Potential Mechanisms Linking Atherosclerosis and Increased Cardiovascular Risk in COPD: Focus On Sirtuins

 ,
, 
Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Endothelial Dysfunction and Inflammation
3. COPD and CVD
4. Sirtuins and Atherosclerosis
5. Sirtuins and COPD
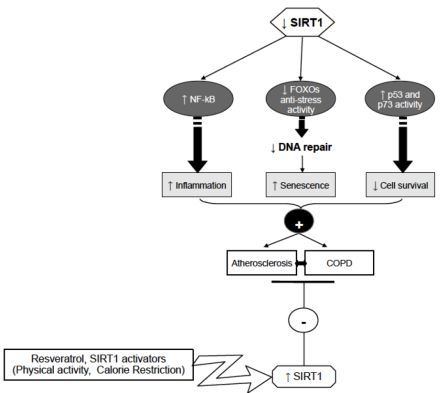
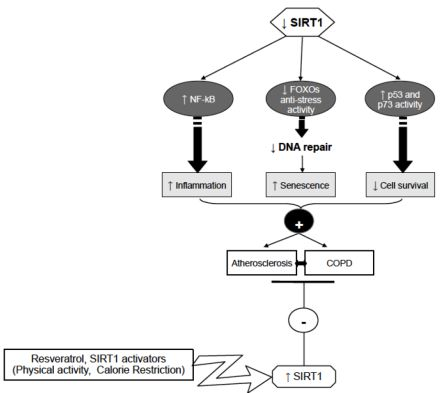
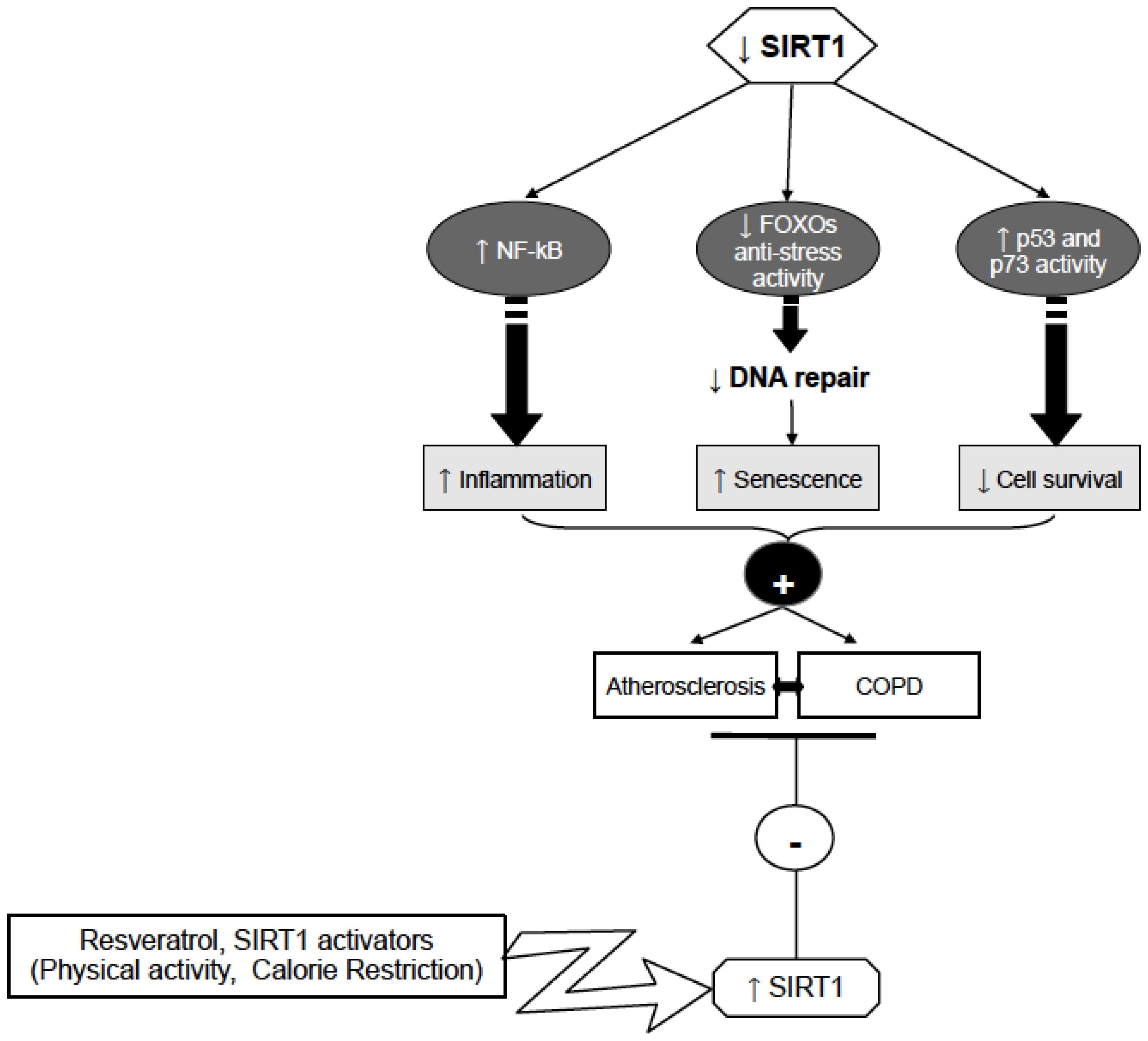
6. Conclusions
Conflict of Interest
References
- Corbi, G.; Conti, V.; Scapagnini, G.; Filippelli, A.; Ferrara, N. Role of sirtuins, calorie restriction and physical activity in aging. Front. Biosci 2012, 4, 768–778. [Google Scholar]
- Rattan, S.I. Hormesis in aging. Ageing Res. Rev 2008, 7, 63–78. [Google Scholar]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci 2000, 908, 244–254. [Google Scholar]
- Rahman, I.; Kinnula, V.L.; Gorbunova, V.; Yao, H. SIRT1 as a therapeutic target in inflammaging of the pulmonary disease. Prev. Med 2012, 54, S20–S28. [Google Scholar]
- Nussbaumer-Ochsner, Y.; Rabe Klaus, F. Systemic manifestations of COPD. Chest 2011, 139, 165–173. [Google Scholar]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar]
- Brandes, R.P. Activating SIRT1: A new strategy to prevent atherosclerosis? Cardiovasc. Res 2008, 80, 163–164. [Google Scholar]
- Aird, W.C. The Endothelium as an Organ. In Endothelial Cells in Health and Disease; Aird, W.C., Ed.; Taylor & Francis Group: Boca Raton, FL, USA, 2005; pp. 1–32. [Google Scholar]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Invest 1994, 94, 2493–2503. [Google Scholar]
- Dollery, C.M.; Owen, C.A.; Sukhova, G.K.; Krettek, A.; Shapiro, S.D.; Libby, P. Neutrophil elastase in human atherosclerotic plaques: Production by macrophages. Circulation 2003, 107, 2829–2836. [Google Scholar]
- Sukhova, G.K.; Shi, G.P.; Simon, D.I.; Chapman, H.A.; Libby, P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J. Clin. Invest 1998, 102, 576–583. [Google Scholar]
- Hautamaki, R.D.; Kobayashi, D.K.; Senior, R.M.; Shapiro, S.D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997, 277, 2002–2004. [Google Scholar]
- Curci, J.A.; Liao, S.; Huffman, M.D.; Shapiro, S.D.; Thompson, R.W. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. J. Clin. Invest 1998, 102, 1900–1910. [Google Scholar]
- Goligorsky, M.S. Endothelial cell dysfunction: Can’t live with it, how to live without it. Am. J. Physiol. Renal Physiol 2005, 288, F871–F880. [Google Scholar]
- Libby, P.; Okamoto, Y.; Rocha, V.Z.; Folco, E. Inflammation in Atherosclerosis: Transition from theory to practice. Circ. J 2010, 74, 213–220. [Google Scholar]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol 2009, 54, 2129–2138. [Google Scholar]
- Campobasso, C.P.; Dell’Erba, A.S.; Addante, A.; Zotti, F.; Marzullo, A.; Colonna, M.F. Sudden cardiac death and myocardial ischemia indicators: A comparative study of four immunohistochemical markers. Am. J. Forensic Med. Pathol 2008, 29, 154–161. [Google Scholar]
- Bianco, A.; Whiteman, S.C.; Sethi, S.K.; Allen, J.T.; Knight, R.A.; Spiteri, M.A. Expression of Intercellular Adhesion Molecule-1 (ICAM-1) in nasal epithelial cells of atopic subjects: A mechanism for increased rhinovirus infection? Clin. Exp. Immunol 2000, 121, 339–345. [Google Scholar]
- Bianco, A.; Sethi, S.K.; Allen, J.T.; Knight, R.A.; Spiteri, M.A. Th2 cytokines exert a dominant influence on epithelial cell expression of the Major group Human Rhinovirus Receptor, ICAM-1. Eur. Respir. J 1998, 12, 619–626. [Google Scholar]
