; Murray, E.W.; Keon, R.G.; Goren, H.J.; Markley, N.A.; Moloney, M.M. Transgenic expression and recovery of biologically active recombinant human insulin from Arabidopsis thaliana seeds. Plant Biotechnol. J 2006, 4, 77–85. [Google Scholar]
- Vine, N.D.; Drake, P.; Hiatt, A.; Ma, J.K.-C. Assembly and plasma membrane targeting of recombinant immunoglobulin chains in plants with a murine immunoglobulin transmembrane sequence. Plant Mol. Biol 2001, 45, 159–167. [Google Scholar]
- Barbi, T.; Drake, P.M.W.; Drever, M.; van Dolleweerd, C.J.; Porter, A.R.; Ma, J.K.-C. Generation of transgenic plants expressing plasma membrane-bound antibodies to the environmental pollutant microcystin-LR. Transgenic Res 2011, 20, 701–707. [Google Scholar]
- Paris, N.; Rogers, S.W.; Jiang, L.W.; Kirsch, T.; Beevers, L.; Phillips, T.E.; Rogers, J.C. Molecular cloning and further characterization of a probable plant vacuolar sorting receptor. Plant Physiol 1997, 115, 29–39. [Google Scholar]
- Miao, Y.S.; Yan, P.K.; Kim, H.; Hwang, I.; Jiang, L.W. Localization of green fluorescent protein fusions with the seven Arabidopsis vacuolar sorting receptors to prevacuolar compartments in tobacco BY-2 cells. Plant Physiol 2006, 142, 945–962. [Google Scholar]
- Niemes, S.; Langhans, M.; Viotti, C.; Scheuring, D.; Yan, M.S.W.; Jiang, R.; Hillmer, S.; Robinson, D.G.; Pimpl, P. Retromer recycles vacuolar sorting receptors from the trans Golgi. Plant J 2010, 61, 102–121. [Google Scholar]
- Fung, K.L.; Yim, Y.F.; Tse, Y.C.; Miao, Y.S.; Sun, S.S.M.; Jiang, L.W. Targeting and processing of membrane-anchored YFP fusion proteins to protein storage vacuoles in transgenic tobacco seeds. Seed Sci. Res 2005, 15, 361–364. [Google Scholar]
- Borgese, N.; Brambillasca, S.; Soffientini, P.; Yabal, M.; Makarow, M. Biogenesis of tail-anchored proteins. Biochem. Soc. Trans 2003, 31, 1238–1242. [Google Scholar]
- Maggio, C.; Barbante, A.; Ferro, F.; Frigerio, L.; Pedrazzini, E. Intracellular sorting of the tail-anchored protein cytochrome b5 in plants: A comparative study using different isoforms from rabbit and Arabidopsis. J. Exp. Bot 2007, 58, 1365–1379. [Google Scholar]
- Barbante, A.; Irons, S.; Hawes, C.; Frigerio, L.; Vitale, A.; Pedrazzini, E. Anchorage to the cytosolic face of the endoplasmic reticulum membrane: A new strategy to stabilize a cytosolic recombinant antigen in plants. Plant Biotechnol. J 2008, 6, 560–575. [Google Scholar]
- Munro, S. An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J 1995, 14, 4695–4704. [Google Scholar]
- Brandizzi, F.; Frangne, N.; Marc-Martin, S.; Hawes, C.; Neuhaus, J.M.; Paris, N. The destination for single-pass membrane proteins is influenced markedly by the length of the hydrophobic domain. Plant Cell 2002, 14, 1077–1092. [Google Scholar]
- Novitsky, V.; Gilbert, P.; Peter, T.; McLane, M.F.; Gaolekwe, S.; Rybak, N.; Thior, I.; Ndung’u, T.; Marlink, R.; Lee, T.H.; et al. Association between virus-specific T-cell responses and plasma viral load in human immunodeficiency virus type 1 subtype C infection. J. Virol 2003, 77, 882–890. [Google Scholar]
- Zuñiga, R.; Lucchetti, A.; Galvan, P.; Sanchez, S.; Sanchez, C.; Hernandez, A.; Sanchez, H.; Frahm, N.; Linde, C.H.; Hewitt, H.S.; et al. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol 2006, 80, 3122–3125. [Google Scholar]
- Zhang, G.G.; Rodrigues, L.; Rovinski, B.; White, K.A. Production of HIV-1 p24 protein in transgenic tobacco plants. Mol. Biotechnol 2002, 20, 131–136. [Google Scholar]
- Lindh, I.; Kalbina, I.; Thulin, S.; Scherbak, N.; Savenstrand, H.; Brave, A.; Hinkula, J.; Strid, A.; Andersson, S. Feeding of mice with Arabidopsis thaliana expressing the HIV-1 subtype C p24 antigen gives rise to systemic immune responses. Acta Pathol. Microbiol. Immunol. Scand 2008, 116, 985–994. [Google Scholar]
