The Melatonergic System in Mood and Anxiety Disorders and the Role of Agomelatine: Implications for Clinical Practice

 ,
, 

Abstract
:1. Anatomy and Physiology of the Brain Melatonergic System
2. Interactions between Melatonergic System and Monoaminergic Systems
2.1. Serotonin Is the Main Controller of Circadian Clocks
2.2. Norepinephrine Controls Limiting Steps of Enzymatic Melatonin Production
2.3. Melatonin-Dopamine Reciprocal Interactions: Molecular Bases for Neuropsychiatric Disorder Pathophysiology
3. Circadian Disturbances in Depression
4. Chronobiotic Properties of Agomelatine
5. Pharmacodynamics and Pharmacokinetics of Agomelatine
6. Agomelatine in the Treatment of Major Depressive Disorder
6.1. Materials and Methods of Literature Review
6.2. Acute Phase Trials with Agomelatine versus Placebo
6.3. Antidepressant Efficacy in Active Comparator Trials
6.4. Anhedonia in Major Depressive Disorder
6.5. Sleep in Major Depressive Disorder
6.6. Sexual Function
6.7. Anxiety Symptoms within Depression
6.8. Discontinuation Symptoms
6.9. Responders, Remitters and Relapse Prevention
6.10. Serum Transaminases
6.11. Limitations of Agomelatine Trials in Major Depressive Disorder
7. Conclusions
References
- Wurtman, R.J.; Larin, F.; Axelrod, J.; Shein, H.M.; Rosasco, K. Formation of melatonin and 5-hydroxyindole acetic acid from 14C-tryptophan by rat pineal glands in organ culture. Nature 1968, 217, 953–954. [Google Scholar]
- Cardinali, D.P.; Pévet, P. Basic aspects of melatonin action. Sleep. Med. Rev 1998, 2, 175–190. [Google Scholar]
- Hardeland, R.; Cardinali, D.P.; Srinivasan, V.; Spence, D.W.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin—A pleiotropic, orchestrating regulator molecule. Prog. Neurobiol 2011, 93, 350–384. [Google Scholar]
- Pévet, P. Melatonin. Dialogues Clin. Neurosci 2002, 4, 57–72. [Google Scholar]
- Stehle, J.H.; von Gall, C.; Korf, H.W. Melatonin: A clock-output, a clock-input. J. Neuroendocrinol 2003, 15, 383–389. [Google Scholar]
- Hardeland, R. Melatonin metabolism in the central nervous system. Curr. Neuropharmacol 2010, 8, 168–181. [Google Scholar]
- Dubocovich, M.L.; Markowska, M. Functional MT1 and MT2 melatonin receptors in mammals. Endocrine 2005, 27, 101–110. [Google Scholar]
- Nosjean, O.; Ferro, M.; Coge, F.; Beauverger, P.; Henlin, J.M.; Lefoulon, F.; Fauchere, J.L.; Delagrange, P.; Canet, E.; Boutin, J.A. Identification of the melatonin-binding site MT3 as the quinone reductase 2. J. Biol. Chem 2000, 275, 31311–31317. [Google Scholar]
- Ferry, G.; Hecht, S.; Berger, S.; Moulharat, N.; Coge, F.; Guillaumet, G.; Leclerc, V.; Yous, S.; Delagrange, P.; Boutin, J.A. Old and new inhibitors of quinone reductase 2. Chem. Biol. Interact 2010, 186, 103–109. [Google Scholar]
- Macias, M.; Escames, G.; Leon, J.; Coto, A.; Sbihi, Y.; Osuna, A.; Acuna-Castroviejo, D. Calreticulin-melatonin. An unexpected relationship. Eur. J. Biochem 2003, 270, 832–840. [Google Scholar]
- Benitez-King, G. Melatonin as a cytoskeletal modulator: Implications for cell physiology and disease. J. Pineal Res 2006, 40, 1–9. [Google Scholar]
- Carrillo-Vico, A.; Guerrero, J.M.; Lardone, P.J.; Reiter, R.J. A review of the multiple actions of melatonin on the immune system. Endocrine 2005, 27, 189–200. [Google Scholar]
- Hardeland, R. Melatonin: Signaling mechanisms of a pleiotropic agent. Biofactors 2009, 35, 183–192. [Google Scholar]
- Wu, Y.H.; Zhou, J.N.; Balesar, R.; Unmehopa, U.; Bao, A.; Jockers, R.; van Heerikhuize, J.; Swaab, D.F. Distribution of MT1 melatonin receptor immunoreactivity in the human hypothalamus and pituitary gland: Colocalization of MT1 with vasopressin, oxytocin, and corticotropin-releasing hormone. J. Comp. Neurol 2006, 499, 897–910. [Google Scholar]
- Brunner, P.; Sozer-Topcular, N.; Jockers, R.; Ravid, R.; Angeloni, D.; Fraschini, F.; Eckert, A.; Muller-Spahn, F.; Savaskan, E. Pineal and cortical melatonin receptors MT1 and MT2 are decreased in Alzheimer’s disease. Eur. J. Histochem 2006, 50, 311–316. [Google Scholar]
- Masson-Pevet, M.; Gauer, F.; Schuster, C.; Guerrero, H.Y. Photic regulation of mt(1) melatonin receptors and 2-iodomelatonin binding in the rat and Siberian hamster. Biol. Signals Recept 2000, 9, 188–196. [Google Scholar]
- Masana, M.I.; Benloucif, S.; Dubocovich, M.L. Circadian rhythm of mt1 melatonin receptor expression in the suprachiasmatic nucleus of the C3H/HeN mouse. J. Pineal Res 2000, 28, 185–192. [Google Scholar]
- Brydon, L.; Roka, F.; Petit, L.; de Coppet, P.; Tissot, M.; Barrett, P.; Morgan, P.J.; Nanoff, C.; Strosberg, A.D.; Jockers, R. Dual signaling of human Mel1a melatonin receptors via G(i2), G(i3), and G(q/11) proteins. Mol. Endocrinol 1999, 13, 2025–2038. [Google Scholar]
- Schuster, C.; Williams, L.M.; Morris, A.; Morgan, P.J.; Barrett, P. The human MT1 melatonin receptor stimulates cAMP production in the human neuroblastoma cell line SH-SY5Y cells via a calcium-calmodulin signal transduction pathway. J. Neuroendocrinol 2005, 17, 170–178. [Google Scholar]
- Jones, M.P.; Melan, M.A.; Witt-Enderby, P.A. Melatonin decreases cell proliferation and transformation in a melatonin receptor-dependent manner. Cancer Lett 2000, 151, 133–143. [Google Scholar]
- Rimler, A.; Jockers, R.; Lupowitz, Z.; Zisapel, N. Gi and RGS proteins provide biochemical control of androgen receptor nuclear exclusion. J. Mol. Neurosci 2007, 31, 1–12. [Google Scholar]
- Boutin, J.A.; Audinot, V.; Ferry, G.; Delagrange, P. Molecular tools to study melatonin pathways and actions. Trends Pharmacol. Sci 2005, 26, 412–419. [Google Scholar]
