A Novel Low Temperature PCR Assured High-Fidelity DNA Amplification

Abstract
:1. Introduction
2. Results and Discussion
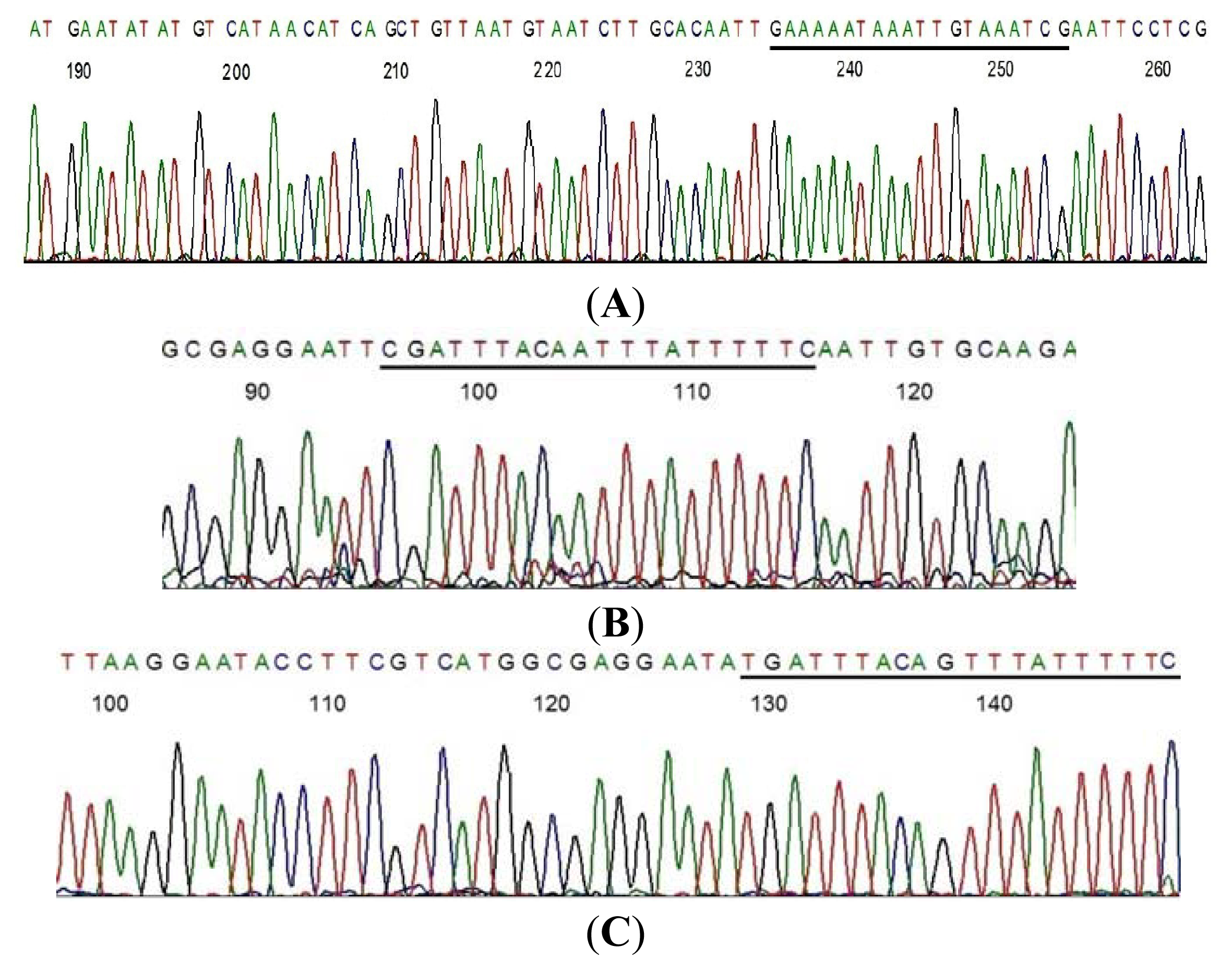
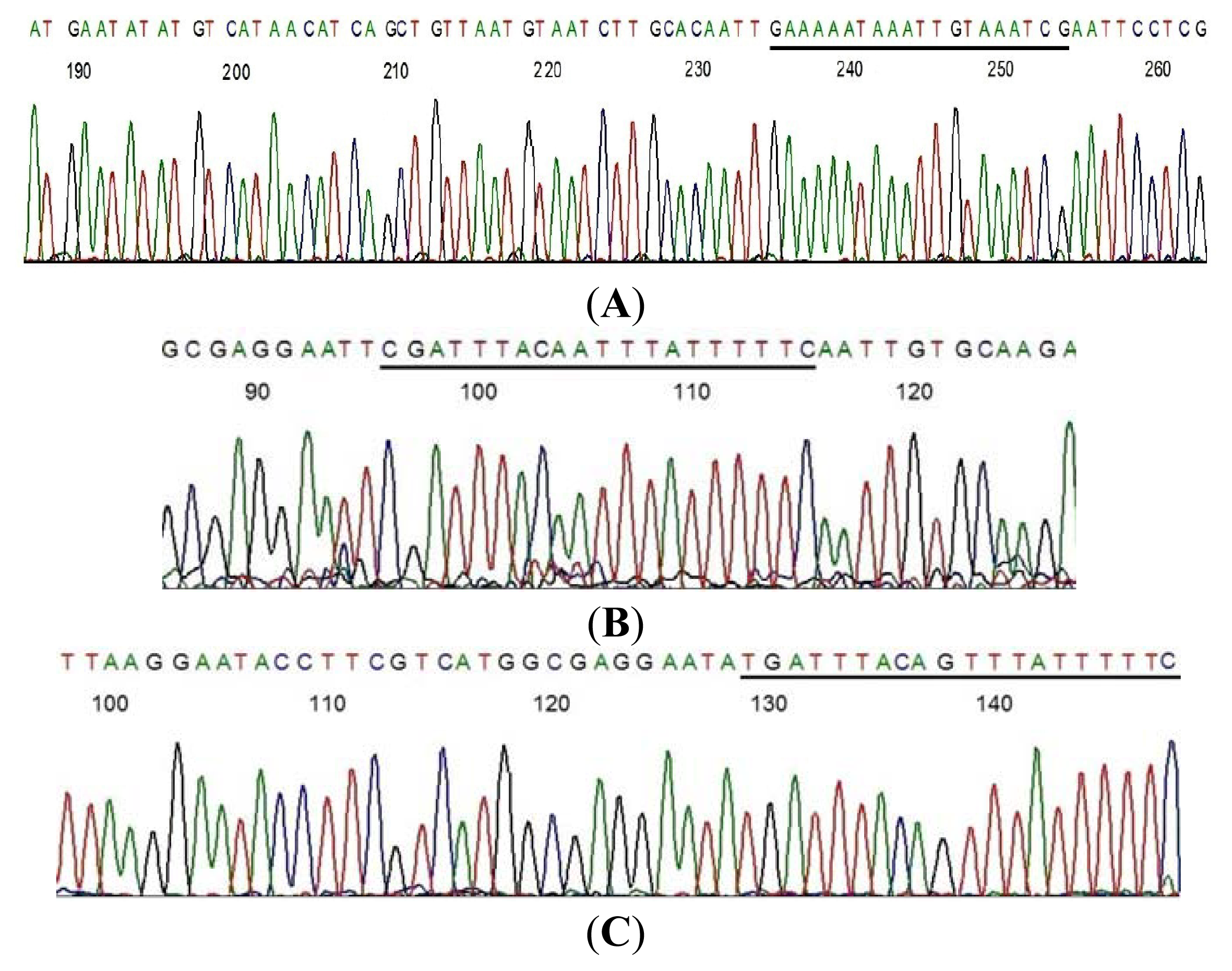
2.1. LoTemp PCR Extension of Primer with Mismatched Base at the 3′-Terminus
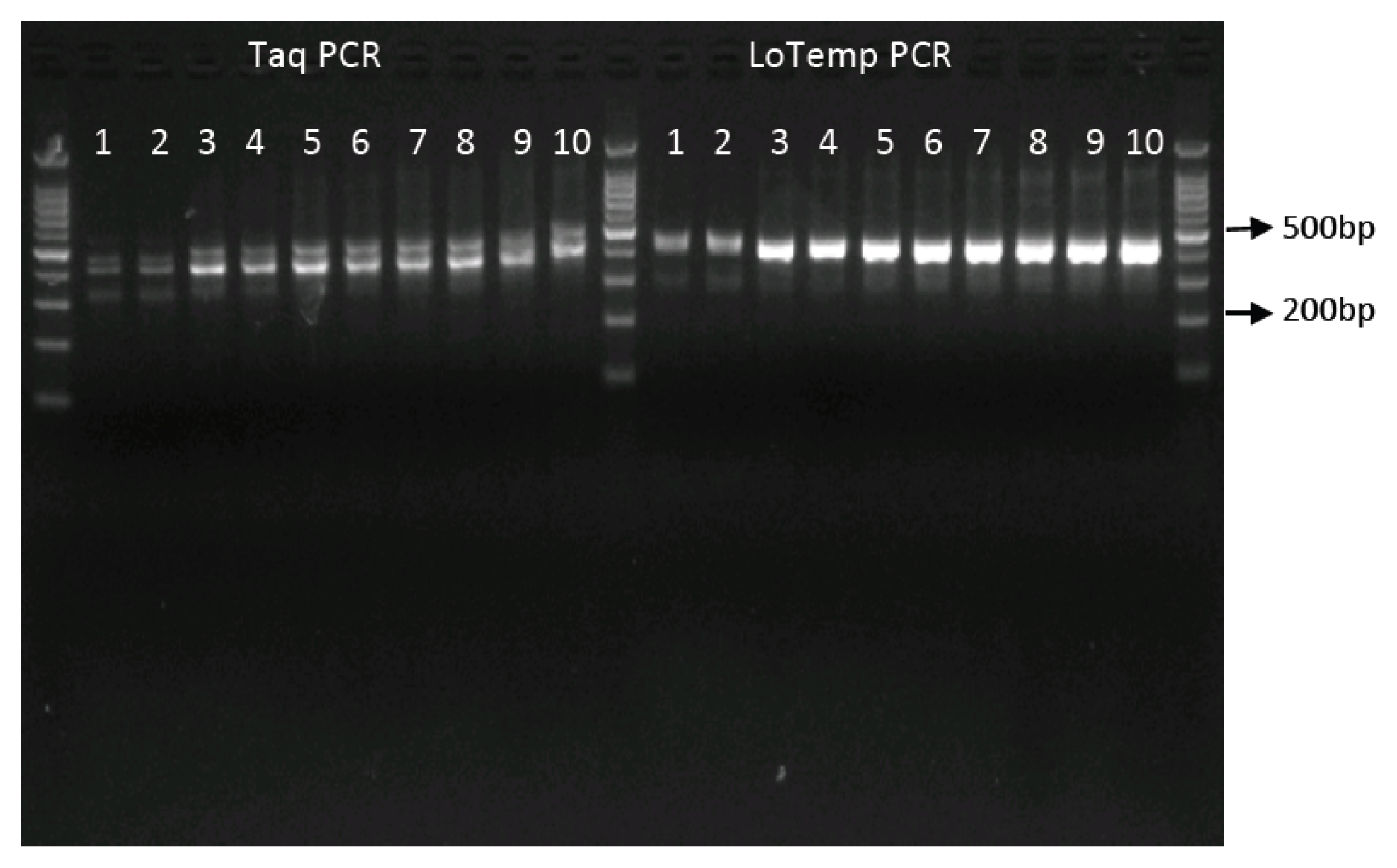
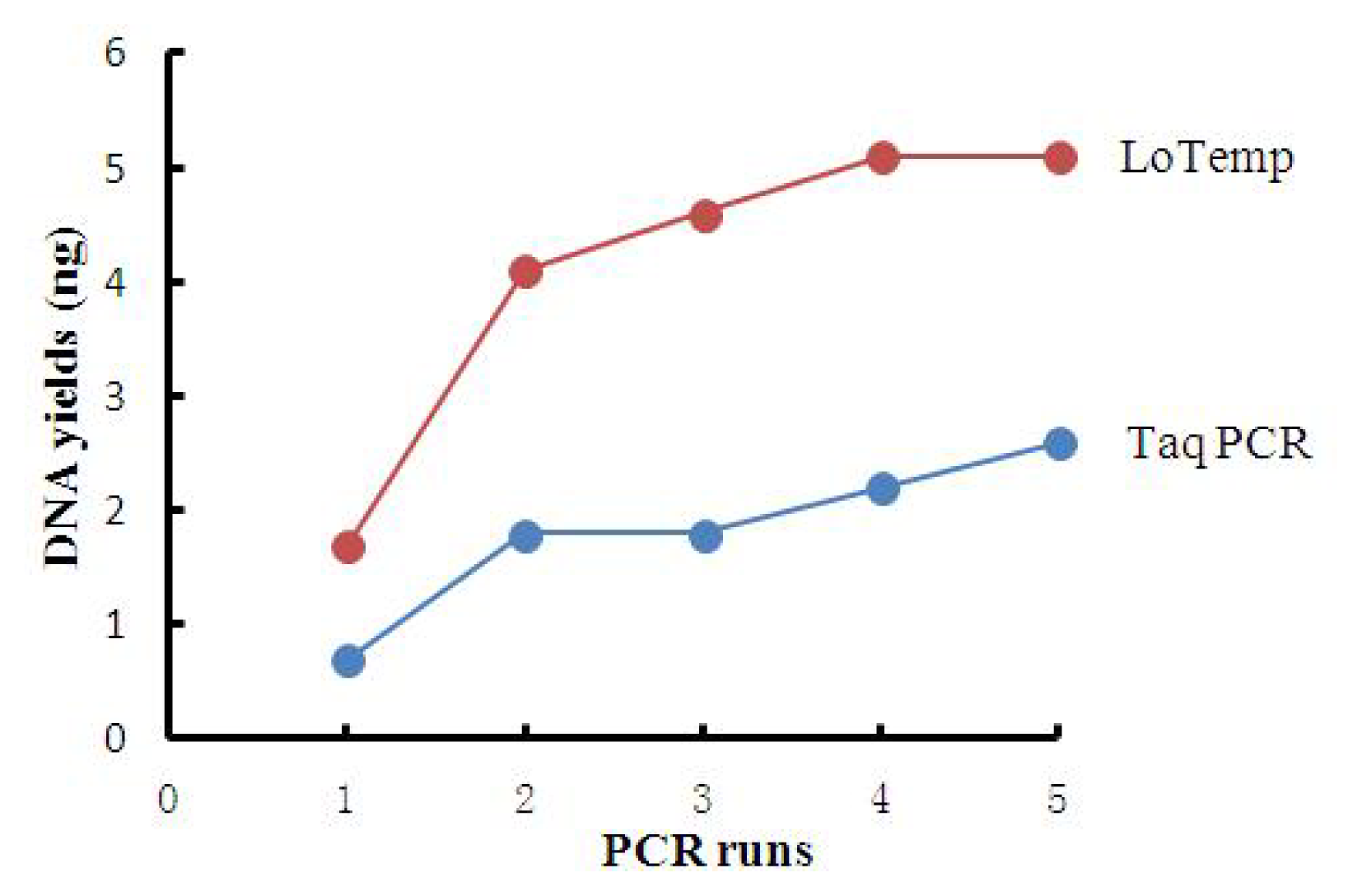
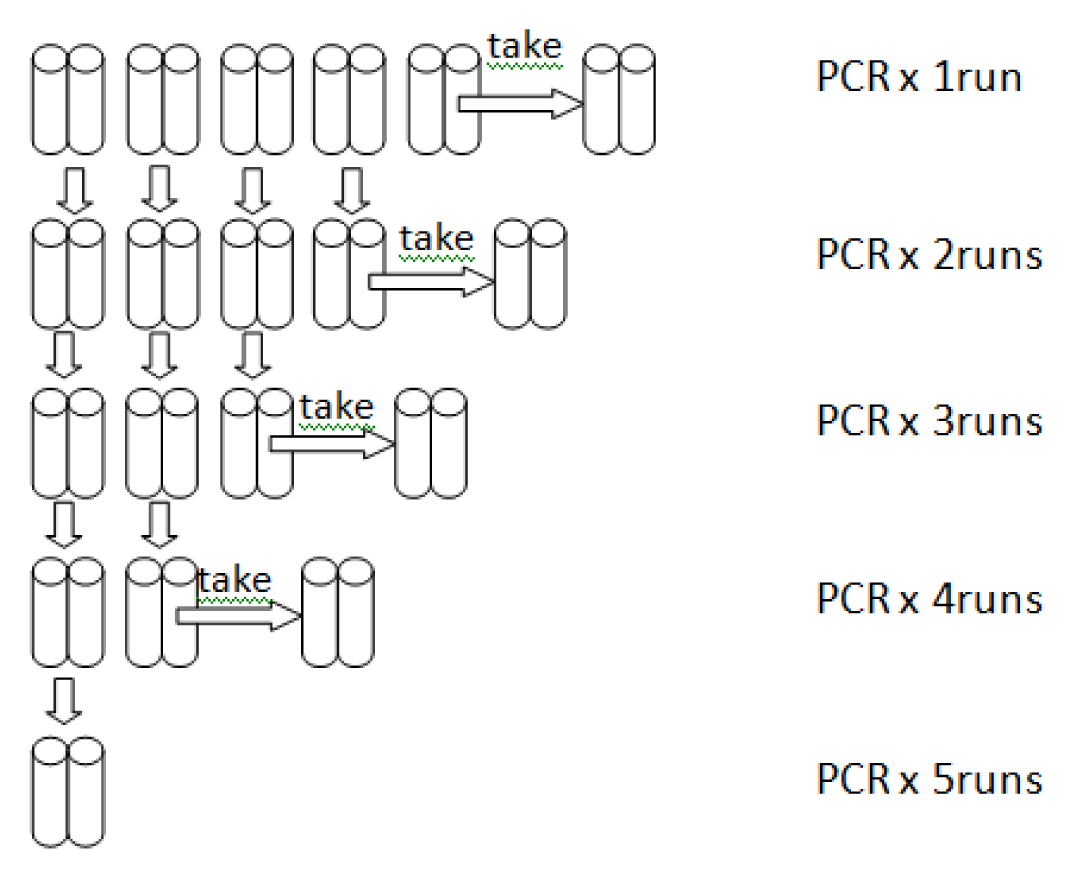
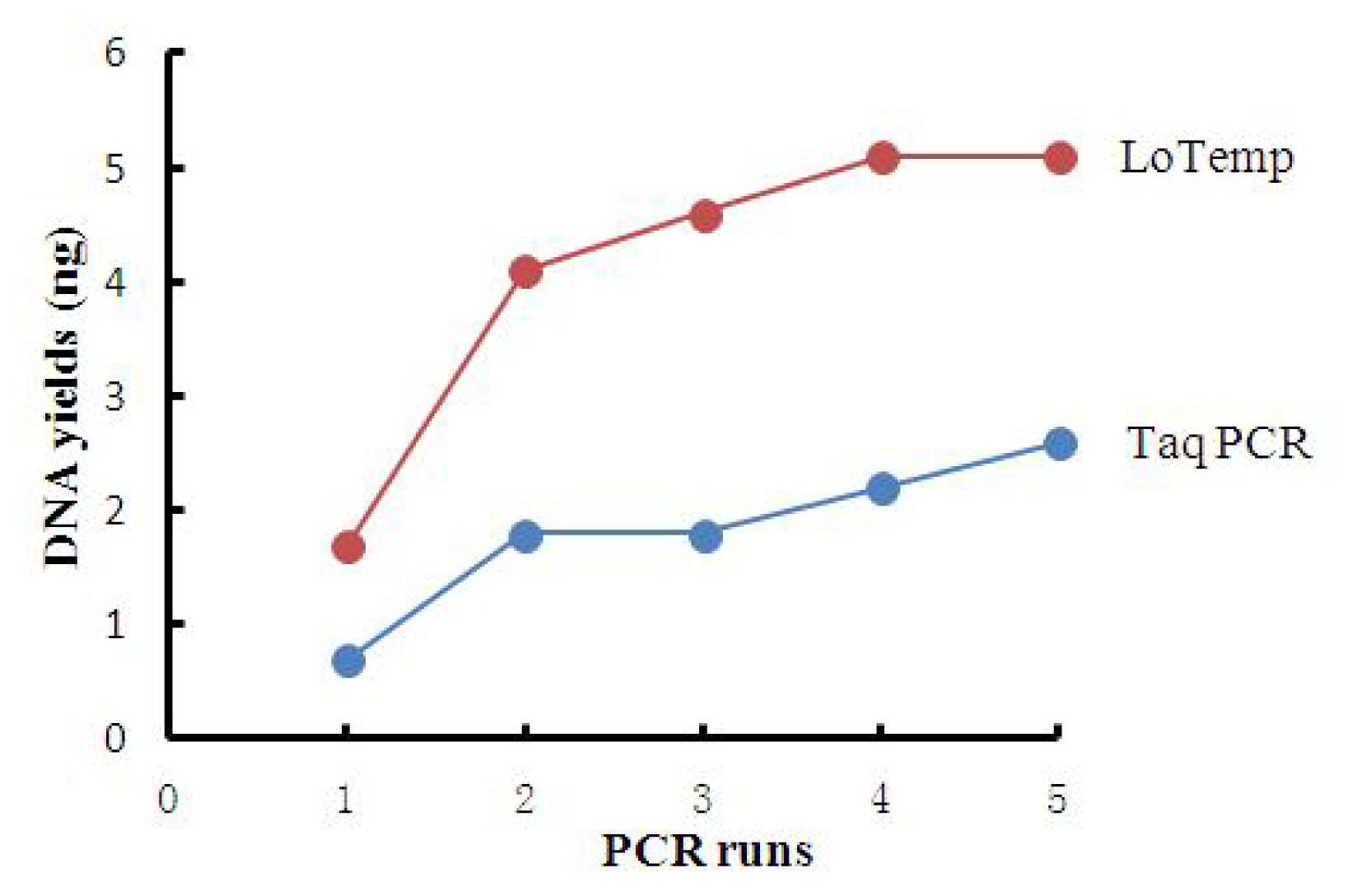
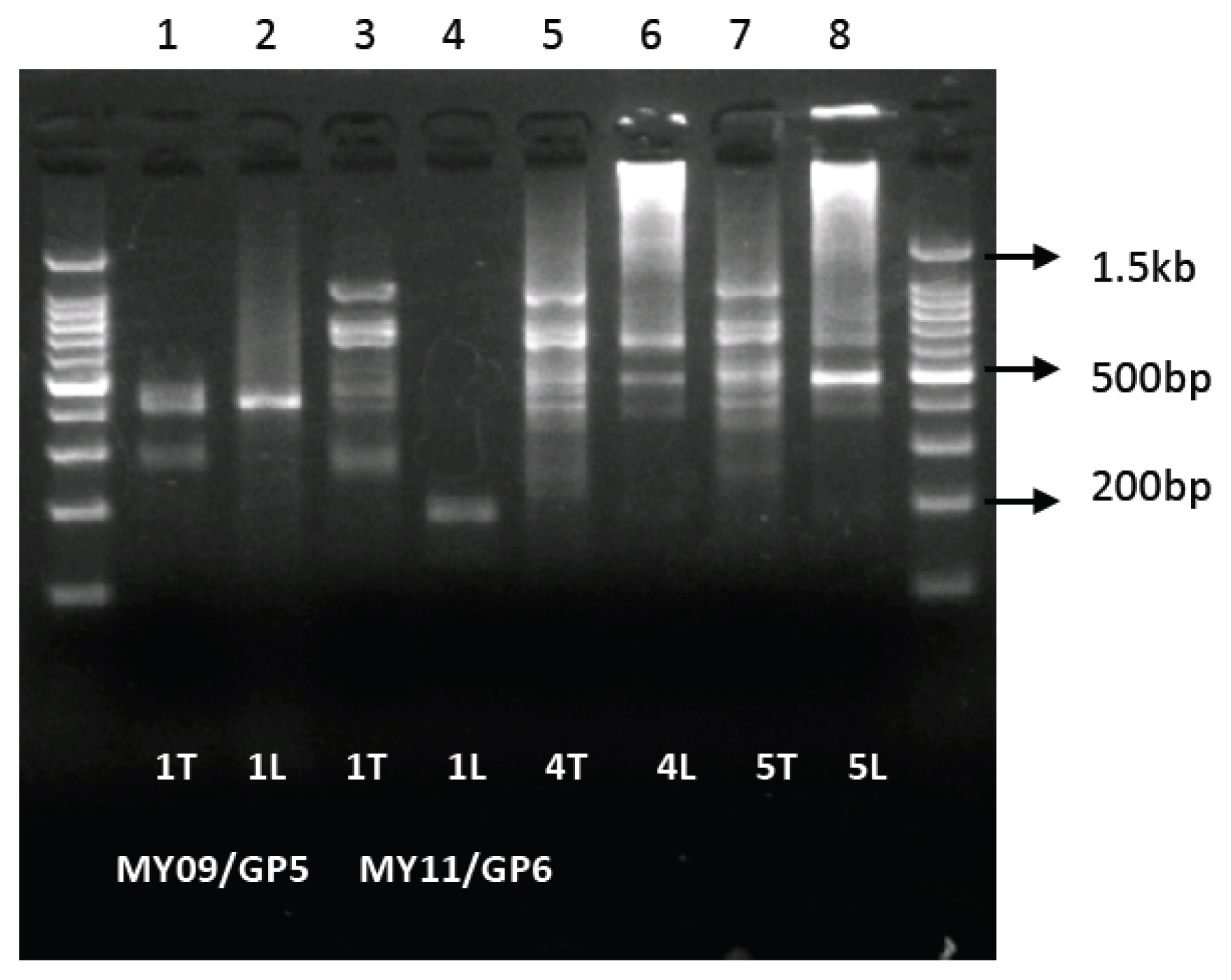
2.2. High Amplicon Yield by LoTemp PCR
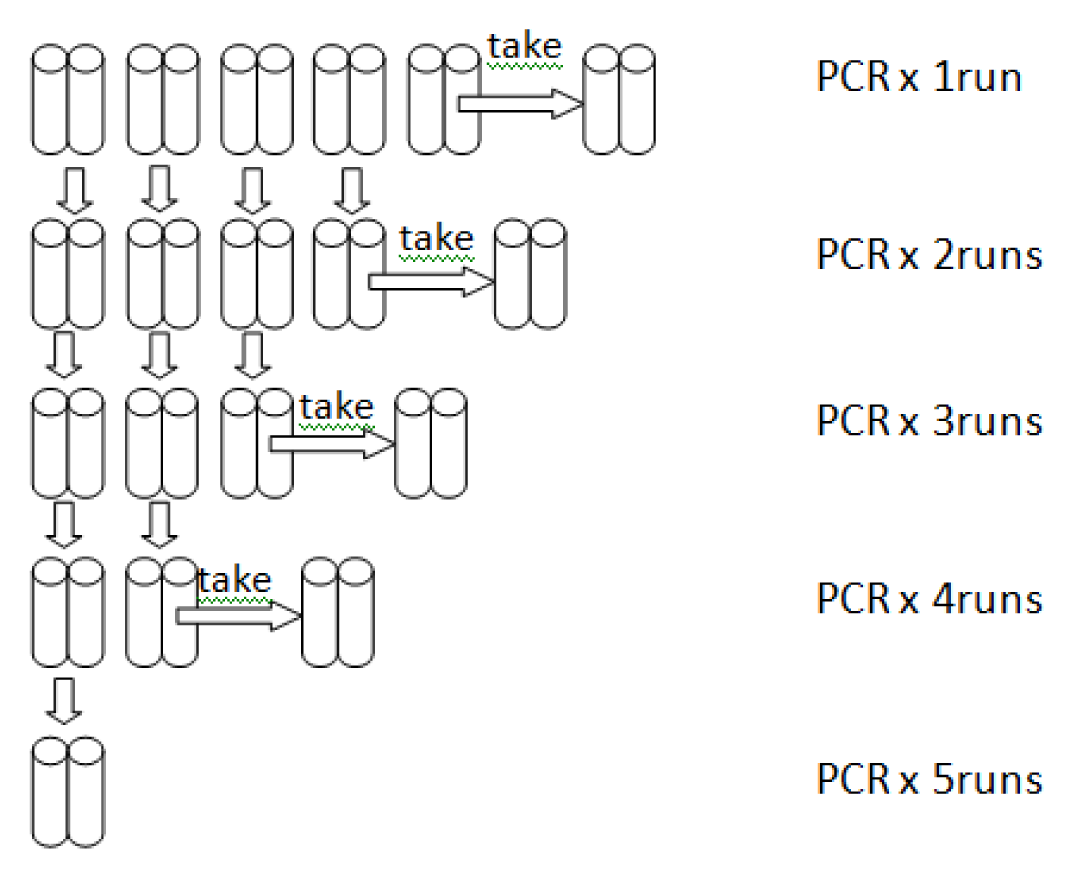
2.3. High Processivity of LoTemp PCR
3. Experimental Section
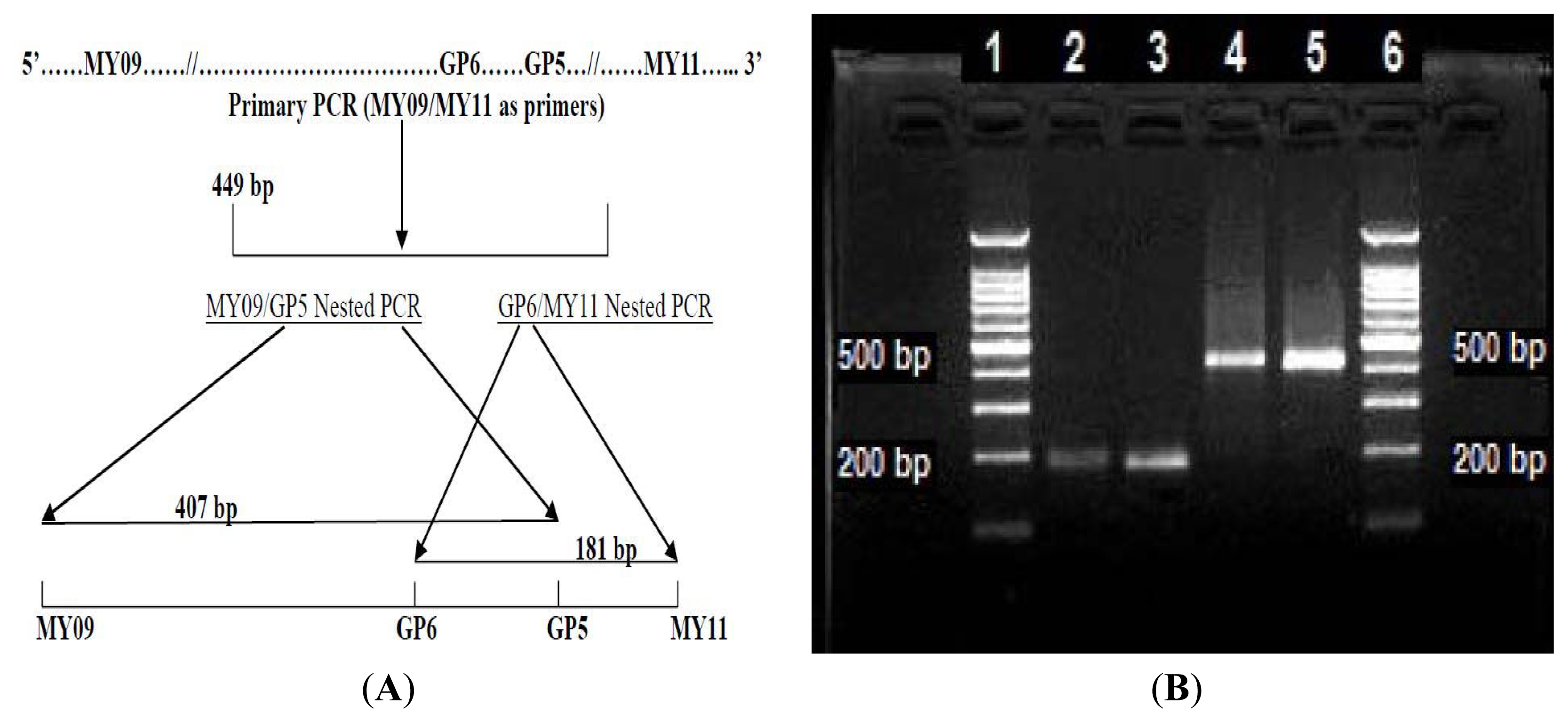
3.1. Preparation of the 449 bp HPV-52 L1 Gene MY09/MY11 Primary PCR Amplicon
3.2. LoTemp Heminested PCR Amplicon as Template for DNA Sequencing