- McCabe, M.S.; Klaas, M.; Gonzalez-Rabade, N.; Poage, M.; Badillo-Corona, J.A.; Zhou, F.; Karcher, D.; Bock, R.; Gray, J.C.; Dix, P.J. Plastid transformation of high-biomass tobacco variety Maryland Mammoth for production of human immunodeficiency virus type 1 (HIV-1) p24 antigen. Plant Biotechnol. J 2008, 6, 914–929. [Google Scholar]
- Zhou, F.; Badillo-Corona, J.A.; Karcher, D.; Gonzalez-Rabade, N.; Piepenburg, K.; Borchers, A.M.; Maloney, A.P.; Kavanagh, T.A.; Gray, J.C.; Bock, R. High-level expression of human immunodeficiency virus antigens from the tobacco and tomato plastid genomes. Plant Biotechnol. J 2008, 6, 897–913. [Google Scholar]
- Mainieri, D.; Rossi, M.; Archinti, M.; Bellucci, M.; De Marchis, F.; Vavassori, S.; Pompa, A.; Arcioni, S.; Vitale, A. Zeolin. A new recombinant storage protein constructed using maize gamma-zein and bean phaseolin. Plant Physiol 2004, 136, 3447–3456. [Google Scholar]
- Torrent, M.; Llompart, B.; Lasserre-Ramassamy, S.; Llop-Tous, I.; Bastida, M.; Marzabal, P.; Westerholm-Parvinen, A.; Saloheimo, M.; Heifetz, P.B.; Ludevid, M.D. Eukaryotic protein production in designed storage organelles. BMC Biol 2009, 7, 5. [Google Scholar]
- Vitale, A.; Smaniotto, E.; Longhi, R.; Galante, E. Reduced soluble proteins associated with maize endosperm protein bodies. J. Exp. Bot 1982, 33, 439–448. [Google Scholar]
- Kogan, M.J.; Dalcol, I.; Gorostiza, P.; Lopez-Iglesias, C.; Pons, R.; Miquel Pons, M.; Sanz, F.; Giralt, E. Supramolecular properties of the proline-rich γ-zein N-terminal domain. Biophys. J 2002, 83, 1194–1204. [Google Scholar]
- Burr, B.; Burr, F.A.; Rubinstein, I.; Simon, M.N. Purification and translation of zein messenger RNA from maize endosperm protein bodies. Proc. Natl. Acad. Sci. USA 1978, 75, 696–700. [Google Scholar]
- Bellucci, M.; De Marchis, F.; Mannucci, R.; Bock, R.; Arcioni, S. Cytoplasm and chloroplasts are not suitable subcellular locations for β-zein accumulation in transgenic plants. J. Exp. Bot 2005, 56, 1205–1212. [Google Scholar]
- Bellucci, M.; De Marchis, F.; Nicoletti, I.; Arcioni, S. Zeolin is a recombinant storage protein with different solubility and stability properties according to its localization in the endoplasmic reticulum or in the chloroplast. J. Biotechnol 2007, 131, 97–105. [Google Scholar]
- Brandizzi, F.; Hanton, S.; DaSilva, L.L.; Boevink, P.; Evan, D.; Oparka, K.; Denecke, J.; Hawes, C. ER quality control can lead to retrograde transport from the ER lumen to the cytosol and the nucleoplasm in plants. Plant J 2003, 34, 269–281. [Google Scholar]
- daSilva, L.L.; Taylor, J.P.; Hadlington, J.L.; Hanton, S.L.; Snowden, C.J.; Fox, S.J.; Foresti, O.; Brandizzi, F.; Denecke, J. Receptor salvage from the prevacuolar compartment is essential for efficient vacuolar protein targeting. Plant Cell 2005, 17, 132–148. [Google Scholar]
- Pedrazzini, E.; Giovinazzo, G.; Bielli, A.; de Virgilio, M.; Frigerio, L.; Pesca, M.; Faoro, F.; Bollini, R.; Ceriotti, A.; Vitale, A. Protein quality control along the route to the plant vacuole. Plant Cell 1997, 10, 1869–1880. [Google Scholar]
- Marusic, C.; Nuttall, J.; Buriani, G.; Lico, C.; Lombardi, R.; Baschieri, S.; Benvenuto, E.; Frigerio, L. Expression, intracellular targeting and purification of HIV Nef variants in tobacco cells. BMC Biotechnol 2007, 7, 12. [Google Scholar]
- Bar-Peled, M.; Da Silva Conceição, A.; Frigerio, L.; Raikhel, N.V. Expression and regulation of aERD2, a gene encoding the KDEL receptor homolog in plants, and other genes encoding protein involved in ER-Golgi vesicular trafficking. Plant Cell 1995, 7, 667–676. [Google Scholar]
- Denecke, J.; Carlsson, L.E.; Vidal, S.; Hoglund, A.S.; Ek, B.; van Zeijl, M.J.; Sinjorgo, K.M.; Palva, E.T. The tobacco homolog of mammalian calreticulin is present in protein complexes in vivo. Plant Cell 1995, 7, 391–406. [Google Scholar]