- Roka, F.; Brydon, L.; Waldhoer, M.; Strosberg, A.D.; Freissmuth, M.; Jockers, R.; Nanoff, C. Tight association of the human Mel(1a)-melatonin receptor and G(i): Precoupling and constitutive activity. Mol. Pharmacol 1999, 56, 1014–1024. [Google Scholar]
- Brown, T.M.; Piggins, H.D. Electrophysiology of the suprachiasmatic circadian clock. Prog. Neurobiol 2007, 82, 229–255. [Google Scholar]
- Liu, C.; Weaver, D.R.; Jin, X.; Shearman, L.P.; Pieschl, R.L.; Gribkoff, V.K.; Reppert, S.M. Molecular dissection of two distinct actions of melatonin on the suprachiasmatic circadian clock. Neuron 1997, 19, 91–102. [Google Scholar]
- Gerdin, M.J.; Masana, M.I.; Rivera-Bermudez, M.A.; Hudson, R.L.; Earnest, D.J.; Gillette, M.U.; Dubocovich, M.L. Melatonin desensitizes endogenous MT2 melatonin receptors in the rat suprachiasmatic nucleus: Relevance for defining the periods of sensitivity of the mammalian circadian clock to melatonin. FASEB J 2004, 18, 1646–1656. [Google Scholar]
- Roy, D.; Angelini, N.L.; Fujieda, H.; Brown, G.M.; Belsham, D.D. Cyclical regulation of GnRH gene expression in GT1–7 GnRH-secreting neurons by melatonin. Endocrinology 2001, 142, 4711–4720. [Google Scholar]
- Witt-Enderby, P.A.; Jarzynka, M.J.; Krawitt, B.J.; Melan, M.A. Knock-down of RGS4 and β-tubulin in CHO cells expressing the human MT1 melatonin receptor prevents melatonin-induced receptor desensitization. Life Sci 2004, 75, 2703–2715. [Google Scholar]
- Jarzynka, M.J.; Passey, D.K.; Ignatius, P.F.; Melan, M.A.; Radio, N.M.; Jockers, R.; Rasenick, M.M.; Brydon, L.; Witt-Enderby, P.A. Modulation of melatonin receptors and G-protein function by microtubules. J. Pineal Res 2006, 41, 324–336. [Google Scholar]
- Musshoff, U.; Riewenherm, D.; Berger, E.; Fauteck, J.D.; Speckmann, E.J. Melatonin receptors in rat hippocampus: Molecular and functional investigations. Hippocampus 2002, 12, 165–173. [Google Scholar]
- Wang, L.M.; Suthana, N.A.; Chaudhury, D.; Weaver, D.R.; Colwell, C.S. Melatonin inhibits hippocampal long-term potentiation. Eur. J. Neurosci 2005, 22, 2231–2237. [Google Scholar]
- Larson, J.; Jessen, R.E.; Uz, T.; Arslan, A.D.; Kurtuncu, M.; Imbesi, M.; Manev, H. Impaired hippocampal long-term potentiation in melatonin MT2 receptor-deficient mice. Neurosci. Lett 2006, 393, 23–26. [Google Scholar]
- Conboy, L.; Tanrikut, C.; Zoladz, P.R.; Campbell, A.M.; Park, C.R.; Gabriel, C.; Mocaër, E.; Sandi, C.; Diamond, D.M. The antidepressant agomelatine blocks the adverse effects of stress on memory and enables spatial learning to rapidly increase neural cell adhesion molecule (NCAM) expression in the hippocampus of rats. Int. J. Neuropsychopharmacol 2009, 12, 329–341. [Google Scholar]
- Bertaina-Anglade, V.; Drieu-La-Rochelle, C.; Mocaër, E.; Seguin, L. Memory facilitating effects of agomelatine in the novel object recognition memory paradigm in the rat. Pharmacol. Biochem. Behav 2011, 98, 511–517. [Google Scholar]
- Dubocovich, M.L.; Rivera-Bermudez, M.A.; Gerdin, M.J.; Masana, M.I. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front. Biosci 2003, 8, d1093–d1108. [Google Scholar]
- Fuller, P.M.; Gooley, J.J.; Saper, C.B. Neurobiology of the sleep-wake cycle: Sleep architecture, circadian regulation, and regulatory feedback. J. Biol. Rhythms 2006, 21, 482–493. [Google Scholar]
- Jan, J.E.; Reiter, R.J.; Wasdell, M.B.; Bax, M. The role of the thalamus in sleep, pineal melatonin production, and circadian rhythm sleep disorders. J. Pineal Res 2009, 46, 1–7. [Google Scholar]
- Ochoa Sanchez, R.; Comai, S.; Lacoste, B.; Bambico, F.R.; Lopez-Dominguez, S.; Rivara, S.; Mor, M.; Bedini, A.L.; Spadoni, G.; Fraschini, F.; et al. Promotion of non-rapid eye movement sleep and activation of reticular thalamic neurons by a novel MT2 melatonin receptor ligand. J. Neurosci 2011, 14, 18439–18452. [Google Scholar]
- Ochoa Sanchez, R.; Comai, S.; Rainer, Q.; Rivara, S.; Mor, M.; Bedini, A.L.; Spadoni, G.; Fraschini, F.; Tarzia, G.; Gobbi, G. Anxiolytic effects of the melatonin MT2 receptor partial agonist UCM765: Comparison with melatonin and diazepam. Prog. Neuropsychopharmacol. Biol. Psychiatr 2012, 39, 318–325. [Google Scholar]
- Comai, S.; Ochoa-Sanchez, R.; Gobbi, G. Sleep-wake characterization of double MT1 and MT2 receptor knockout mice and comparison with MT1 and MT2 receptor knockout mice. Behav. Brain Res 2013, 243, 231–238. [Google Scholar]
- Reiter, R.J. Pineal melatonin: Cell biology of its synthesis and of its physiological interactions. Endocr. Rev 1991, 12, 151–180. [Google Scholar]
- Esteban, S.; Garau, C.; Aparicio, S.; Moranta, D.; Barcelo, P.; Fiol, M.A.; Rial, R. Chronic melatonin treatment and its precursor l-tryptophan improve the monoaminergic neurotransmission and related behavior in the aged rat brain. J. Pineal Res 2010, 48, 170–177. [Google Scholar]
- Jiang, Z.G.; Teshima, K.; Yang, Y.; Yoshioka, T.; Allen, C.N. Pre- and postsynaptic actions of serotonin on rat suprachiasmatic nucleus neurons. Brain Res 2000, 866, 247–256. [Google Scholar]
- Morin, L.P. Serotonin and the regulation of mammalian circadian rhythmicity. Ann. Med 1999, 31, 12–33. [Google Scholar]
- Edgar, D.M.; Miller, J.D.; Prosser, R.A.; Dean, R.R.; Dement, W.C. Serotonin and the mammalian circadian system: II. Phase-shifting rat behavioral rhythms with serotonergic agonists. J. Biol. Rhythms 1993, 8, 17–31. [Google Scholar]
- Prosser, R.A.; Heller, H.C.; Miller, J.D. Serotonergic phase advances of the mammalian circadian clock involve protein kinase A and K+ channel opening. Brain Res 1994, 644, 67–73. [Google Scholar]