3.3. Direct DNA Sequencing
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Jones, W.; Pappu, S. Increased sensitivity and specificity of Borrelia burgdorferi 16S ribosomal DNA detection. Am. J. Clin. Path 2010, 133, 569–576. [Google Scholar]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Jones, W.; Williams, J.; Walshon, J. Early lyme disease with spirochetemia—Diagnosed by DNA sequencing. BMC Res. Notes 2010, 3, 273–280. [Google Scholar]
- Lee, S.H.; Vigliotti, V.S.; Vigliotti, J.S.; Pappu, S. Routine human papillomavirus genotyping by DNA sequencing in community hospital laboratories. Infect. Agent. Cancer 2007, 2, 11–21. [Google Scholar]
- Lee, S.H. Guidelines for the use of molecular tests for the detection and genotyping of human papilloma virus from clinical specimens. Methods Mol. Biol 2012, 903, 65–101. [Google Scholar]
- Lee, S.H.; Vigliotti, V.S.; Pappu, S. DNA sequencing validation of Chlamydia trachomatis and Neisseria gonorrhoeae nucleic acid tests. Am. J. Clin. Pathol 2008, 129, 852–859. [Google Scholar]
- Lee, S.H.; Vigliotti, V.S.; Pappu, S. Molecular tests for human papillomavirus (HPV), Chlamydia trachomatis and Neisseria gonorrhoeae in liquid-based cytology specimen. BMC Women’s Health 2009, 9, 8–17. [Google Scholar]
- Lee, S.H. Detection of human papillomavirus L1 gene DNA fragments in postmortem blood and spleen after Gardasil® vaccination-A case report. Adv. Biosci. Biotechnol 2012, 3, 1214–1224. [Google Scholar]
- Lee, S.H. Detection of human papillomavirus (HPV) L1 gene DNA possibly bound to particulate aluminum adjuvant in the HPV vaccine Gardasil®. J. Inorg. Biochem 2012, 117, 85–92. [Google Scholar]