- Whiteman, S.C.; Bianco, A.; Knight, R.A.; Spiteri, M.A. Human rhinovirus selectively modulates membranous and soluble forms of its Intercellular Adhesion Molecule-1 (ICAM-1) receptor to promote epithelial cell infectivity. J. Biol. Chem 2003, 278, 11954–11961. [Google Scholar]
- Murray, C.J.; Lopez, A.D. Alternative projections of mortality and disability by cause 1990–2020: Global burden of disease study. Lancet 1997, 349, 1498–1504. [Google Scholar]
- Man, S.F.; Connett, J.E.; Anthonisen, N.R.; Wise, R.A.; Tashkin, D.P.; Sin, D.D. C-reactive protein and mortality in mild to moderate chronic obstructive pulmonary disease. Thorax 2006, 61, 849–853. [Google Scholar]
- Camilli, A.E.; Robbins, D.R.; Lebowitz, M.D. Death certificate reporting of confirmed airways obstructive disease. Am. J. Epidemiol 1991, 133, 795–800. [Google Scholar]
- Anthonisen, N.R.; Connett, J.E.; Enright, P.L.; Manfreda, J. Lung health study research group. hospitalizations and mortality in the lung health study. Am. J. Respir. Crit. Care Med 2002, 166, 333–339. [Google Scholar]
- Sin, D.D.; Anthonisen, N.R.; Soriano, J.B.; Agusti, A.G. Mortality in COPD: Role of comorbidities. Eur. Respir. J 2006, 28, 1245–1257. [Google Scholar]
- Calverley, P.M.; Anderson, J.A.; Celli, B.; Ferguson, G.T.; Jenkins, C.; Jones, P.W.; Yates, J.C.; Vestbo, J. TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N. Engl. J. Med 2007, 356, 775–789. [Google Scholar]
- De Laurentiis, G.; Paris, D.; Melck, D.; Montuschi, P.; Maniscalco, M.; Bianco, A.; Sofia, M.; Motta, A. Separating smoking-related diseases using NMR-based metabolomics of exhaled breath condensate. J. Proteome Res 2013, 12, 1502–1511. [Google Scholar]
- Campobasso, C.P.; Procacci, R.; Caligara, M. Fatal adverse reaction to ketorolac tromethamine in asthmatic patient. Am. J. Forensic Med. Pathol 2008, 29, 358–363. [Google Scholar]
- Baldacci, S.; Maio, S.; Simoni, M.; Cerrai, S.; Sarno, G.; Silvi, P.; di Pede, F.; Borbotti, M.; Pala, A.P.; Bresciani, M.; Viegi, G. ARGA study group. The ARGA study with general practitioners: Impact of medical education on asthma/rhinitis management. Respir. Med 2012, 106, 777–785. [Google Scholar]
- Braido, F.; Comaschi, M.; Valle, I.; Delgado, L.; Coccini, A.; Guerreras, P.; Stagi, E.; Canonica, G.W. ARGA Study Group, EAACI/CME Committee. Knowledge and health care resource allocation: CME/CPD course guidelines-based efficacy. Eur. Ann. Allergy Clin. Immunol 2012, 44, 193–199. [Google Scholar]
- Maio, S.; Baldacci, S.; Simoni, M.; Angino, A.; Martini, F.; Cerrai, S.; Sarno, G.; Pala, A.; Bresciani, M.; Paggiaro, P.; et al. Impact of asthma and comorbid allergic rhinitis on quality of life and control in patients of Italian general practitioners. J. Asthma 2012, 49, 854–861. [Google Scholar]
- Mazzarella, G.; Ferraraccio, F.; Prati, M.V.; Annunziata, S.; Bianco, A.; Mezzogiorno, A.; Liguori, G.; Angelillo, I.F.; Cazzola, M. Effects of diesel exhaust particles on human lung epithelial cells: An in vitro study. Respir. Med 2007, 101, 1155–1162. [Google Scholar]
- Mazzarella, G.; Esposito, V.; Bianco, A.; Ferraraccio, F.; Prati, M.V.; Lucariello, A.; Manente, L.; Mezzogiorno, A.; de Luca, A. Inflammatory effects on human lung epithelial cells after exposure to diesel exhaust micron sub particles (PM1.0) and pollen allergens. Environ. Pollut 2012, 161, 64–69. [Google Scholar]