- Prosser, R.A.; Lee, H.M.; Wehner, A. Serotonergic pre-treatments block in vitro serotonergic phase shifts of the mouse suprachiasmatic nucleus circadian clock. Neuroscience 2006, 142, 547–555. [Google Scholar]
- Meyer-Bernstein, E.L.; Morin, L.P. Differential serotonergic innervation of the suprachiasmatic nucleus and the intergeniculate leaflet and its role in circadian rhythm modulation. J. Neurosci 1996, 16, 2097–2111. [Google Scholar]
- Yan, L.; Takekida, S.; Shigeyoshi, Y.; Okamura, H. Per1 and Per2 gene expression in the rat suprachiasmatic nucleus: Circadian profile and the compartment-specific response to light. Neuroscience 1999, 94, 141–150. [Google Scholar]
- Caldelas, I.; Challet, E.; Saboureau, M.; Pevet, P. Light and melatonin inhibit in vivo serotonergic phase advances without altering serotonergic-induced decrease of per expression in the hamster suprachiasmatic nucleus. J. Mol. Neurosci 2005, 25, 53–63. [Google Scholar]
- Mendoza, J.; Clesse, D.; Pevet, P.; Challet, E. Serotonergic potentiation of dark pulse-induced phase-shifting effects at midday in hamsters. J. Neurochem 2008, 106, 1404–1414. [Google Scholar]
- Gannon, R.L.; Millan, M.J. Evaluation of serotonin, noradrenaline and dopamine reuptake inhibitors on light-induced phase advances in hamster circadian activity rhythms. Psychopharmacology 2007, 195, 325–332. [Google Scholar]
- Cuesta, M.; Clesse, D.; Pevet, P.; Challet, E. New light on the serotonergic paradox in the rat circadian system. J. Neurochem 2009, 110, 231–243. [Google Scholar]
- Ciarleglio, C.M.; Resuehr, H.E.; McMahon, D.G. Interactions of the serotonin and circadian systems: Nature and nurture in rhythms and blues. Neuroscience 2011, 197, 8–16. [Google Scholar]
- Dominguez-Lopez, S.; Mahar, I.; Bambico, F.R.; Labonte, B.; Ochoa-Sanchez, R.; Leyton, M.; Gobbi, G. Short-term effects of melatonin and pinealectomy on serotonergic neuronal activity across the light-dark cycle. J. Psychopharmacol 2012, 26, 830–844. [Google Scholar]
- Klein, D.C.; Coon, S.L.; Roseboom, P.H.; Weller, J.L.; Bernard, M.; Gastel, J.A.; Zatz, M.; Iuvone, P.M.; Rodriguez, I.R.; Begay, V.; et al. The melatonin rhythm-generating enzyme: Molecular regulation of serotonin N-acetyltransferase in the pineal gland. Recent. Prog. Horm. Res 1997, 52, 307–357, ; discussion 357–308.. [Google Scholar]
- Maronde, E.; Saade, A.; Ackermann, K.; Goubran-Botros, H.; Pagan, C.; Bux, R.; Bourgeron, T.; Dehghani, F.; Stehle, J.H. Dynamics in enzymatic protein complexes offer a novel principle for the regulation of melatonin synthesis in the human pineal gland. J. Pineal Res 2011, 51, 145–155. [Google Scholar]
- Ho, A.K.; Mackova, M.; Price, L.; Chik, C.L. Diurnal variation in p42/44 mitogen-activated protein kinase in the rat pineal gland. Mol. Cell. Endocrinol 2003, 208, 23–30. [Google Scholar]
- Ho, A.K.; Chik, C.L. Modulation of Aanat gene transcription in the rat pineal gland. J. Neurochem 2010, 112, 321–331. [Google Scholar]
- Moller, M.; Liu, W. Innervation of the rat pineal gland by nerve fibres originating in the sphenopalatine, otic and trigeminal ganglia. A retrograde in vivo neuronal tracing study. Reprod. Nutr. Dev 1999, 39, 345–353. [Google Scholar]
- Moller, M.; Phansuwan-Pujito, P.; Govitrapong, P.; Schmidt, P. Indications for a central innervation of the bovine pineal gland with substance P-immunoreactive nerve fibers. Brain Res 1993, 611, 347–351. [Google Scholar]
- Mukda, S.; Chetsawang, B.; Govitrapong, P.; Schmidt, P.T.; Hay-Schmidt, A.; Moller, M. Tachykinins and tachykinin-receptors in the rat pineal gland. Eur. J. Neurosci 2005, 21, 2743–2751. [Google Scholar]
- Mukda, S.; Moller, M.; Ebadi, M.; Govitrapong, P. The modulatory effect of substance P on rat pineal norepinephrine release and melatonin secretion. Neurosci. Lett 2009, 461, 258–261. [Google Scholar]
- Koch, M.; Dehghani, F.; Habazettl, I.; Schomerus, C.; Korf, H.W. Cannabinoids attenuate norepinephrine-induced melatonin biosynthesis in the rat pineal gland by reducing arylalkylamine N-acetyltransferase activity without involvement of cannabinoid receptors. J. Neurochem 2006, 98, 267–278. [Google Scholar]
- Koch, M.; Habazettl, I.; Dehghani, F.; Korf, H.W. The rat pineal gland comprises an endocannabinoid system. J. Pineal Res 2008, 45, 351–360. [Google Scholar]
- Garcia, R.A.; Afeche, S.C.; Scialfa, J.H.; do Amaral, F.G.; dos Santos, S.H.; Lima, F.B.; Young, M.E.; Cipolla-Neto, J. Insulin modulates norepinephrine-mediated melatonin synthesis in cultured rat pineal gland. Life Sci 2008, 82, 108–114. [Google Scholar]
- Bailey, M.J.; Coon, S.L.; Carter, D.A.; Humphries, A.; Kim, J.S.; Shi, Q.; Gaildrat, P.; Morin, F.; Ganguly, S.; Hogenesch, J.B.; et al. Night/day changes in pineal expression of >600 genes: Central role of adrenergic/cAMP signaling. J. Biol. Chem 2009, 284, 7606–7622. [Google Scholar]
- Kim, J.S.; Bailey, M.J.; Weller, J.L.; Sugden, D.; Rath, M.F.; Moller, M.; Klein, D.C. Thyroid hormone and adrenergic signaling interact to control pineal expression of the dopamine receptor D4 gene (Drd4). Mol. Cell. Endocrinol 2010, 314, 128–135. [Google Scholar]
- Gonzalez, S.; Moreno-Delgado, D.; Moreno, E.; Perez-Capote, K.; Franco, R.; Mallol, J.; Cortes, A.; Casado, V.; Lluis, C.; Ortiz, J.; et al. Circadian-related heteromerization of adrenergic and dopamine D(4) receptors modulates melatonin synthesis and release in the pineal gland. PLoS Biol 2012, 10, e1001347. [Google Scholar]
- Zisapel, N.; Egozi, Y.; Laudon, M. Inhibition of dopamine release by melatonin: Regional distribution in the rat brain. Brain Res 1982, 246, 161–163. [Google Scholar]
