An Overview of Biological Macromolecule Crystallization





Abstract
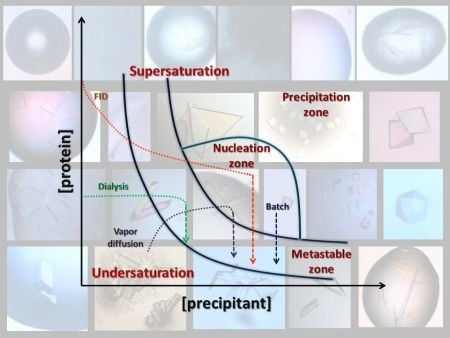
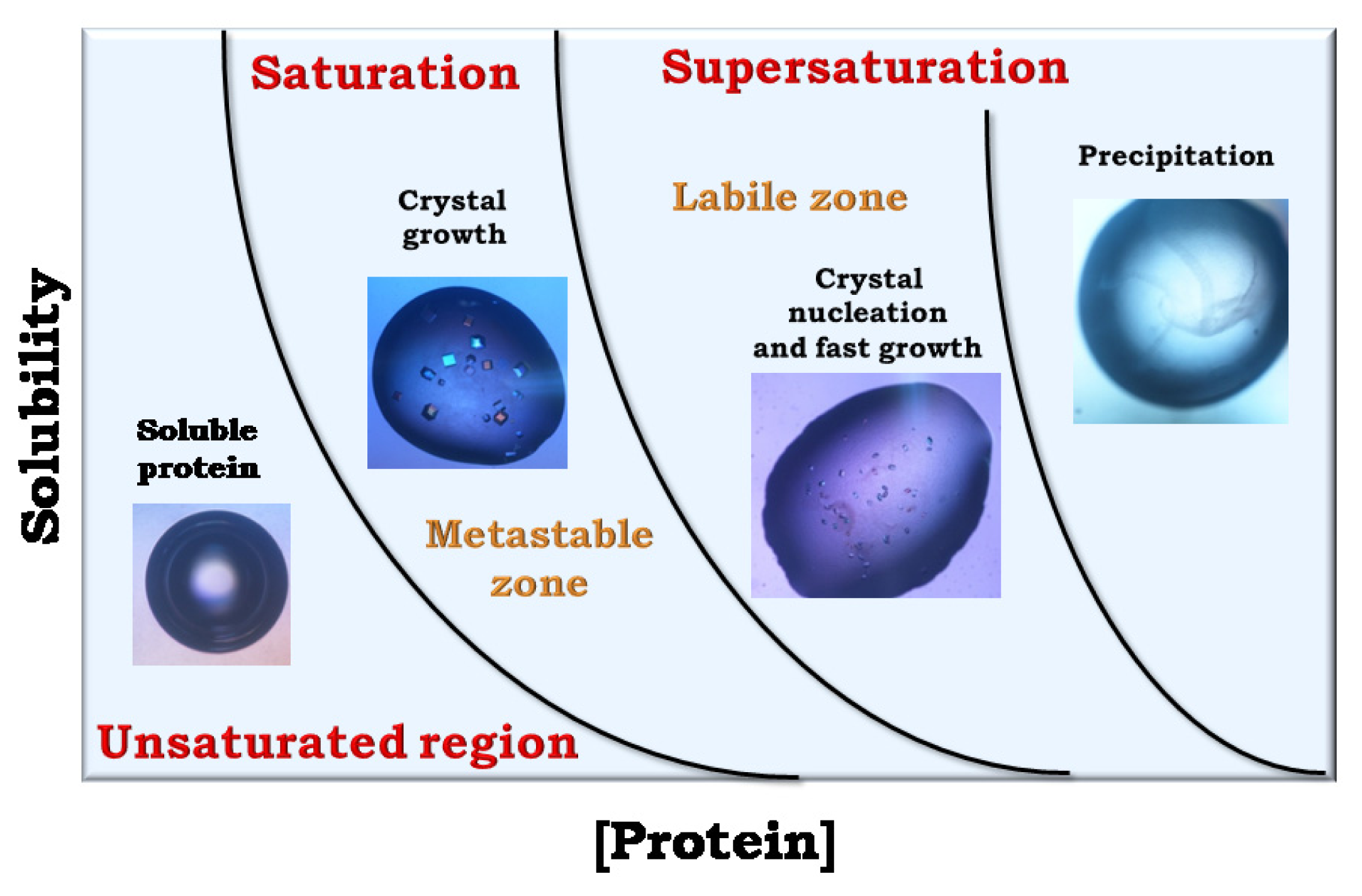
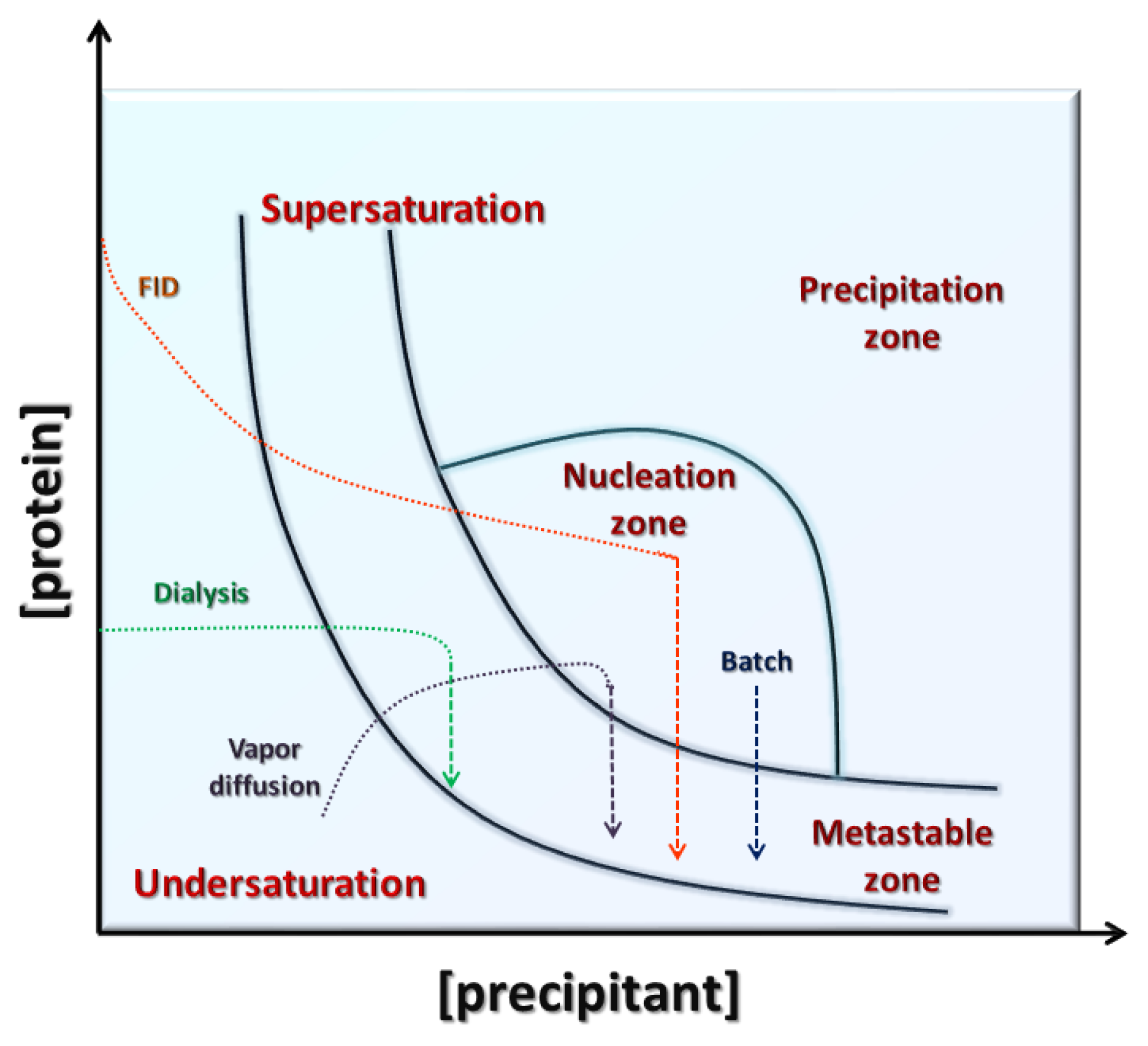