- Esposito, V.; Lucariello, A.; Savarese, L.; Cinelli, M.P.; Ferraraccio, F.; Bianco, A.; de Luca, A.; Mazzarella, G. Morphology changes in human lung epithelial cells after exposure to diesel exhaust micron sub particles (PM1.0) and pollen allergens. Environ. Pollut 2012, 171, 162–167. [Google Scholar]
- Corsonello, A.; Antonelli Incalzi, R.; Pistelli, R.; Pedone, C.; Bustacchini, S.; Lattanzio, F. Comorbidities of chronic obstructive pulmonary disease. Curr. Opin. Pulm. Med 2011, 17, S21–S28. [Google Scholar]
- Agusti, A. Systemic effects of chronic obstructive pulmonary disease. What we know and what we don’t know (but should). Proc. Am. Thorac. Soc 2007, 4, 522–525. [Google Scholar]
- Yao, H.; Rahman, I. Current concepts on the role of inflammation in COPD and lung cancer. Curr. Opin. Pharmacol 2009, 9, 375–383. [Google Scholar]
- Iwamoto, H.; Yokoyama, A.; Kitahara, Y.; Ishikawa, N.; Haruta, Y.; Yamane, K.; Hattori, N.; Hara, H.; Kohno, N. Airflow limitation in smokers is associated with subclinical atherosclerosis. Am. J. Respir. Crit. Care Med 2009, 179, 35–40. [Google Scholar]
- Rabe, K.F.; Hurd, S.; Anzueto, A.; Barnes, P.J.; Buist, S.A.; Calverley, P.; Fukuchi, Y.; Jenkins, C.; Rodriguez-Roisin, R.; van Weel, C.; et al. Global strategy for the diagnosis, management.; and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med 2007, 176, 532–555. [Google Scholar]
- Rodríguez-Roisin, R.; Soriano, J.B. Chronic obstructive pulmonary disease with lung cancer and/or cardiovascular disease. Proc. Am. Thorac. Soc 2008, 5, 842–847. [Google Scholar]
- Sin, D.D.; Wu, L.; Man, S.F. The relationship between reduced lung function and cardiovascular mortality: A population-based study and a systematic review of the literature. Chest 2005, 127, 1952–1959. [Google Scholar]
- Donaldson, G.C.; Hurst, J.R.; Smith, C.J.; Hubbard, R.B.; Wedzicha, J.A. Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest 2010, 137, 1091–1097. [Google Scholar]
- Rodríguez, L.A.; Wallander, M.A.; Martín-Merino, E.; Johansson, S. Heart failure, myocardial infarction, lung cancer and death in COPD patients: A UK primary care study. Respir. Med 2010, 104, 1691–1699. [Google Scholar]
- Danesh, J.; Whincup, P.; Walker, M.; Lennon, L.; Thomson, A.; Appleby, P.; Gallimore, J.R.; Pepys, M.B. Low grade inflammation and coronary heart disease: Prospective study and updated meta-analyses. BMJ 2000, 321, 199–204. [Google Scholar]
- Blake, G.J.; Ridker, P.M. C-reactive protein and other inflammatory risk markers in acute coronary syndromes. J. Am. Coll. Cardiol 2003, 41, S37–S42. [Google Scholar]
- Ridker, P.M.; Bassuk, S.S.; Toth, P.P. C-reactive protein and risk of cardiovascular disease: Evidence and clinicial application. Curr. Atheroscler. Rep 2003, 5, 341–349. [Google Scholar]
- Hancox, R.J.; Poulton, R.; Greene, J.M.; Filsell, S.; McLachlan, C.R.; Rasmussen, F.; Taylor, D.R.; Williams, M.J.; Williamson, A.; Sears, M.R. Systemic inflammation and lung function in young adults. Thorax 2007, 62, 1064–1068. [Google Scholar]
- Dahl, M.; Vestbo, J.; Lange, P.; Bojesen, S.E.; Tybjaerg-Hansen, A.; Nordestaarg, B.G. C-reactive protein as a predictor of prognosis in chronic pulmonary disease. Am. J. Respir. Crit. Care Med 2007, 175, 250–255. [Google Scholar]
- Duvoix, A.; Dickens, J.; Haq, I.; Mannino, D.; Miller, B.; Tal-Singer, R.; Lomas, D.A. Blood fibrinogen as a biomarker of chronic obstructive pulmonary disease. Thorax 2012. [Google Scholar] [CrossRef]