- Alexiuk, N.A.; Uddin, M.; Vriend, J. Melatonin increases the in situ activity of tyrosine hydroxylase in the mediobasal hypothalamus of male Syrian hamsters. Life Sci 1996, 59, 687–694. [Google Scholar]
- Escames, G.; Acuna Castroviejo, D.; Vives, F. Melatonin-dopamine interaction in the striatal projection area of sensorimotor cortex in the rat. Neuroreport 1996, 7, 597–600. [Google Scholar]
- Di Chiara, G.; Morelli, M.; Consolo, S. Modulatory functions of neurotransmitters in the striatum: ACh/dopamine/NMDA interactions. Trends Neurosci 1994, 17, 228–233. [Google Scholar]
- Hamdi, A. Melatonin administration increases the affinity of D2 dopamine receptors in the rat striatum. Life Sci 1998, 63, 2115–2120. [Google Scholar]
- Iuvone, P.M.; Gan, J. Functional interaction of melatonin receptors and D1 dopamine receptors in cultured chick retinal neurons. J. Neurosci 1995, 15, 2179–2185. [Google Scholar]
- Al-Ghoul, W.M.; Herman, M.D.; Dubocovich, M.L. Melatonin receptor subtype expression in human cerebellum. Neuroreport 1998, 9, 4063–4068. [Google Scholar]
- Poirel, V.J.; Cailotto, C.; Streicher, D.; Pevet, P.; Masson-Pevet, M.; Gauer, F. MT1 melatonin receptor mRNA tissular localization by PCR amplification. Neuro Endocrinol. Lett 2003, 24, 33–38. [Google Scholar]
- Mazzucchelli, C.; Pannacci, M.; Nonno, R.; Lucini, V.; Fraschini, F.; Stankov, B.M. The melatonin receptor in the human brain: Cloning experiments and distribution studies. Brain Res. Mol. Brain Res 1996, 39, 117–126. [Google Scholar]
- Kurtuncu, M.; Arslan, A.D.; Akhisaroglu, M.; Manev, H.; Uz, T. Involvement of the pineal gland in diurnal cocaine reward in mice. Eur J. Pharmacol 2004, 489, 203–205. [Google Scholar]
- Sircar, R. Effect of melatonin on cocaine-induced behavioral sensitization. Brain Res 2000, 857, 295–299. [Google Scholar]
- Imbesi, M.; Uz, T.; Yildiz, S.; Arslan, A.D.; Manev, H. Drug- and region-specific effects of protracted antidepressant and cocaine treatment on the content of melatonin MT(1) and MT(2) receptor mRNA in the mouse brain. Int. J. Neuroprot. Neuroregener 2006, 2, 185–189. [Google Scholar]
- Sharma, R.; McMillan, C.R.; Niles, L.P. Neural stem cell transplantation and melatonin treatment in a 6-hydroxydopamine model of Parkinson’s disease. J. Pineal Res 2007, 43, 245–254. [Google Scholar]
- Lin, C.H.; Huang, J.Y.; Ching, C.H.; Chuang, J.I. Melatonin reduces the neuronal loss, downregulation of dopamine transporter, and upregulation of D2 receptor in rotenone-induced parkinsonian rats. J. Pineal Res 2008, 44, 205–213. [Google Scholar]
- Lai, I.C.; Chen, M.L.; Wang, Y.C.; Chen, J.Y.; Liao, D.L.; Bai, Y.M.; Lin, C.C.; Chen, T.T.; Liou, Y.J. Analysis of genetic variations in the human melatonin receptor (MTNR1A, MTNR1B) genes and antipsychotics-induced tardive dyskinesia in schizophrenia. World J. Biol. Psychiatr 2011, 12, 143–148. [Google Scholar]
- Courtet, P.; Olié, E. Circadian dimension and severity of depression. Eur. Neuropsychopharmacol 2012, 22, S476–S481. [Google Scholar]
- Wirz-Justice, A. Diurnal variation of depressive symptoms. Dialogues Clin. Neurosci 2008, 10, 337–343. [Google Scholar]
- Dallaspezia, S.; Benedetti, F. Chronobiological therapy for mood disorders. Expert Rev. Neurother 2011, 11, 961–970. [Google Scholar]
- Coogan, A.N.; Thome, J. Chronotherapeutics and psychiatry: Setting the clock to relieve the symptoms. World J. Biol. Psychiatr 2011, 12, 40–43. [Google Scholar]
- Barden, N.; Shink, E.; Labbé, M.; Vacher, R.; Rochford, J.; Mocaër, E. Antidepressant action of agomelatine (S 20098) in a transgenic mouse model. Prog. Neuropsychopharmacol. Biol. Psychiatr 2005, 29, 908–916. [Google Scholar]
- Leproult, R.; Van Onderbergen, A.; L’hermite-Balériaux, M.; van Cauter, E.; Copinschi, G. Phase-shifts of 24 h rhythms of hormonal release and body temperature following early evening administration of the melatonin agonist agomelatine in healthy older men. Clin. Endocrinol 2005, 63, 298–304. [Google Scholar]
- Audinot, V.; Mailliet, F.; Lahaye-Brasseur, C.; Bonnaud, A.; Le Gall, A.; Amossé, C.; Dromaint, S.; Rodriguez, M.; Nagel, N.; Galizzi, J.P.; et al. New selective ligands of human cloned melatonin MT1 and MT2 receptors. Naunyn Schmiedebergs Arch. Pharmacol 2003, 6, 553–561. [Google Scholar]
- Sharpley, A.L.; Rawlings, N.B.; Brain, S.; McTavish, S.F.; Cowen, P.J. Does agomelatine block 5-HT2C receptors in humans? Psychopharmacology 2011, 213, 653–655. [Google Scholar]
- Norman, T.R. The effect of agomelatine on 5HT(2C) receptors in humans: A clinically relevant mechanism? Psychopharmacology 2012, 221, 177–178. [Google Scholar]
- Millan, M.J.; Gobert, A.; Lejeune, F.; Dekeyne, A.; Newman-Tancredi, A.; Pasteau, V.; Rivet, J.M.; Cussac, D. The novel melatonin agonist agomelatine (S20098) is an antagonist at 5-hydroxytryptamine2C receptors, blockade of which enhances the activity of frontocortical dopaminergic and adrenergic pathways. J. Pharmacol. Exp. Ther 2003, 306, 954–964. [Google Scholar]
- Dubocovich, M.L. Drug evaluation: Agomelatine targets a range of major depressive disorder symptoms. Curr. Opin. Investig. Drugs 2006, 7, 670–680. [Google Scholar]
- Palazidou, E.; Papadopoulos, A.; Ratcliff, H.; Dawling, S.; Checkley, S.A. NE uptake inhibition increases melatonin secretion, a measure of noradrenergic neurotransmission, in depressed patients. Psychol. Med 1992, 22, 309–315. [Google Scholar]
- Mitchell, H.A.; Weinshenker, D. Good night and good luck: Norepinephrine in sleep pharmacology. Biochem. Pharmacol 2009, 79, 801–809. [Google Scholar]
- Kripke, D.F.; Youngstedt, S.D.; Rex, K.M.; Klauber, M.R.; Elliott, J.A. Melatonin excretion with affect disorders over age 60. Psychiatr. Res 2003, 118, 47–54. [Google Scholar]