:
1. Introduction
2. Factors Affecting Crystallization of Bio-Macromolecules
2.1. Sample Purity and Homogeneity
2.2. Temperature
2.3. pH
2.4. Thermal Stability
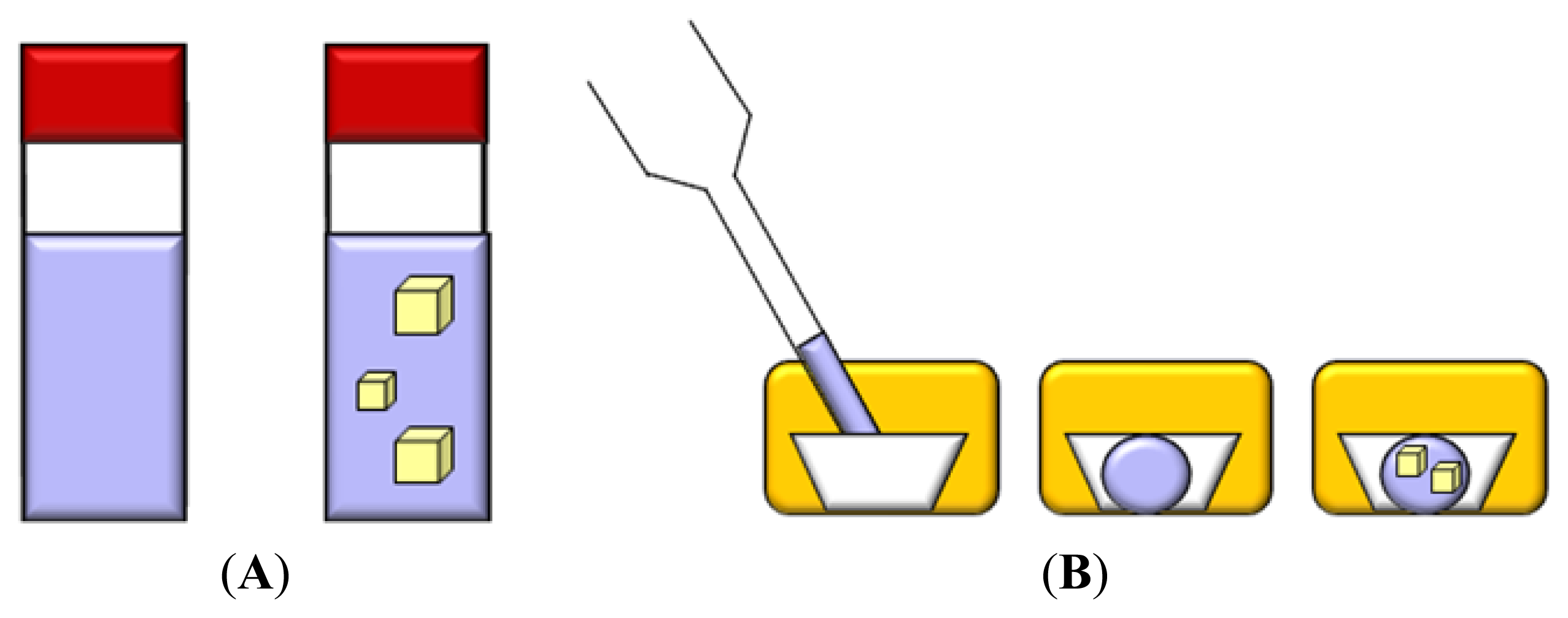