- Huang, J.T.; Chaudhuri, R.; Albarbarawi, O.; Barton, A.; Grierson, C.; Rauchhaus, P.; Weir, C.J.; Messow, M.; Stevens, N.; McSharry, C.; et al. Clinical validity of plasma and urinary desmosine as biomarkers for chronic obstructive pulmonary disease. Thorax 2012, 67, 502–508. [Google Scholar]
- Costopoulos, C.; Liew, T.V.; Bennett, M. Ageing and atherosclerosis: Mechanisms and therapeutic options. Biochem. Pharmacol 2008, 75, 1251–1261. [Google Scholar]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res 2012, 111, 245–259. [Google Scholar]
- Cardus, A.; Uryga, A.K.; Walters, G.; Erusalimsky, J.D. SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc. Res 2013, 97, 571–579. [Google Scholar]
- Burnett, C.; Valentini, S.; Cabreiro, F.; Goss, M.; Somogyvári, M.; Piper, M.D.; Hoddinott, M.; Sutphin, G.L.; Leko, V.; McElwee, J.J.; et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 2011, 477, 482–485. [Google Scholar]
- Frye, R.A. Phylogenic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun 2000, 273, 793–798. [Google Scholar]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell Cardiol 2007, 43, 571–579. [Google Scholar]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol 2007, 8, 729–740. [Google Scholar]
- Zu, Y.; Liu, L.; Lee, M.Y.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 promotes proliferation and prevents senescence through targeting LKB1 in primary porcine aortic endothelial cells. Circ. Res 2010, 106, 1384–1393. [Google Scholar]
- Potente, M.; Ghaeni, L.; Baldessari, D.; Mostoslavsky, R.; Rossig, L.; Dequiedt, F.; Haendeler, J.; Mione, M.; Dejana, E.; Alt, F.W.; et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev 2007, 21, 2644–2658. [Google Scholar]
- Guarani, V.; Deflorian, G.; Franco, C.A.; Kruger, M.; Phng, L.K.; Bentley, K.; Toussaint, L.; Dequiedt, F.; Mostoslavsky, R.; Schmidt, M.H.; et al. Acetylationdependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 2011, 473, 234–238. [Google Scholar]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar]
- Murayama, A.; Ohmori, K.; Fujimura, A.; Minami, H.; Yasuzawa-Tanaka, K.; Kuroda, T.; Oie, S.; Daitoku, H.; Okuwaki, M.; Nagata, K.; et al. Epigenetic control of rDNA loci in response to intracellular energy status. Cell 2008, 133, 627–639. [Google Scholar]
- Vaquero, A.; Scher, M.; Erdjument-Bromage, H.; Tempst, P.; Serrano, L.; Reinberg, D. SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 2007, 450, 440–444. [Google Scholar]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar]
- Nadtochiy, S.M.; Yao, H.; McBurney, M.W.; Gu, W.; Guarente, L.; Rahman, I.; Brookes, P.S. SIRT1-mediated acute cardioprotection. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H1506–H1512. [Google Scholar]
- Li, L.; Zhang, H.N.; Chen, H.Z.; Gao, P.; Zhu, L.H.; Li, H.L.; Lv, X.; Zhang, Q.J.; Zhang, R.; Wang, Z.; et al. SIRT1 acts as a modulator of neointima formation following vascular injury in mice. Circ. Res 2011, 108, 1180–1189. [Google Scholar]
- Stein, S.; Matter, C.M. Protective roles of SIRT1 in atherosclerosis. Cell Cycle 2011, 10, 640–647. [Google Scholar]
- Alcendor, R.R.; Kirshenbaum, L.A.; Imai, S.; Vatner, S.F.; Sadoshima, J. Silent information regulator 2alpha; a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ. Res 2004, 95, 971–980. [Google Scholar]
- Pillai, J.B.; Isbatan, A.; Imai, S.; Gupta, M.P. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J. Biol. Chem 2005, 280, 43121–43130. [Google Scholar]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell 2007, 28, 91–106. [Google Scholar]