- Lee, S.H. Topological conformational changes of human papillomavirus (HPV) DNA bound to an insoluble aluminum salt—A study by low temperature PCR. Adv. Biol. Chem 2013, 3, 76–85. [Google Scholar]
- Ahn, J.H.; Kim, B.Y.; Song, J.; Weon, H.Y. Effects of PCR cycle number and DNA polymerase type on the 16S rRNA gene pyrosequencing analysis of bacterial communities. J. Microbiol 2012, 50, 1071–1074. [Google Scholar]
- Qiu, X.; Wu, L.; Huang, H.; McDonel, P.E.; Palumbo, A.V.; Tiedje, J.M.; Zhou, J. Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl. Environ. Microbiol 2001, 67, 880–887. [Google Scholar]
- Acinas, S.G.; Sarma-Rupavtarm, R.; Klepac-Ceraj, V.; Polz, M.F. PCR-induced sequence artifacts and bias: Insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol 2005, 71, 8966–8969. [Google Scholar]
- Zylstra, P.; Rothenfluh, H.S.; Weiller, G.F.; Blanden, R.V.; Steele, E.J. PCR amplification of murine immunoglobulin germline V genes: Strategies for minimization of recombination artefacts. Immunol. Cell Biol. 1998, 395–405. [Google Scholar]
- Okayama, H.; Curiel, D.T.; Brantly, M.L.; Holmes, M.D.; Crystal, R.G. Rapid, nonradioactive detection of mutations in the human genome by allele-specific amplification. J. Lab. Clin. Med 1989, 114, 105–113. [Google Scholar]
- Wu, D.Y.; Ugozzoli, L.; Pal, B.K.; Wallace, R.B. Allele-specific enzymatic amplification of β-globin genomic DNA for diagnosis of sickle cell anemia. Proc. Natl. Acad. Sci. USA 1989, 86, 2757–2760. [Google Scholar]
- Manos, M.M.; Ting, Y.; Wright, D.K.; Lewis, A.J.; Broker, T.R.; Wolinsky, S.M. Use of polymerase chain reaction amplification for the detection of genital human papillomaviruses. Cancer Cells 1989, 7, 209–214. [Google Scholar]