2.5. Precipitant
2.6. Additives
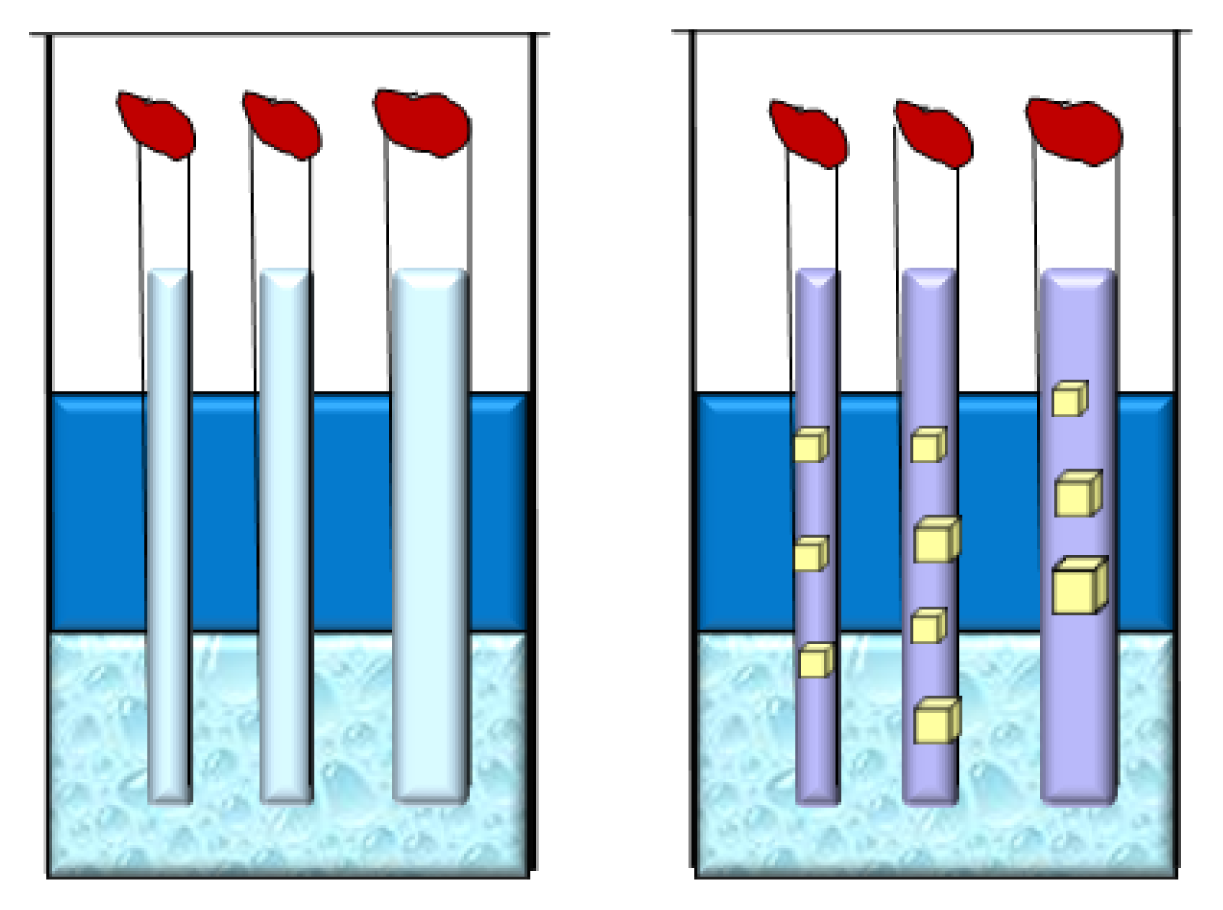
2.7. Gravity
2.8. Magnetic Field
2.9. Electric Field
2.10. Stirring
3. Conventional Crystallization Methods