- El-Mowafy, A.M.; Alkhalaf, M.; El-Kashef, H.A. Resveratrol reverses hydrogen peroxide-induced proliferative effects in human coronary smooth muscle cells: A novel signaling mechanism. Arch. Med. Res 2008, 39, 155–161. [Google Scholar]
- Chong, Z.Z.; Wang, S.; Shang, Y.C.; Maiese, K. Targeting cardiovascular disease with novel SIRT1 pathways. Fut. Cardiol 2012, 8, 89–100. [Google Scholar]
- Breitenstein, A.; Stein, S.; Holy, E.W.; Camici, G.G.; Lohmann, C.; Akhmedov, A.; Spescha, R.; Elliott, P.J.; Westphal, C.H.; Matter, C.M.; et al. Sirt1 inhibition induces in vivo arterial thrombosis and arterial thrombosis and tissue factor expression in activated human endothelial cells. Cardiovasc. Res 2011, 89, 464–472. [Google Scholar]
- Cardellini, M.; Menghini, R.; Martelli, E.; Casagrande, V.; Marino, A.; Rizza, S.; Porzio, O.; Mauriello, A.; Solini, A.; Ippoliti, A.; et al. TIMP3 is reduced in atherosclerotic plaques from subjects with type 2 diabetes and increased by SirT1. Diabetes 2009, 58, 2396–2401. [Google Scholar]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S. B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest 2009, 119, 2758–2771. [Google Scholar]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res 2008, 102, 703–710. [Google Scholar]
- Totsikas, C.; Röhm, J.; Kantartzis, K.; Thamer, C.; Rittig, K.; Machann, J.; Schick, F.; Hansel, J.; Niess, A.; Fritsche, A.; et al. Cardiorespiratory fitness determines the reduction in blood pressure and insulin resistance during lifestyle intervention. J. Hypertens 2011, 29, 1220–1227. [Google Scholar]
- Lim, S.; Despres, J.P.; Koh, K.K. Prevention of atherosclerosis in overweight/obese patients.—In need of novel multi-targeted approaches. Circ. J 2011, 75, 1019–1027. [Google Scholar]
- Corbi, G.; Carbone, S.; Ziccardi, P.; Giugliano, G.; Marfella, R.; Nappo, F.; Paolisso, G.; Esposito, K.; Giugliano, D. FFAs and QT intervals in obese women with visceral adiposity: Effects of sustained weight loss over 1 year. J. Clin. Endocrinol. Metab 2002, 87, 2080–2083. [Google Scholar]
- Vitale, G.; Galderisi, M.; Colao, A.; Innelli, P.; Guerra, G.; Guerra, E.; Dini, F.L.; Orio, F., Jr; Soscia, A.; de Divitiis, O.; et al. Circulating IGF-I levels are associated with increased biventricular contractility in top-level rowers. Clin. Endocrinol. 2008, 69, 231–236. [Google Scholar]
- Ferrara, N.; Abete, P.; Corbi, G.; Paolisso, G.; Longobardi, G.; Calabrese, C.; Cacciatore, F.; Scarpa, D.; Iaccarino, G.; Trimarco, B.; et al. Insulin-induced changes in β-adrenergic response: An experimental study in the isolated rat papillary muscle. Am. J. Hypertens 2005, 18, 348–353. [Google Scholar]
- Ferrara, N.; Corbi, G.; Bosimini, E.; Cobelli, F.; Furgi, G.; Giannuzzi, P.; Giordano, A.; Pedretti, R.; Scrutinio, D.; Rengo, F. Cardiac rehabilitation in the elderly: Patient selection and outcomes. Am. J. Geriatr. Cardiol 2006, 15, 22–27. [Google Scholar]
- Short, K.R.; Vittone, J.L.; Bigelow, M.L.; Proctor, D.N.; Nair, K.S. Age and aerobic exercise training effects on whole body and muscle protein metabolism. Am. J. Physiol. Endocrinol. Metab 2004, 286, E92–E101. [Google Scholar]
- Wu, H.; Kanatous, S.B.; Thurmond, F.A.; Gallardo, T.; Isotani, E.; Bassel-Duby, R.; Williams, R.S. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 2002, 296, 349–352. [Google Scholar]