- Carvalho, L.A.; Gorenstein, C.; Moreno, R.A.; Markus, R.P. Melatonin levels in drug-free patients with major depression from the southern hemisphere. Psychoneuroendocrinology 2006, 31, 761–768. [Google Scholar]
- Rubin, R.T.; Heist, E.K.; McGeoy, S.S.; Hanada, K.; Lesser, I.M. Neuroendocrine aspects of primary endogenous depression. XI. Serum melatonin measures in patients and matched control subjects. Arch. Gen. Psychiatr 1992, 49, 558–567. [Google Scholar]
- Shafii, M.; MacMillan, D.R.; Key, M.P.; Derrick, A.M.; Kaufman, N.; Nahinsky, I.D. Nocturnal serum melatonin profile in major depression in children and adolescents. Arch. Gen. Psychiatr 1996, 53, 1009–1013. [Google Scholar]
- Sekula, L.K.; Lucke, J.F.; Heist, E.K.; Czambel, R.K.; Rubin, R.T. Neuroendocrine aspects of primary endogenous depression. XV: Mathematical modeling of nocturnal melatonin secretion in major depressives and normal controls. Psychiatr. Res 1997, 69, 143–153. [Google Scholar]
- Crasson, M.; Kjiri, S.; Colin, A.; Kjiri, K.; L’Hermite-Baleriaux, M.; Ansseau, M.; Legros, J.J. Serum melatonin and urinary 6- sulfatoxymelatonin in major depression. Psychoneuroendocrinology 2004, 29, 1–12. [Google Scholar]
- Wetterberg, L. Clinical importance of melatonin. Prog. Brain Res 1979, 52, 539–547. [Google Scholar]
- Srinivasan, V.; de Berardis, D.; Shillcutt, S.D.; Brzezinski, A. Role of melatonin in mood disorders and the antidepressant effects of agomelatine. Expert Opin. Investig. Drugs 2012, 21, 1503–1522. [Google Scholar]
- Pompili, M.; Serafini, G.; Innamorati, M.; Venturini, P.; Fusar-Poli, P.; Sher, L.; Amore, M.; Girardi, P. Agomelatine, a novel intriguing antidepressant option enhancing neuroplasticity: A critical review. World J. Biol. Psychiatr. 2013, in press. [Google Scholar]
- Dagytė, G.; Crescente, I.; Postema, F.; Seguin, L.; Gabriel, C.; Mocaër, E.; Boer, J.A.; Koolhaas, J.M. Agomelatine reverses the decrease in hippocampal cell survival induced by chronic mild stress. Behav. Brain Res 2011, 218, 121–128. [Google Scholar]
- Tardito, D.; Molteni, R.; Popoli, M.; Racagni, G. Synergistic mechanisms involved in the antidepressant effects of agomelatine. Eur. Neuropsychopharmacol 2012, 22, 482–486. [Google Scholar]
- San, L.; Arranz, B. Agomelatine: A novel mechanism of antidepressant action involving the melatonergic and the serotonergic system. Eur. Psychiatr 2008, 23, 396–402. [Google Scholar]
- European Medicines Agency. Evaluation of Medicines for Human Use CHMP Assessment Report for Valdoxan. Available online: http://www.emea.europa.eu/humandocs/PDFs/EPAR/valdoxan/H-915-en6.pdf (accessed on 26 December 2009).
- Servier Laboratories Ltd. Valdoxan (agomelatine) Summary of Product Characteristics. 2009. Available online: http://emc.medicines.org.uk/medicine/21830/SPC/Valdoxan/ (accessed on 29 January 2010).
- Dolder, C.R.; Nelson, M.; Snider, M. Agomelatine treatment of major depressive disorder. Ann. Pharmacother 2008, 42, 1822–1831. [Google Scholar]
- Sadock, B.J.; Sadock, V.A. Kaplan Sadock’s Synopsis of Psychiatry: Behavioral Sciences; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Merikangas, K.R.; Walters, E.E. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatr 2005, 62, 593–602. [Google Scholar]
- Kessler, R.C.; Chiu, W.T.; Demler, O.; Merikangas, K.R.; Walters, E.E. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatr 2005, 62, 617–627. [Google Scholar]
- De Berardis, D.; Serroni, N.; Campanella, D.; Carano, A.; Gambi, F.; Valchera, A.; Conti, C.; Sepede, G.; Scali, M.; Fulcheri, M.; et al. Alexithymia and its relationships with C-reactive protein and serum lipid levels among drug naïve adult outpatients with major depression. Prog. Neuropsychopharmacol. Biol. Psychiatr 2008, 32, 1982–1986. [Google Scholar]
- Murray, C.J.; Lopez, A.D. Alternative projections of mortality and disability by cause 1990–2020: Global burden of disease study. Lancet 1997, 349, 1498–1504. [Google Scholar]
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757. [Google Scholar]
- Fava, M. Diagnosis and definition of treatment-resistant depression. Biol. Psychiatr 2003, 15, 649–659. [Google Scholar]
- Little, A. Treatment-resistant depression. Am. Fam. Physician 2009, 80, 167–172. [Google Scholar]
- Parker, G.; Brotchie, H. Do the old psychostimulant drugs have a role in managing treatment-resistant depression? Acta Psychiatr. Scand 2009, 121, 308–314. [Google Scholar]
- Hickie, I. Is depression overdiagnosed? No. BMJ 2007, 335, 329. [Google Scholar]
- Hall, W.D.; Mant, A.; Mitchell, P.B.; Rendle, V.A.; Hickie, I.B.; McManus, P. Association between antidepressant prescribing and suicide in Australia, 1991–2000: Trend analysis. BMJ 2003, 326, 1008. [Google Scholar]
- Loo, H.; Hale, A.; D’Haenen, H. Determination of the dose of agomelatine, a melatoninergic agonist and selective 5-HT(2C) antagonist, in the treatment of major depressive disorder: A placebo-controlled dose range study. Int. Clin. Psychopharmacol 2002, 17, 239–247. [Google Scholar]
- Kennedy, S.H.; Emsley, R. Placebo-controlled trial of agomelatine in the treatment of major depressive disorder. Eur. Neuropsychopharmacol 2006, 16, 93–100. [Google Scholar]
- Olie, J.P.; Kasper, S. Efficacy of agomelatine, a MT1/MT2 receptor agonist with 5-HT2C antagonistic properties, in major depressive disorder. Int. J. Neuropsychopharmacol 2007, 10, 661–673. [Google Scholar]
- Lemoine, P.; Guilleminault, C.; Alvarez, E. Improvement in subjective sleep in major depressive disorder with a novel antidepressant, agomelatine: Randomized, double-blind comparison with venlafaxine. J. Clin. Psychiatr 2007, 68, 1723–1732. [Google Scholar]