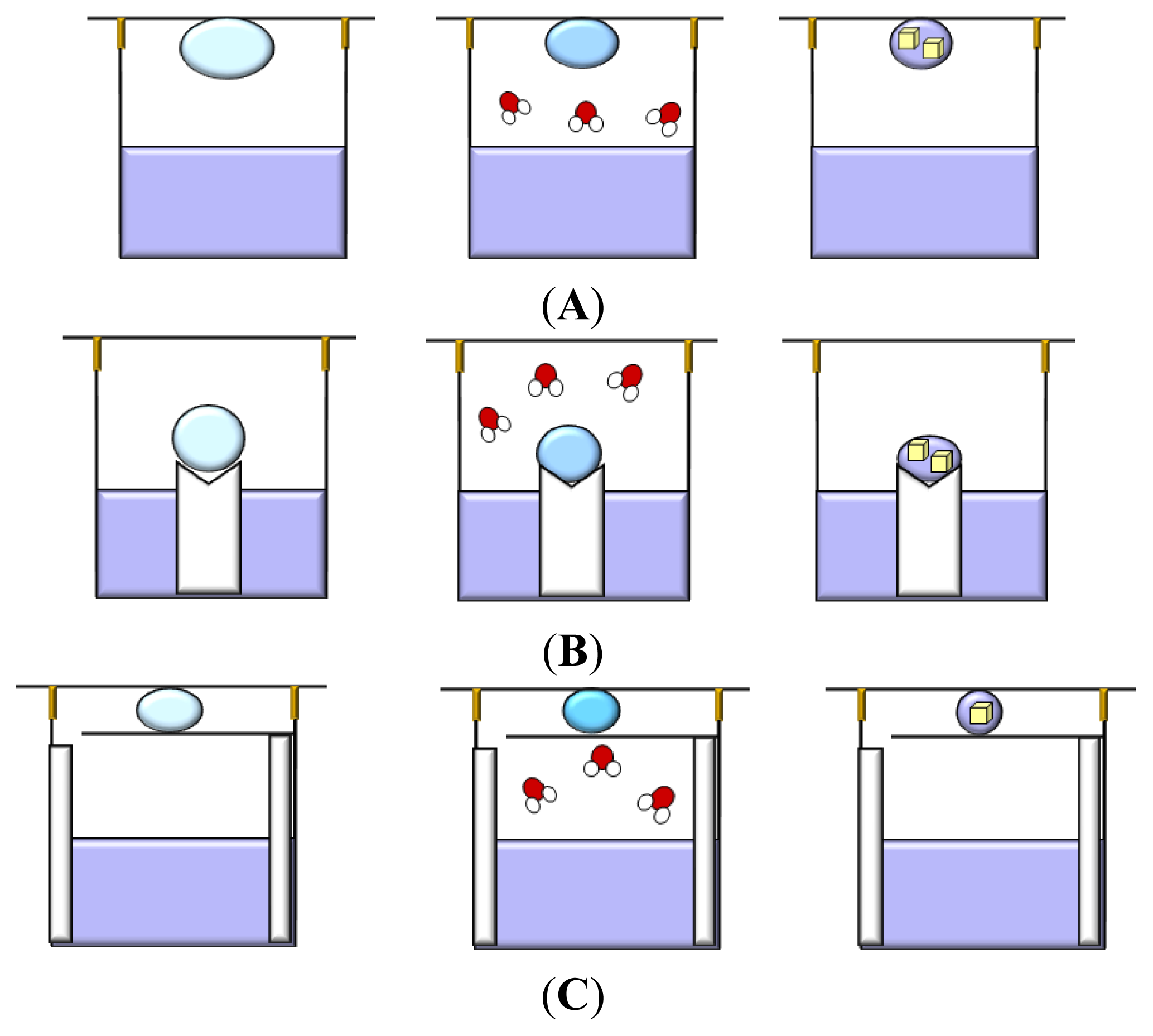
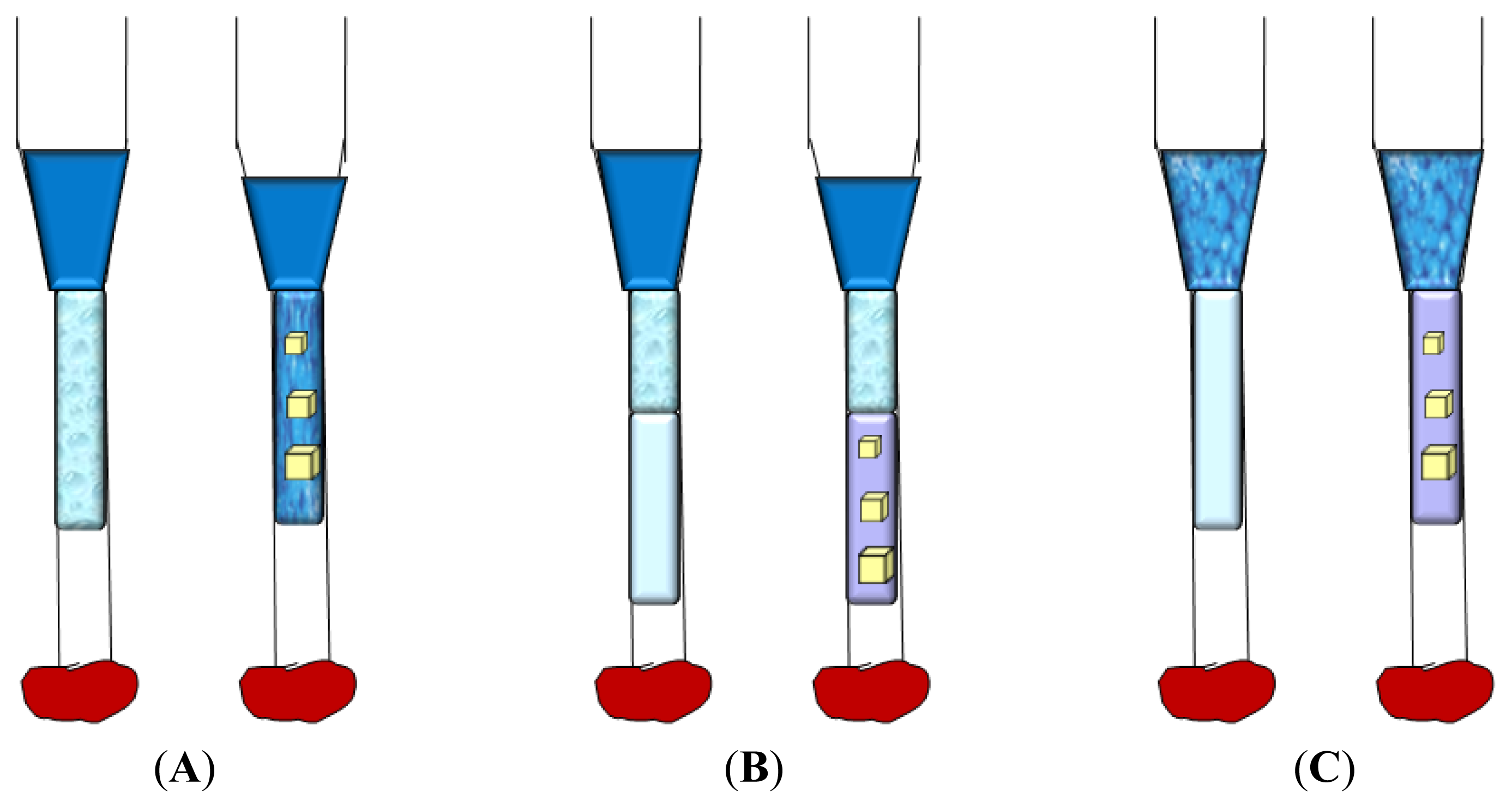
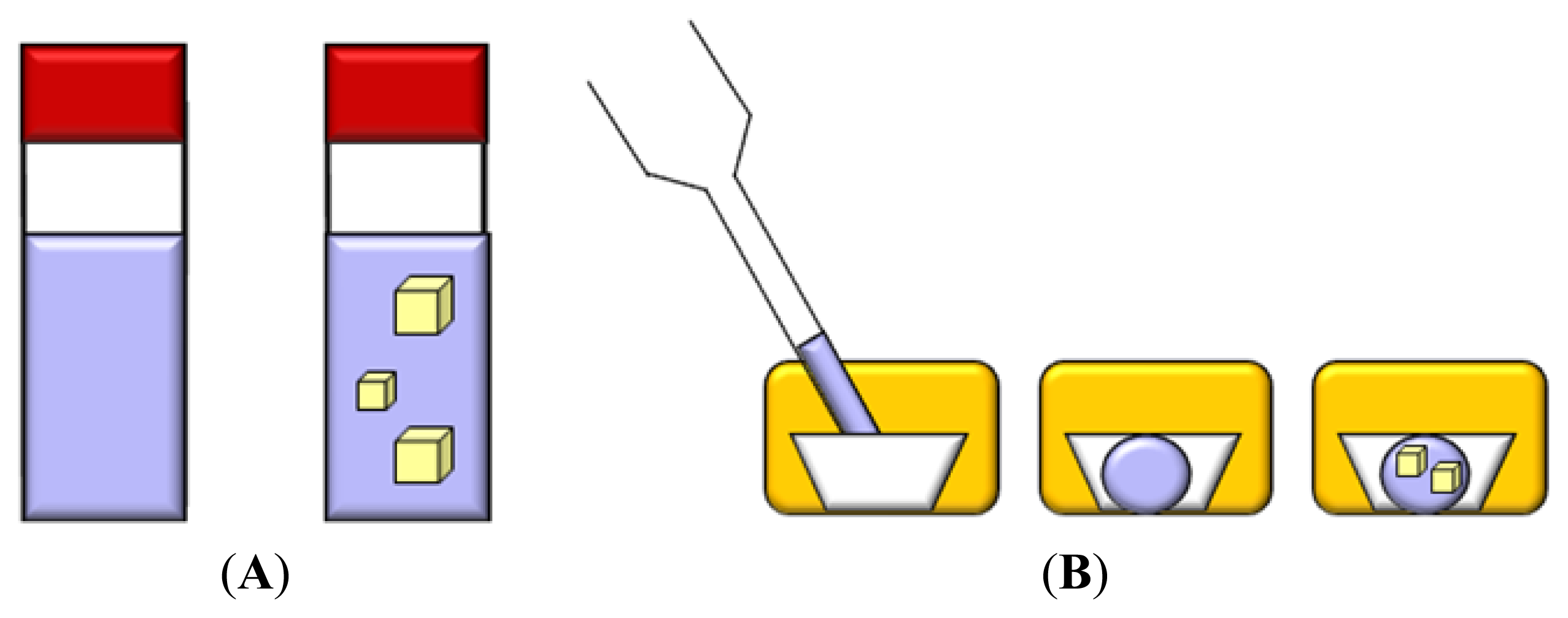
3.1. Vapor Diffusion
3.2. Batch
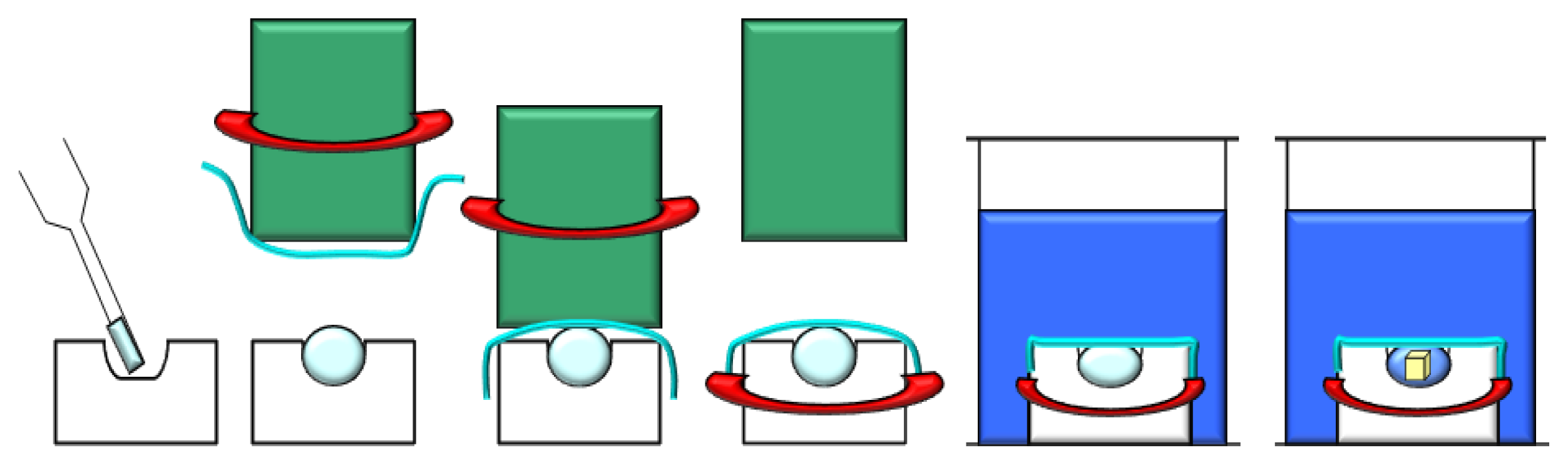