- Durante, P.E.; Mustard, K.J.; Park, S.H.; Winder, W.W.; Hardie, D.G. Effects of endurance training on activity and expression of AMP-activated protein kinase isoforms in rat muscles. Am. J. Physiol. Endocrinol. Metab 2002, 283, E178–E186. [Google Scholar]
- Frøsig, C.; Jørgensen, S.B.; Hardie, D.G.; Richter, E.A.; Wojtaszewski, J.F. 5′-AMP-activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab 2004, 286, E411–E417. [Google Scholar]
- Winder, W.W.; Holmes, B.F.; Rubink, D.S.; Jensen, E.B.; Chen, M.; Holloszy, J.O. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J. Appl. Physiol 2000, 88, 2219–2226. [Google Scholar]
- Chen, D.; Steele, A.D.; Lindquist, S.; Guarente, L. Increase in activity during calorie restriction requires Sirt1. Science 2005, 310, 1641. [Google Scholar]
- Fulco, M.; Schiltz, R.L.; Iezzi, S.; King, M.T.; Zhao, P.; Kashiwaya, Y.; Hoffman, E.; Veech, R.L.; Sartorelli, V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol. Cell 2003, 12, 51–62. [Google Scholar]
- Robergs, R.A.; Ghiasvand, F.; Parker, D. Biochemistry of exercise-induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol 2004, 287, R502–R516. [Google Scholar]
- Koltai, E.; Szabo, Z.; Atalay, M.; Boldogh, I.; Naito, H.; Goto, S.; Nyakas, C.; Radak, Z. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech. Ageing Dev 2010, 131, 21–28. [Google Scholar]
- Rinaldi, B.; Corbi, G.; Boccuti, S.; Filippelli, W.; Rengo, G.; Leosco, D.; Rossi, F.; Filippelli, A.; Ferrara, N. Exercise training affects age-induced changes in SOD and heat shock protein expression in rat heart. Exp. Gerontol 2006, 41, 764–770. [Google Scholar]
- Ferrara, N.; Rinaldi, B.; Corbi, G.; Conti, V.; Stiuso, P.; Boccuti, S.; Rengo, G.; Rossi, F.; Filippelli, A. Exercise training promotes SIRT1 activity in aged rats. Rejuvenation Res 2008, 11, 139–150. [Google Scholar]
- Conti, V.; Corbi, G.; Russomanno, G.; Simeon, V.; Ferrara, N.; Filippelli, W.; Limongelli, F.; Canonico, R.; Grasso, C.; Stiuso, P.; et al. Oxidative stress effects on endothelial cells treated with different athletes; sera. Med. Sci. Sports Exerc 2012, 44, 39–49. [Google Scholar]
- Conti, V.; Corbi, G.; Russomanno, G.; Ferrara, N.; Filippelli, A. Cell redox homeostasis: Reading Conti et al. data from a blood-centric perspective: Response. Med. Sci. Sports Exerc 2012, 44, 191. [Google Scholar]
- Conti, V.; Russomanno, G.; Corbi, G.; Guerra, G.; Grasso, C.; Filippelli, W.; Paribello, V.; Ferrara, N.; Filippelli, A. Aerobic training workload affects human endothelial cells redox homeostasis. Med. Sci. Sports Exerc 2013, 45, 644–653. [Google Scholar]
- Corbi, G.; Conti, V.; Russomanno, G.; Rengo, G.; Vitulli, P.; Ciccarelli, A.L.; Filippelli, A.; Ferrara, N. Is physical activity able to modify oxidative damage in cardiovascular aging? Oxid. Med. Cell. Longev 2012, 2012, 728547. [Google Scholar]
- Stein, S.; Schäfer, N.; Breitenstein, A.; Besler, C.; Winnik, S.; Lohmann, C.; Heinrich, K.; Brokopp, C.E.; Handschin, C.; Landmesser, U.; et al. SIRT1 reduces endothelial activation without affecting vascular function in ApoE−/− mice. Aging 2010, 2, 353–360. [Google Scholar]
- Schug, T.T.; Xu, Q.; Gao, H.; Peres-da-Silva, A.; Draper, D.W.; Fessler, M.B.; Purushotham, A.; Li, X. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol. Cell Biol 2010, 30, 4712–4721. [Google Scholar]
- Stein, S.; Lohmann, C.; Schäfer, N.; Hofmann, J.; Rohrer, L.; Besler, C.; Rothgiesser, K.M.; Becher, B.; Hottiger, M.O.; Borén, J.; et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur. Heart J 2010, 31, 2301–2309. [Google Scholar]