- Martinotti, G.; Sepede, G.; Gambi, F.; di Iorio, G.; de Berardis, D.; di Nicola, M.; Onofrj, M.; Janiri, L.; di Giannantonio, M. Agomelatine versus venlafaxine XR in the treatment of anhedonia in major depressive disorder: A pilot study. J. Clin. Psychopharmacol 2012, 32, 487–491. [Google Scholar]
- Kennedy, S.H.; Rizvi, S.; Fulton, K.; Rasmussen, J. A double-blind comparison of sexual functioning, antidepressant efficacy, and tolerability between agomelatine and venlafaxine XR. J. Clin. Psychopharmacol 2008, 28, 329–333. [Google Scholar]
- Kasper, S.; Hajak, G.; Wulff, K.; Hoogendijk, W.J.; Montejo, A.L.; Smeraldi, E.; Rybakowski, J.K.; Quera-Salva, M.A.; Wirz-Justice, A.M.; Picarel-Blanchot, F.; et al. Efficacy of the novel antidepressant agomelatine on the circadian rest-activity cycle and depressive and anxiety symptoms in patients with major depressive disorder: A randomized, double-blind comparison with sertraline. J. Clin. Psychiatr 2010, 71, 109–120. [Google Scholar]
- Hale, A.; Corral, R.M.; Mencacci, C.; Ruiz, J.S.; Severo, C.A.; Gentil, V. Superior antidepressant efficacy results of agomelatine versus fluoxetine in severe MDD patients: A randomized, double-blind study. Int. Clin. Psychopharmacol 2010, 25, 305–314. [Google Scholar]
- Quera-Salva, M.A.; Hajak, G.; Philip, P.; Montplaisir, J.; Keufer-Le Gall, S.; Laredo, J.; Guilleminault, C. Comparison of agomelatine and escitalopram on nighttime sleep and daytime condition and efficacy in major depressive disorder patients. Int. Clin. Psychopharmacol 2011, 26, 252–262. [Google Scholar]
- Karaiskos, D.; Tzavellas, E.; Ilias, I.; Liappas, I.; Paparrigopoulos, T. Agomelatine and sertraline for the treatment of depression in type 2 diabetes mellitus. Int. J. Clin. Pract 2013, 67, 257–260. [Google Scholar]
- Spijker, J.; Bijl, R.V.; de Graaf, R.; Nolen, W.A. Determinants of poor 1-year outcome of DSM-III-R major depression in the general population: Results of the Netherlands Mental Health Survey and Incidence Study (NEMESIS). Acta Psychiatr. Scand 2001, 103, 122–130. [Google Scholar]
- Taylor, D.J.; Walters, H.M.; Vittengl, J.R.; Krebaum, S.; Jarret, R.B. Which depressive symptoms remain after response to cognitive therapy of depression and predict relapse and recurrence? J. Affect. Disord 2010, 123, 181–187. [Google Scholar]
- Keedwell, P.A.; Andrew, C.; Williams, S.C.; Brammer, M.J.; Phillips, M.L. The neural correlates of anhedonia in major depressive disorder. Biol. Psychiatr 2005, 58, 843–853. [Google Scholar]
- Di Giannantonio, M.; Martinotti, G. Anhedonia and major depression: The role of agomelatine. Eur. Neuropsychopharmacol 2012, 22, 505–510. [Google Scholar]
- Di Giannantonio, M.; di Iorio, G.; Guglielmo, R.; de Berardis, D.; Conti, C.M.; Acciavatti, T.; Cornelio, M.; Martinotti, G. Major depressive disorder, anhedonia and agomelatine: An open-label study. J. Biol. Regul. Homeost. Agents 2011, 25, 109–114. [Google Scholar]
- Quera Salva, M.A.; Vanier, B.; Laredo, J.; Hartley, S.; Chapotot, F.; Moulin, C.; Lofaso, F.; Guilleminault, C. Major depressive disorder, sleep EEG and agomelatine: An open-label study. Int. J. Neuropsychopharmacol 2007, 10, 691–696. [Google Scholar]
- Lopes, M.C.; Quera-Salva, M.A.; Guilleminault, C. Non-REM sleep instability in patients with major depressive disorder: Subjective improvement and improvement of non-REM sleep instability with treatment (agomelatine). Sleep Med 2007, 9, 33–41. [Google Scholar]
- Montejo, A.; Prieto, N.; Terleira, A.; Matias, J.; Alonso, S.; Paniagua, G.; Naval, S.; Parra, D.G.; Gabriel, C.; Mocaër, E.; et al. Better sexual acceptability of agomelatine (25 and 50 mg) compared with paroxetine (20 mg) in healthy male volunteers: An 8-week, placebo-controlled study using the PRSEXDQ-SALSEX scale. J. Psychopharmacol 2010, 24, 111–120. [Google Scholar]
- Montgomery, S.A.; Kennedy, S.H.; Burrows, G.D.; Lejoyeux, M.; Hindmarch, I. Absence of discontinuation symptoms with agomelatine and occurrence of discontinuation symptoms with paroxetine: A randomized, double-blind, placebo-controlled discontinuation study. Int. Clin. Psychopharmacol 2004, 19, 271–280. [Google Scholar]
- Sparshatt, A.; McAllister Williams, R.H.; Baldwin, D.S.; Haddad, P.M.; Bazire, S.; Weston, E.; Taylor, P.; Taylor, D. A naturalistic evaluation and audit database of agomelatine: Clinical outcome at 12 weeks. Acta Psychiatr. Scand. 2012, in press. [Google Scholar]
- Goodwin, G.; Emsley, R.; Rembry, S.; Rouillon, F. Agomelatine Study Group. Agomelatine prevents relapse in patients with major depressive disorder, without evidence of a discontinuation syndrome. J. Clin. Psychiatr 2009, 70, 1128–1237. [Google Scholar]
- Servier Laboratories. Valdoxan. Available online: http://www.valdoxan.com/index.php/valdoxan-package-leaflet-information-forthe-user (accessed on 3 May 2011).
- Zajecka, J.; Schatzberg, A.; Stahl, S.; Shah, A.; Caputo, A.; Post, A. Efficacy and safety of agomelatine in the treatment of major depressive disorder: A multicenter, randomized, double-blind, placebo-controlled trial. J. Clin. Psychopharmacol 2010, 30, 135–144. [Google Scholar]
- Stahl, S.M.; Fava, M.; Trivedi, M.H.; Caputo, A.; Shah, A.; Post, A. Agomelatine in the treatmentof major depressive disorder: An 8-week, multicenter, randomized, placebo-controlled trial. J. Clin. Psychiatr 2010, 71, 616–626. [Google Scholar]
- European Medicines Agency, CHMP Assessment Report for Valdoxan. In Document Reference EMEA/655251/2008; European Medicines Agency: London, UK, 2008.