- Pedrazzini, E.; Komarova, N.; Rentsch, D.; Vitale, A. Traffic routes and signals for the tonoplast. Traffic 2013, 14, 622–628. [Google Scholar]
- Srivastava, R.; Chen, Y.; Deng, Y.; Brandizzi, F.; Howell, S.H. Elements proximal to and within the transmembrane domain mediate the organelle-to-organelle movement of bZIP28 under ER stress conditions. Plant J 2012, 70, 1033–1042. [Google Scholar]
- Pedrazzini, E. Tail-anchored proteins in plants. J. Plant Biol 2009, 52, 88–101. [Google Scholar]
- Pompa, A.; Vitale, A. Retention of a bean phaseolin/maize γ-zein fusion in the endoplasmic reticulum depends on disulfide bond formation. Plant Cell 2006, 18, 2608–2621. [Google Scholar]
- Foresti, O.; De Marchis, F.; de Virgilio, M.; Klein, E.M.; Arcioni, S.; Bellucci, M.; Vitale, A. Protein domains involved in assembly in the endoplasmic reticulum promote vacuolar delivery when fused to secretory GFP, indicating a protein quality control pathway for degradation in the plant vacuole. Mol. Plant 2008, 1, 1067–1076. [Google Scholar]
- Ma, J.K.-C.; Hiatt, A.; Hein, M.; Vine, N.D.; Wang, F.; Stabila, P.; von Dolleweerd, C.; Mostov, K.; Lehner, T. Generation and assembly of secretory antibodies in plants. Science 1995, 268, 716–719. [Google Scholar]
- Scotti, N.; Alagna, F.; Ferraiolo, E.; Formisano, G.; Sannino, L.; Buonaguro, L.; De Stradis, A.; Vitale, A.; Monti, L.; Grillo, S.; et al. High-level expression of the HIV-1 Pr55(gag) polyprotein in transgenic tobacco chloroplasts. Planta 2009, 229, 1109–1122. [Google Scholar]
- De Marchis, F.; Pompa, A.; Bellucci, M. Plastid proteostasis and heterologous protein accumulation in transplastomic plants. Plant Physiol 2012, 160, 571–581. [Google Scholar]
- Viotti, A.; Abildsten, D.; Pogna, N.; Sala, E.; Pirrotta, V. Multiplicity and diversity of cloned zein cDNA sequences and their chromosomal localisation. EMBO J 1982, 1, 53–58. [Google Scholar]
- Adamus, G.; Arendt, A.; Hargrave, P.A. Genetic control of antibody response to bovine rhodopsin in mice: epitope mapping of rhodopsin structure. J. Neuroimmunol 1991, 34, 89–97. [Google Scholar]
- Bubeck, J.; Scheuring, D.; Hummel, E.; Langhans, M.; Viotti, C.; Foresti, O.; Denecke, J.; Banfield, D.K.; Robinson, D.G. The syntaxins SYP31 and SYP81 control ER-Golgi trafficking in the plant secretory pathway. Traffic 2008, 9, 1629–1652. [Google Scholar]





© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Virgili-López, G.; Langhans, M.; Bubeck, J.; Pedrazzini, E.; Gouzerh, G.; Neuhaus, J.-M.; Robinson, D.G.; Vitale, A. Comparison of Membrane Targeting Strategies for the Accumulation of the Human Immunodeficiency Virus p24 Protein in Transgenic Tobacco. Int. J. Mol. Sci. 2013, 14, 13241-13265. https://doi.org/10.3390/ijms140713241
Virgili-López G, Langhans M, Bubeck J, Pedrazzini E, Gouzerh G, Neuhaus J-M, Robinson DG, Vitale A. Comparison of Membrane Targeting Strategies for the Accumulation of the Human Immunodeficiency Virus p24 Protein in Transgenic Tobacco. International Journal of Molecular Sciences. 2013; 14(7):13241-13265. https://doi.org/10.3390/ijms140713241
Chicago/Turabian StyleVirgili-López, Goretti, Markus Langhans, Julia Bubeck, Emanuela Pedrazzini, Guillaume Gouzerh, Jean-Marc Neuhaus, David G. Robinson, and Alessandro Vitale. 2013. "Comparison of Membrane Targeting Strategies for the Accumulation of the Human Immunodeficiency Virus p24 Protein in Transgenic Tobacco" International Journal of Molecular Sciences 14, no. 7: 13241-13265. https://doi.org/10.3390/ijms140713241
APA StyleVirgili-López, G., Langhans, M., Bubeck, J., Pedrazzini, E., Gouzerh, G., Neuhaus, J.-M., Robinson, D. G., & Vitale, A. (2013). Comparison of Membrane Targeting Strategies for the Accumulation of the Human Immunodeficiency Virus p24 Protein in Transgenic Tobacco. International Journal of Molecular Sciences, 14(7), 13241-13265. https://doi.org/10.3390/ijms140713241