- Yang, Z.; Kahn, B.B.; Shi, H.; Xue, B.Z. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem 2010, 285, 19051–19059. [Google Scholar]
- Sequeira, J.; Boily, G.; Bazinet, S.; Saliba, S.; He, X.; Jardine, K.; Kennedy, C.; Staines, W.; Rousseaux, C.; Mueller, R.; et al. sirt1-null mice develop an autoimmune-like condition. Exp. Cell Res 2008, 314, 3069–3074. [Google Scholar]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med 2008, 177, 861–870. [Google Scholar]
- Yao, H.; Rahman, I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol. Appl. Pharmacol 2011, 254, 72–85. [Google Scholar]
- MacNee, W. Accelerated lung aging: A novel pathogenic mechanism of chronic obstructive pulmonary disease (COPD). Biochem. Soc. Trans 2009, 37, 819–823. [Google Scholar]
- Ito, K.; Barnes, P.J. COPD as a disease of accelerated lung aging. Chest 2009, 135, 173–180. [Google Scholar]
- Houben, J.M.; Mercken, E.M.; Ketelslegers, H.B.; Bast, A.; Wouters, E.F.; Hageman, G.J.; Schols, A.M. Telomere shortening in chronic obstructive pulmonary disease. Respir. Med 2009, 103, 230–236. [Google Scholar]
- Morlá, M.; Busquets, X.; Pons, J.; Sauleda, J.; MacNee, W.; Agustí, A.G. Telomere shortening in smokers with and without COPD. Eur. Respir. J 2006, 27, 525–528. [Google Scholar]
- Mui, T.S.; Man, J.M.; McElhaney, J.E.; Sandford, A.J.; Coxson, H.O.; Birmingham, C.L.; Li, Y.; Man, S.F.; Sin, D.D. Telomere length and chronic obstructive pulmonary disease: Evidence of accelerated aging. J. Am. Geriatr. Soc 2009, 57, 2372–2374. [Google Scholar]
- Savale, L.; Chaouat, A.; Bastuji-Garin, S.; Marcos, E.; Boyer, L.; Maitre, B.; Sarni, M.; Housset, B.; Weitzenblum, E.; Matrat, M.; et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med 2009, 179, 566–571. [Google Scholar]
- Yang, S.R.; Wright, J.; Bauter, M.; Seweryniak, K.; Kode, A.; Rahman, I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-κB in macrophages in vitro and in rat lungs in vivo: Implications for chronic inflammation and aging. Am. J. Physiol. Lung Cell. Mol. Physiol 2007, 292, L567–L576. [Google Scholar]
- Yao, H.; Rahman, I. Perspectives on translational and therapeutic aspects of SIRT1 in inflammaging and senescence. Biochem. Pharmacol 2012, 84, 1332–1339. [Google Scholar]
- Summer, R.; Walsh, K.; Medoff, B.D. Obesity and pulmonary arterial hypertension: Is adiponectin the molecular link between these conditions? Pulm. Circ 2011, 1, 440–447. [Google Scholar]
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar]
- Turer, A.T.; Hill, J.A.; Elmquist, J.K.; Scherer, P.E. Adipose tissue biology and cardiomyopathy: Translational implications. Circ. Res 2012, 111, 1565–1577. [Google Scholar]
- Maeda, K.; Okubo, K.; Shimomura, I.; Mizuno, K.; Matsuzawa, Y.; Matsubara, K. Analysis of an expression profile of genes in the human adipose tissue. Gene 1997, 190, 227–235. [Google Scholar]
- Hu, E.; Liang, P.; Spiegelman, B.M. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem 1996, 271, 10697–10703. [Google Scholar]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem 1995, 270, 26746–26749. [Google Scholar]
- Shibata, R.; Ouchi, N.; Murohara, T. Adiponectin and cardiovascular disease. Circ. J 2009, 73, 608–614. [Google Scholar]