- De Berardis, D.; di Iorio, G.; Acciavatti, T.; Conti, C.; Serroni, N.; Olivieri, L.; Cavuto, M.; Martinotti, G.; Janiri, L.; Moschetta, F.S.; et al. The emerging role of melatonin agonists in the treatment of major depression: Focus on agomelatine. CNS Neurol. Disord. Drug Targets 2011, 10, 119–132. [Google Scholar]
- Howland, R.H. A benefit-risk assessment of agomelatine in the treatment of major depression. Drug Saf 2011, 34, 709–731. [Google Scholar]
- Howland, R.H. Publication bias and outcome reporting bias: Agomelatine as a case example. J. Psychosoc. Nurs. Ment. Health Serv 2011, 49, 11–14. [Google Scholar]
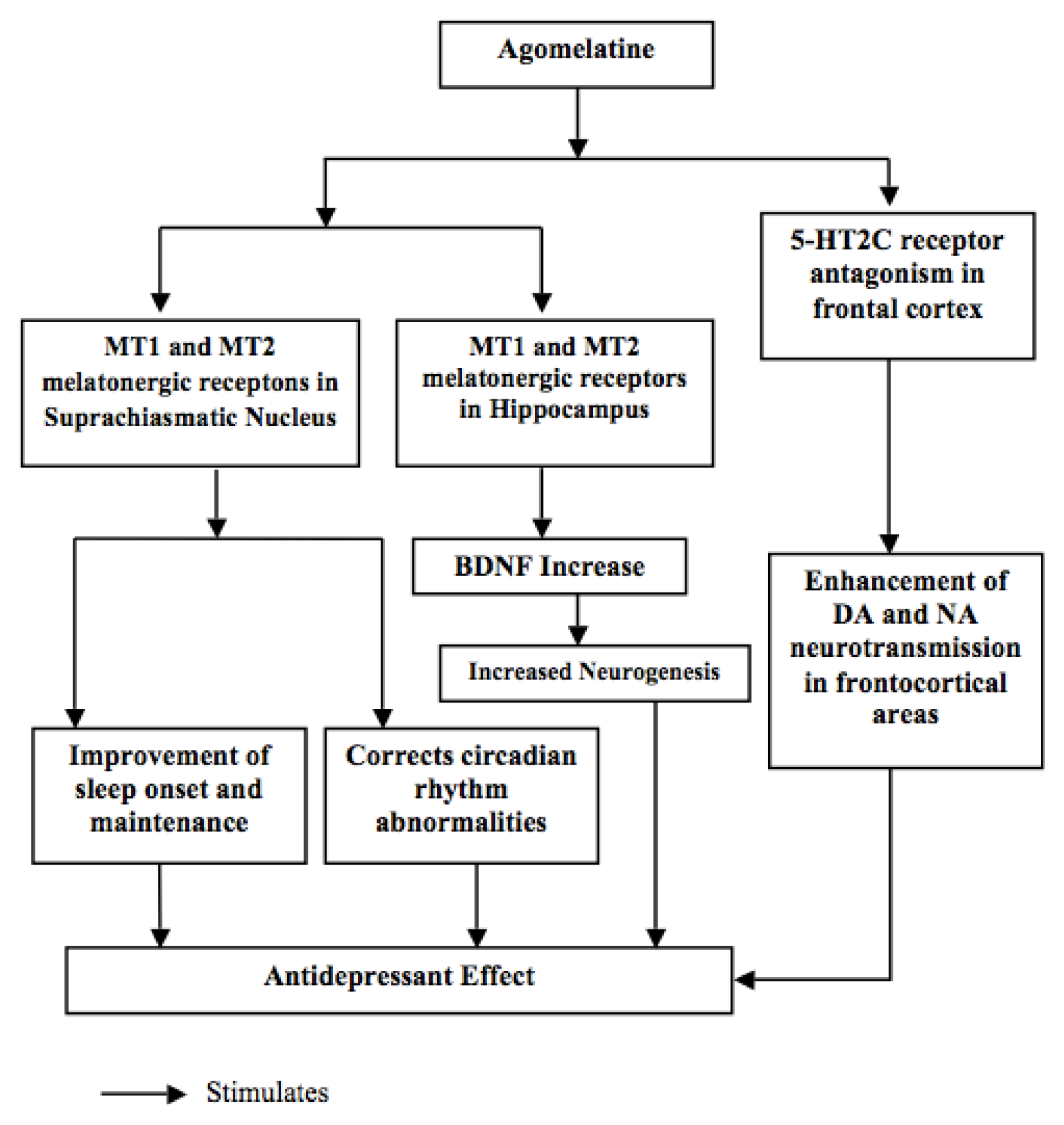

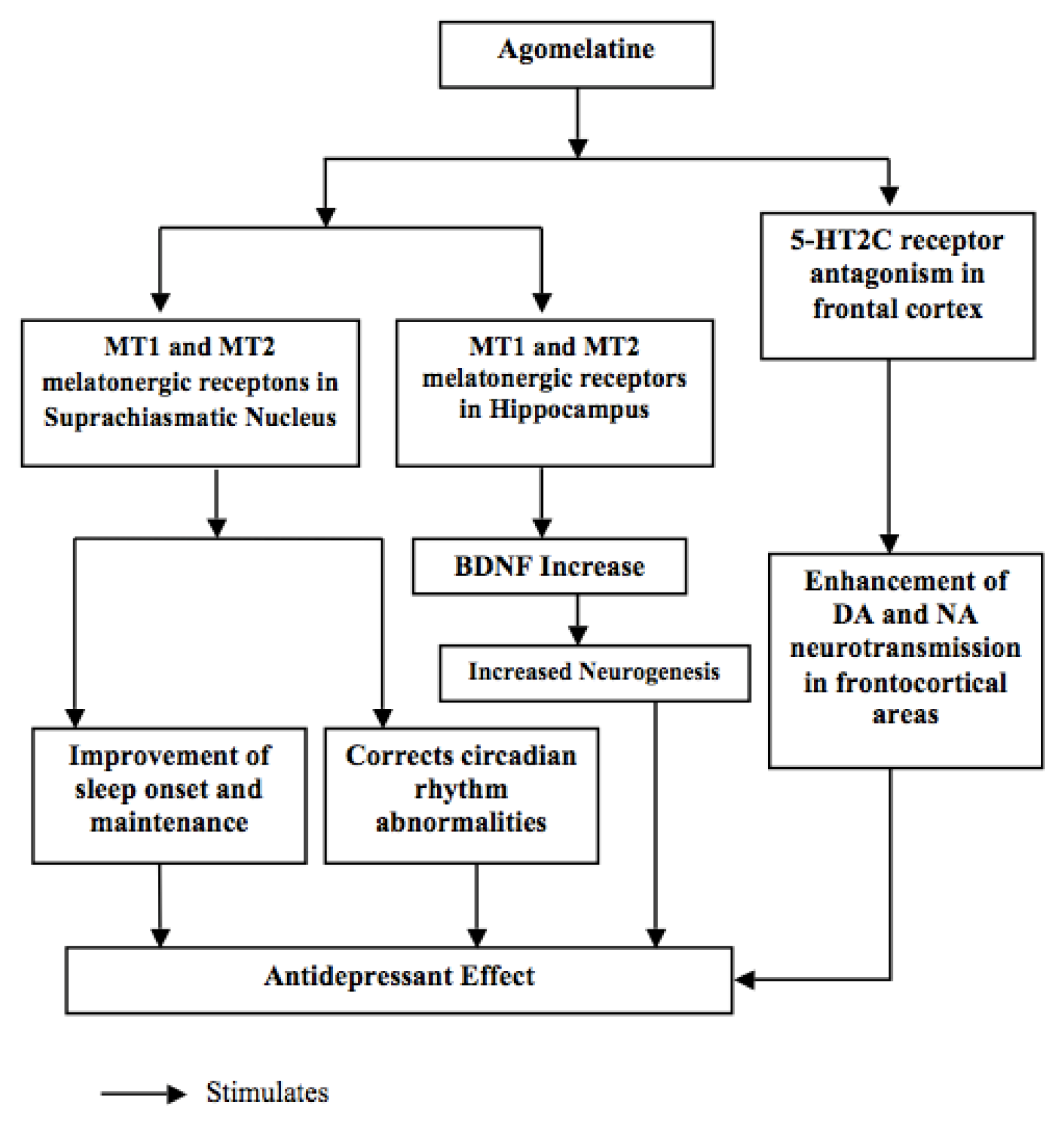{kind=link}
| Authors | Reference | Study Design | Comparator/Active Control | Number of Patients | Duration | Agomelatine Dosage (mg/day) | Results |
|---|---|---|---|---|---|---|---|
| Loo et al., 2002 | 123 | Placebo-controlled dose range study | Placebo/Paroxetine 20 mg/day | 711 | 8 weeks | 1, 5 and 25 | Agomelatine at 25 mg was statistically more effective than placebo |
| Montgomery et al., 2004 | 142 | Randomized, double-blind, placebo-controlled discontinuation study | Placebo/Paroxetine 20 mg/day | 335 | 12 weeks | 25 | Agomelatine was effective and had less potential to cause discontinuation symptoms than paroxetine |
| Kennedy & Emsley, 2006 | 124 | Randomized, double-blind, placebo-controlled study | Placebo | 212 | 6 weeks | 25 and 50 | Agomelatine at 25 mg was effective, but 50 mg may be beneficial for some patients without reducing tolerability |
| Lemoine et al., 2007 | 126 | Randomized, double-blind comparison with venlafaxine study | Venlafaxine 75–150 mg/day | 334 | 6 weeks | 25 and 50 | Agomelatine showed similar antidepressant efficacy earlier and greater efficacy in improving subjective sleep as compared to venlafaxine |
| Olié & Kasper, 2007 | 125 | Double-blind, flexible dose, parallel-group, placebo controlled study | Placebo | 238 | 6 weeks | 25 (with dose adjustment at two weeks to 50 mg/day in patients with insufficient improvement) | Agomelatine was significantly more efficacious than placebo. Agomelatine had a safety profile similar to placebo |
| Kennedy et al., 2008 | 128 | Randomized, double-blind comparison with venlafaxine study | Venlafaxine XR 150 mg/day | 277 | 12 weeks | 50 | Agomelatine showed similar antidepressant efficacy with a superior sexual side effect profile to venlafaxine |
| Goodwin et al., 2009 | 144 | Randomized, double-blind, placebo-controlled study | Placebo | 339 | 24 weeks | 25 and 50 | Agomelatine was more effective than placebo and prevented relapses without evidence of discontinuation symptoms |
| Hale et al., 2010 | 130 | Randomized, double-blind comparison with fluoxetine study on severely depressed patients (HAMD ≥ 25) | Fluoxetine 20–40 mg/day | 515 | 8 weeks | 25 and 50 | Agomelatine was statistically more effective than fluoxetine |