3.3. Dialysis
3.4. Free Interface Diffusion
3.5. Gel Crystallization
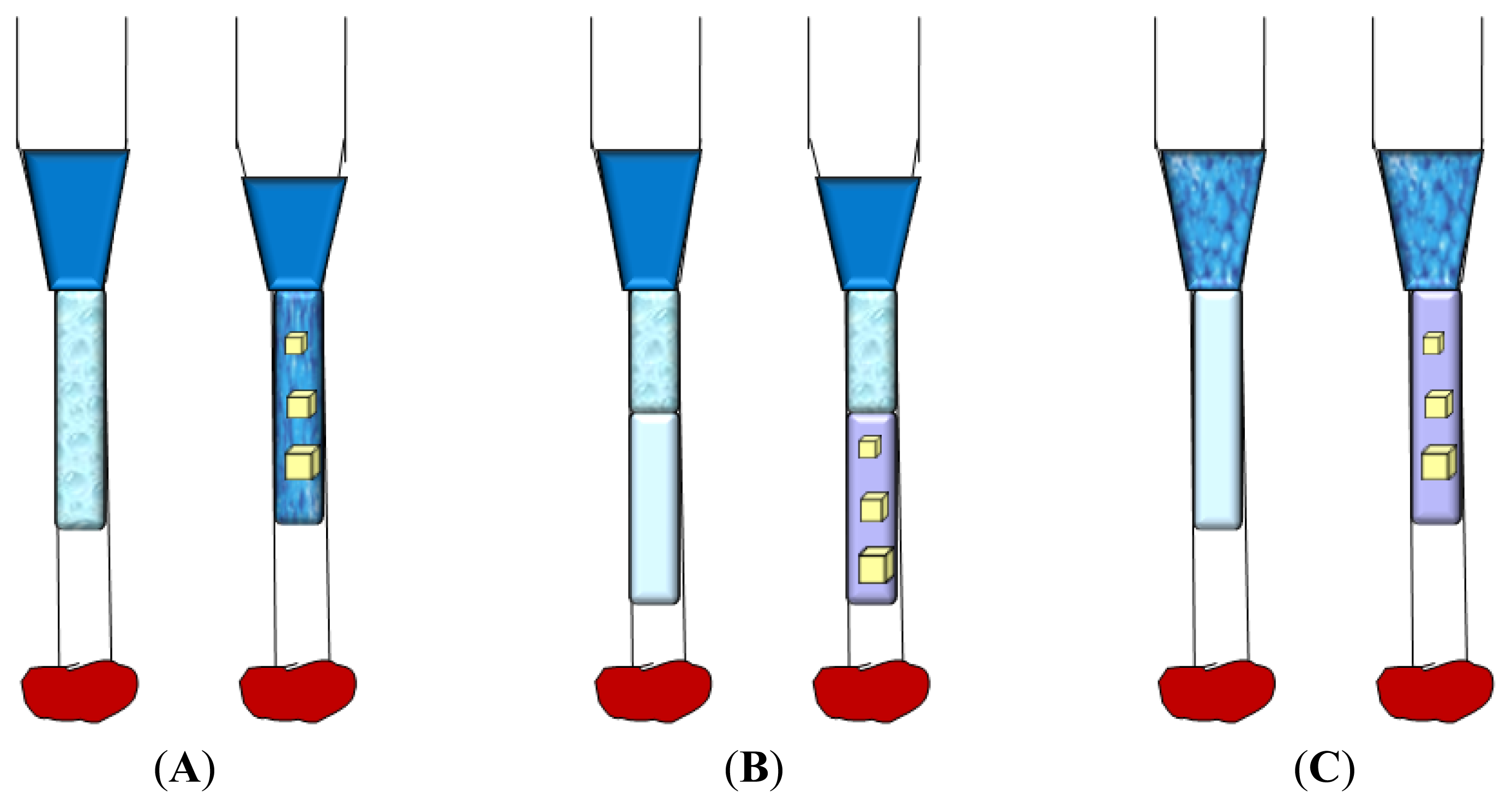
3.6. Counter-Diffusion in Gel
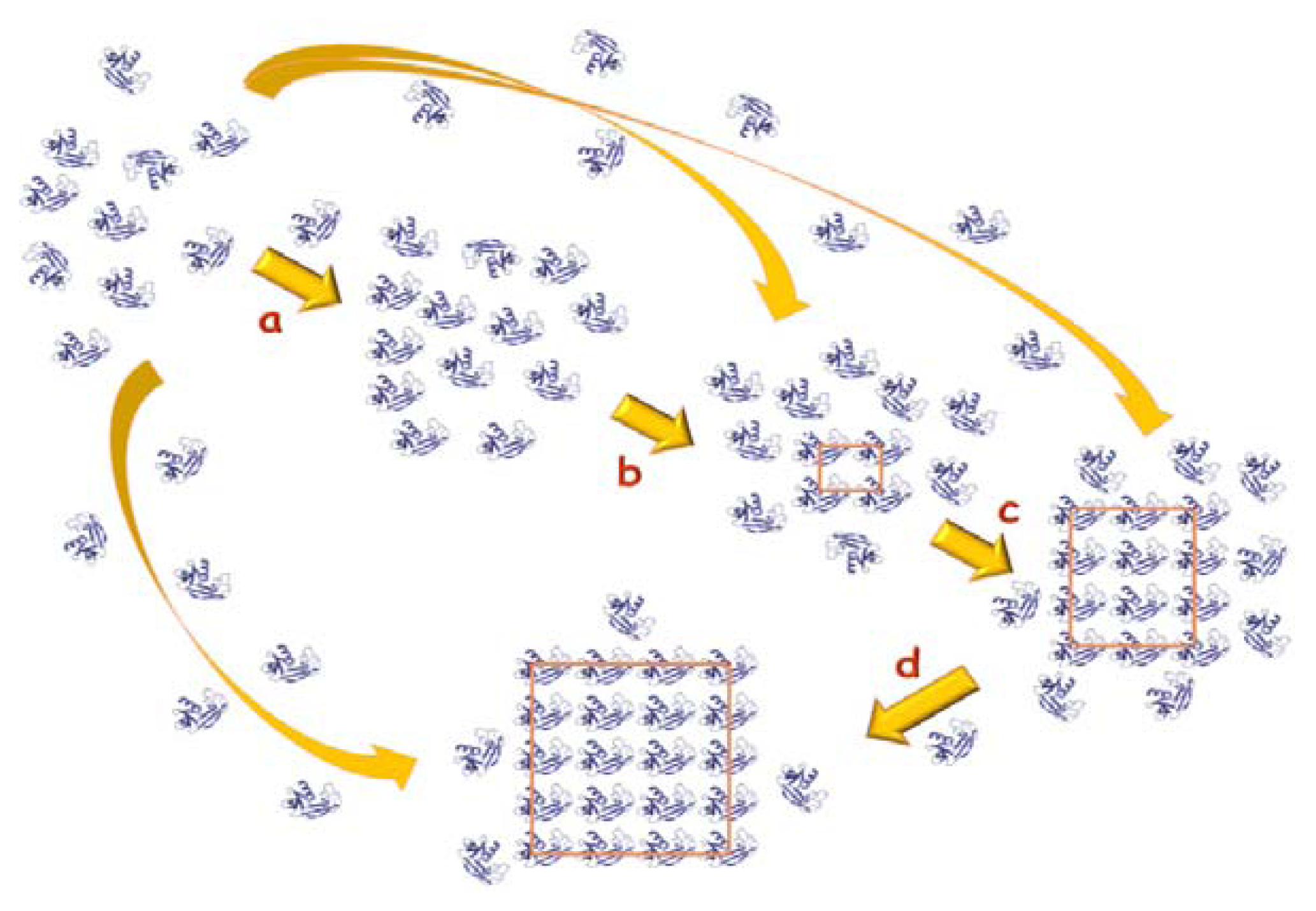