- Okamoto, Y.; Folco, E.J.; Minami, M.; Wara, A.K.; Feinberg, M.W.; Sukhova, G.K.; Colvin, R.A.; Kihara, S.; Funahashi, T.; Luster, A.D.; et al. Adiponectin inhibits the production of CXC receptor 3 chemokine ligands in macrophages and reduces T-lymphocyte recruitment in atherogenesis. Circ. Res 2008, 102, 218–225. [Google Scholar]
- Daniele, A.; de Rosa, A.; de Cristofaro, M.; Monaco, M.L.; Masullo, M.; Porcile, C.; Capasso, M.; Tedeschi, G.; Oriani, G.; di Costanzo, A. Decreased concentration of adiponectin together with a selective reduction of its high molecular weight oligomers is involved in metabolic complications of myotonic dystrophy type 1. Eur. J. Endocrinol 2011, 165, 969–975. [Google Scholar]
- Bianco, A.; Turchiarelli, V.; Fatica, F.; Nigro, E.; Testa, G.; Vitale, C.; Thanassoulas, T.; Scudiero, O.; Daniele, A. COPD and metabolic disorders: Role of Adiponectin. Shortness Breath 2012, 1, 2–6. [Google Scholar]
- Daniele, A.; de Rosa, A.; Nigro, E.; Scudiero, O.; Capasso, M.; Masullo, M.; de Laurentiis, G.; Oriani, G.; Sofia, M.; Bianco, A. Adiponectin oligomerization state and adiponectin receptors airway expression in chronic obstructive pulmonary disease. Int. J. Biochem. Cell. Biol 2012, 44, 563–569. [Google Scholar]
- Nigro, E.; Scudiero, O.; Sarnataro, D.; Mazzarella, G.; Sofia, M.; Bianco, A.; Daniele, A. Adiponectin affects lung epithelial A549 cell viability counteracting TNFα and IL-1ß toxicity through AdipoR1. Int. J. Biochem. Cell. Biol. 2013. [Google Scholar] [CrossRef]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar]
- Donnelly, L.E.; Newton, R.; Kennedy, G.E.; Fenwick, P.S.; Leung, R.H.; Ito, K.; Russell, R.E.; Barnes, P.J. Anti-inflammatory effects of resveratrol in lung epithelial cells: Molecular mechanisms. Am. J. Physiol. Lung. Cell Mol. Physiol 2004, 287, L774–L783. [Google Scholar]
- Xia, L.; Ding, F.; Zhu, J.H.; Fu, G.S. Resveratrol attenuates apoptosis of pulmonary microvascular endothelial cells induced by high shear stress and proinflammatory factors. Hum. Cell 2011, 24, 127–133. [Google Scholar]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar]
- Papaioannou, A.I.; Rossios, C.; Kostikas, K.; Ito, K. Can we delay the accelerated lung aging in COPD? Anti-aging molecules and interventions. Curr. Drug Targets 2013, 14, 149–157. [Google Scholar]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung. Cell. Mol. Physiol 2011, 300, L391–L401. [Google Scholar]
- Gartel, A.L.; Radhakrishnan, S.K. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res 2005, 65, 3980–3985. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Corbi, G.; Bianco, A.; Turchiarelli, V.; Cellurale, M.; Fatica, F.; Daniele, A.; Mazzarella, G.; Ferrara, N. Potential Mechanisms Linking Atherosclerosis and Increased Cardiovascular Risk in COPD: Focus On Sirtuins. Int. J. Mol. Sci. 2013, 14, 12696-12713. https://doi.org/10.3390/ijms140612696
Corbi G, Bianco A, Turchiarelli V, Cellurale M, Fatica F, Daniele A, Mazzarella G, Ferrara N. Potential Mechanisms Linking Atherosclerosis and Increased Cardiovascular Risk in COPD: Focus On Sirtuins. International Journal of Molecular Sciences. 2013; 14(6):12696-12713. https://doi.org/10.3390/ijms140612696
Chicago/Turabian StyleCorbi, Graziamaria, Andrea Bianco, Viviana Turchiarelli, Michele Cellurale, Federica Fatica, Aurora Daniele, Gennaro Mazzarella, and Nicola Ferrara. 2013. "Potential Mechanisms Linking Atherosclerosis and Increased Cardiovascular Risk in COPD: Focus On Sirtuins" International Journal of Molecular Sciences 14, no. 6: 12696-12713. https://doi.org/10.3390/ijms140612696