| Kasper et al., 2010 | 129 | Randomized, double-blind comparison with sertraline study | Sertraline 50–100 mg/day | 313 | 6 weeks | 25 and 50 | Agomelatine was more effective than sertraline. Agomelatine improved the circadian rest-activity cycle more than sertraline |
| Zajecka et al., 2010 | 146 | Multicenter, randomized, double-blind, placebo-controlled study | Placebo | 511 | 8 weeks | 25 and 50 | Agomelatine at 50 mg showed greater and rapid reduction in all core symptoms of depression compared with placebo |
| Stahl et al., 2010 | 147 | Randomized, double-blind, placebo-controlled study | Placebo | 503 | 8 weeks | 25 and 50 | Agomelatine at 25 mg was more effective than placebo over the course of the study, whereas agomelatine at 50 mg provided evidence for its antidepressant efficacy until week six, but not at study end |
| Quera-Salva et al., 2011 | 131 | Randomized, double-blind comparison with escitalopram study | Escitalopram 10–20 mg/day | 138 | 24 weeks | 25 and 50 | Agomelatine was as effective as escitalopram. Treatment with agomelatine improved morning condition and reduced daytime sleepiness compared with escitalopram |
| Martinotti et al. 2012 | 127 | Open-label parallel-group, randomized comparison with venlafaxine study | Venlafaxine 75–150 mg/day | 60 | 8 weeks | 25 and 50 | Agomelatine antidepressant efficacy proved to be similar to that of venlafaxine during an eight-week treatment period |
| Karaiskos et al. 2013 | 132 | Observational open label, randomized, comparison with sertraline study | Sertraline 50–100 mg/day | 40 depressed patients with non-optimally controlled type 2 diabetes mellitus (DM) | 4 months | 25 and 50 | Agomelatine was effective in the treatment of depression and anxiety, as well as in the improvement of health-related behaviors, in depressed patients with non-optimally controlled type 2 DM |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Berardis, D.; Marini, S.; Fornaro, M.; Srinivasan, V.; Iasevoli, F.; Tomasetti, C.; Valchera, A.; Perna, G.; Quera-Salva, M.-A.; Martinotti, G.; et al. The Melatonergic System in Mood and Anxiety Disorders and the Role of Agomelatine: Implications for Clinical Practice. Int. J. Mol. Sci. 2013, 14, 12458-12483. https://doi.org/10.3390/ijms140612458
De Berardis D, Marini S, Fornaro M, Srinivasan V, Iasevoli F, Tomasetti C, Valchera A, Perna G, Quera-Salva M-A, Martinotti G, et al. The Melatonergic System in Mood and Anxiety Disorders and the Role of Agomelatine: Implications for Clinical Practice. International Journal of Molecular Sciences. 2013; 14(6):12458-12483. https://doi.org/10.3390/ijms140612458
Chicago/Turabian StyleDe Berardis, Domenico, Stefano Marini, Michele Fornaro, Venkataramanujam Srinivasan, Felice Iasevoli, Carmine Tomasetti, Alessandro Valchera, Giampaolo Perna, Maria-Antonia Quera-Salva, Giovanni Martinotti, and et al. 2013. "The Melatonergic System in Mood and Anxiety Disorders and the Role of Agomelatine: Implications for Clinical Practice" International Journal of Molecular Sciences 14, no. 6: 12458-12483. https://doi.org/10.3390/ijms140612458