- Snijders, P.J.; van den Brule, A.J.; Schrijnemakers, H.F.; Snow, G.; Meijer, C.J.; Walboomers, J.M. The use of general primers in the polymerase chain reaction permits the detection of a broad spectrum of human papillomavirus genotypes. J. Gen Virol 1990, 71, 173–181. [Google Scholar]
- Ayyadevara, S.; Thaden, J.J.; Shmookler Reis, R.J. Discrimination of primer 3′-nucleotide mismatch by Taq DNA polymerase during polymerase chain reaction. Anal. Biochem 2000, 284, 11–18. [Google Scholar]
- Tabor, S.; Richardson, C.C. DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Proc. Natl. Acad. Sci. USA 1987, 84, 4767–4771. [Google Scholar]
- Ye, S.Y.; Hong, G.F. Heat-stable DNA polymerase I large fragment resolves hairpin structure in DNA sequencing. Sci. Sin., Ser. B 1987, 30, 503–506. [Google Scholar]
- Kunkel, T.A.; Patel, S.S.; Johnson, K.A. Error-prone replication of repeated DNA sequences by T7 DNA polymerase in the absence of its processivity subunit. Proc. Natl. Acad. Sci. USA 1994, 91, 6830–6834. [Google Scholar]
- Wang, Y.; Prosen, D.E.; Mei, L.; Sullivan, J.C.; Finney, M.; Vander Horn, P.B. A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res 2004, 32, 1197–1207. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR runs | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| DNA yields by T | 0.7 | 1.8 | 1.8 | 2.2 | 2.6 |
| DNA yields by L | 1.7 | 4.1 | 4.6 | 5.1 | 5.1 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hong, G.; Lee, S.H.; Ge, S.; Zhou, S. A Novel Low Temperature PCR Assured High-Fidelity DNA Amplification. Int. J. Mol. Sci. 2013, 14, 12853-12862. https://doi.org/10.3390/ijms140612853
Hong G, Lee SH, Ge S, Zhou S. A Novel Low Temperature PCR Assured High-Fidelity DNA Amplification. International Journal of Molecular Sciences. 2013; 14(6):12853-12862. https://doi.org/10.3390/ijms140612853
Chicago/Turabian StyleHong, Guofan, Sin Hang Lee, Shichao Ge, and Shaoxia Zhou. 2013. "A Novel Low Temperature PCR Assured High-Fidelity DNA Amplification" International Journal of Molecular Sciences 14, no. 6: 12853-12862. https://doi.org/10.3390/ijms140612853
APA StyleHong, G., Lee, S. H., Ge, S., & Zhou, S. (2013). A Novel Low Temperature PCR Assured High-Fidelity DNA Amplification. International Journal of Molecular Sciences, 14(6), 12853-12862. https://doi.org/10.3390/ijms140612853