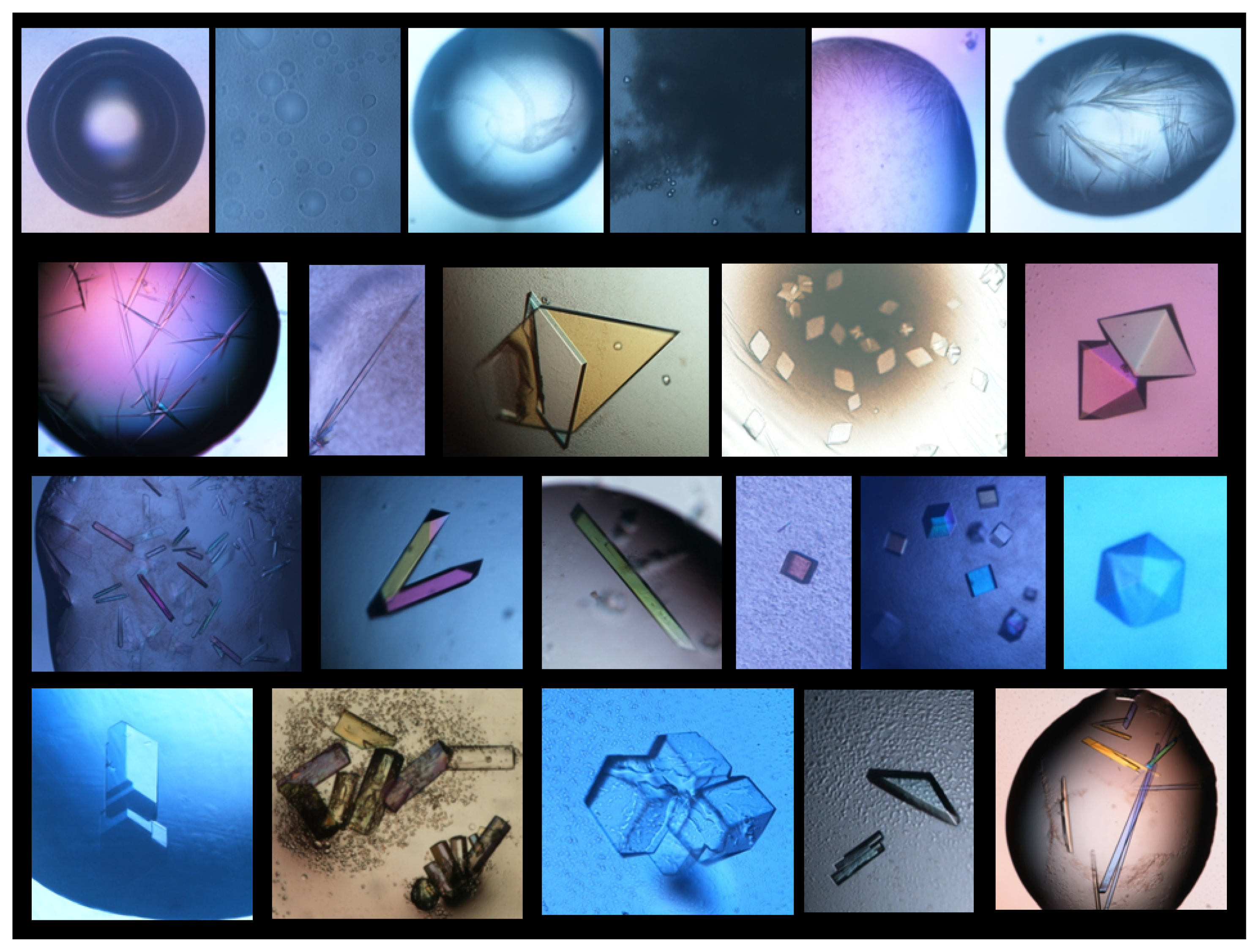
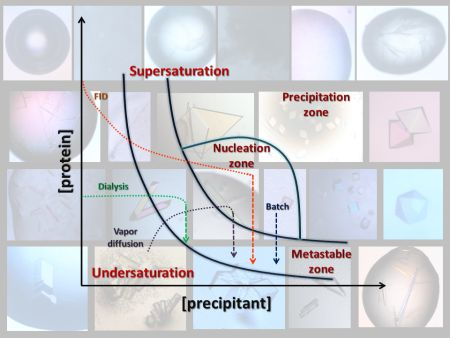
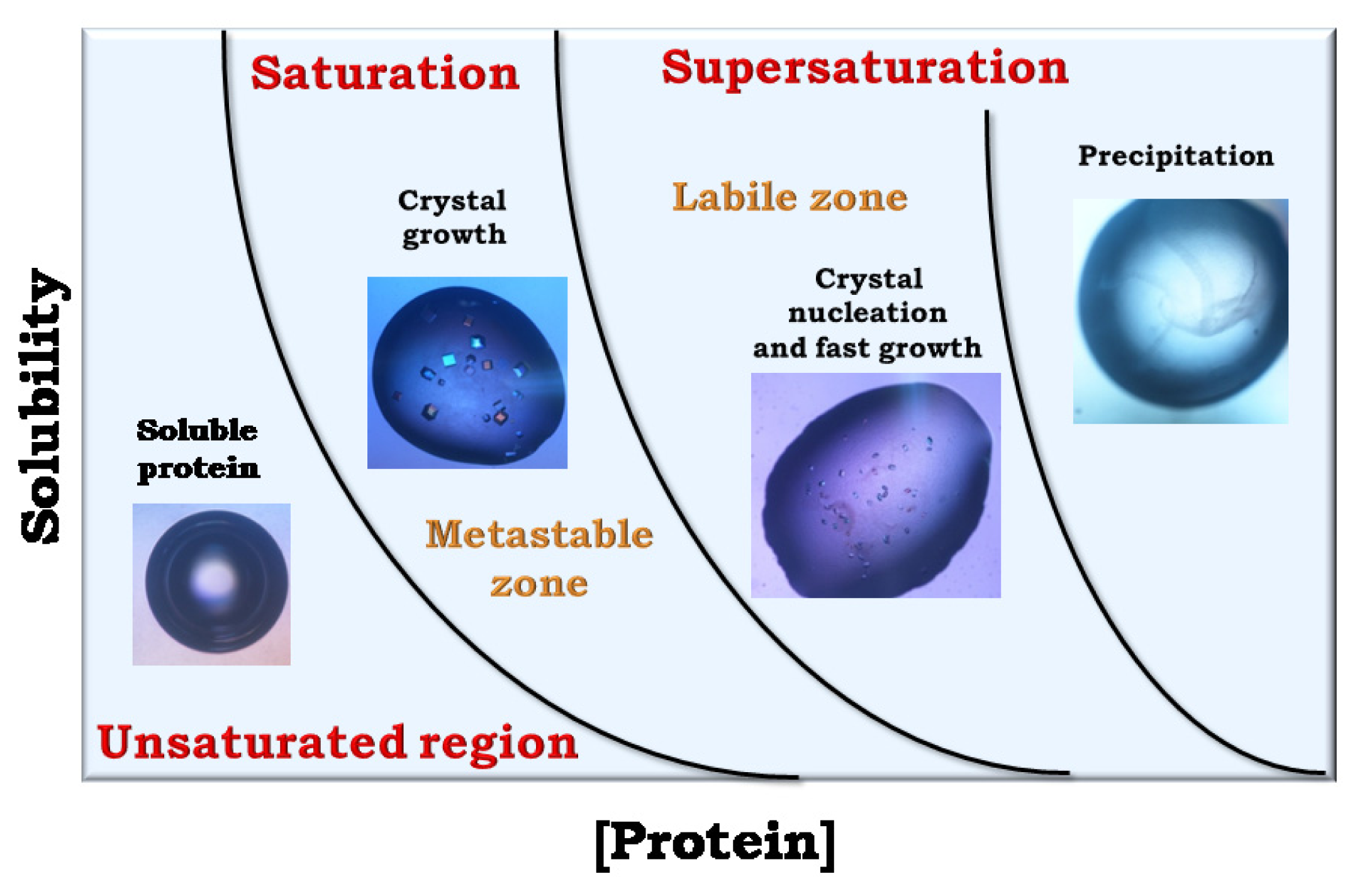
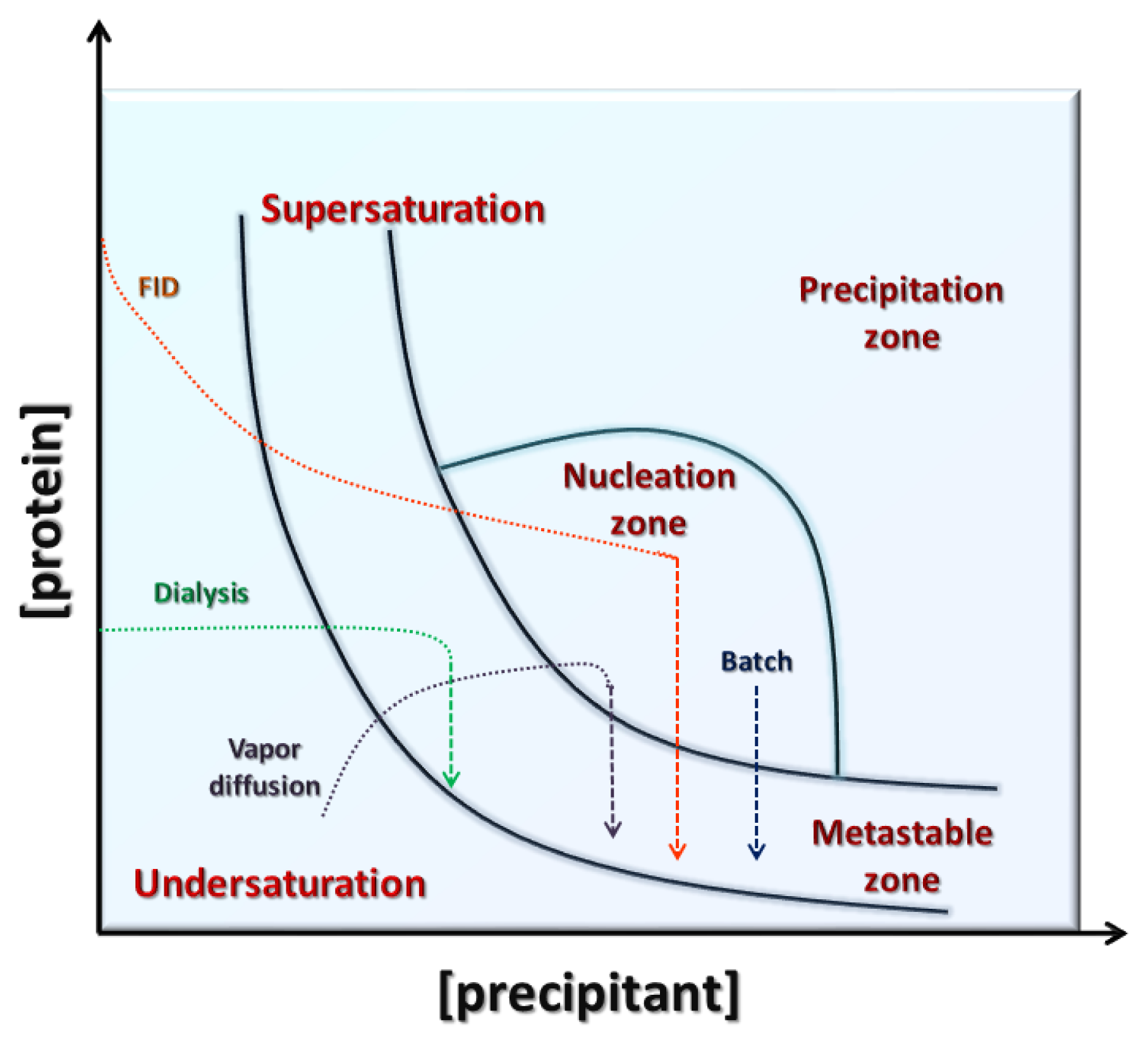
4. Search of Crystallization Conditions
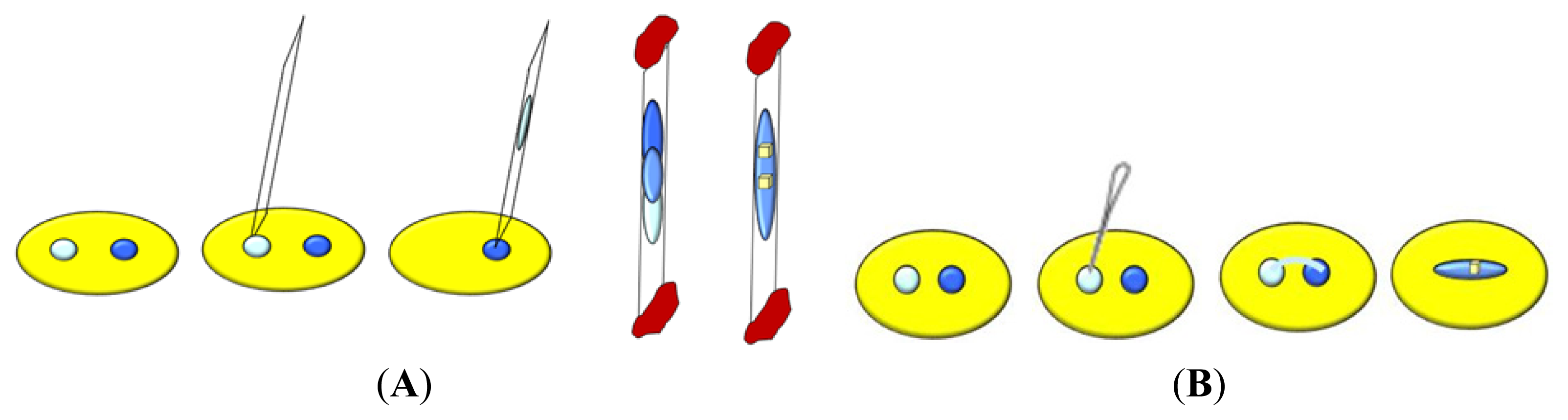
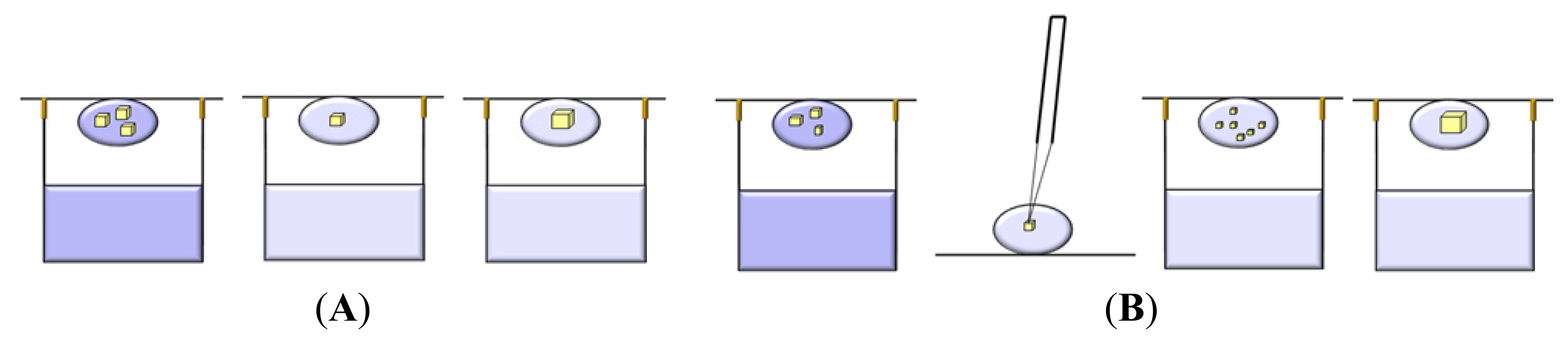
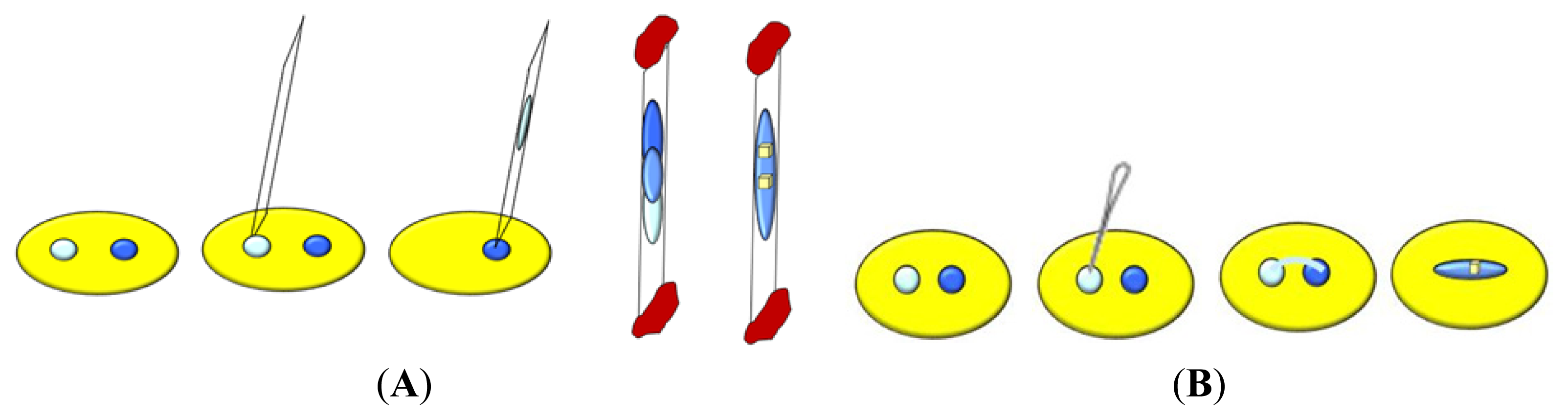
5. Approaches to Induce Nucleation
6. Crystals for Neutron Crystallography
7. Nucleic Acid in Free or Liganded State
7.1. DNA and RNA Quadruplexes as an Example of Nucleic Acid Structures
7.2. Protein-Nucleic Acid Complexes
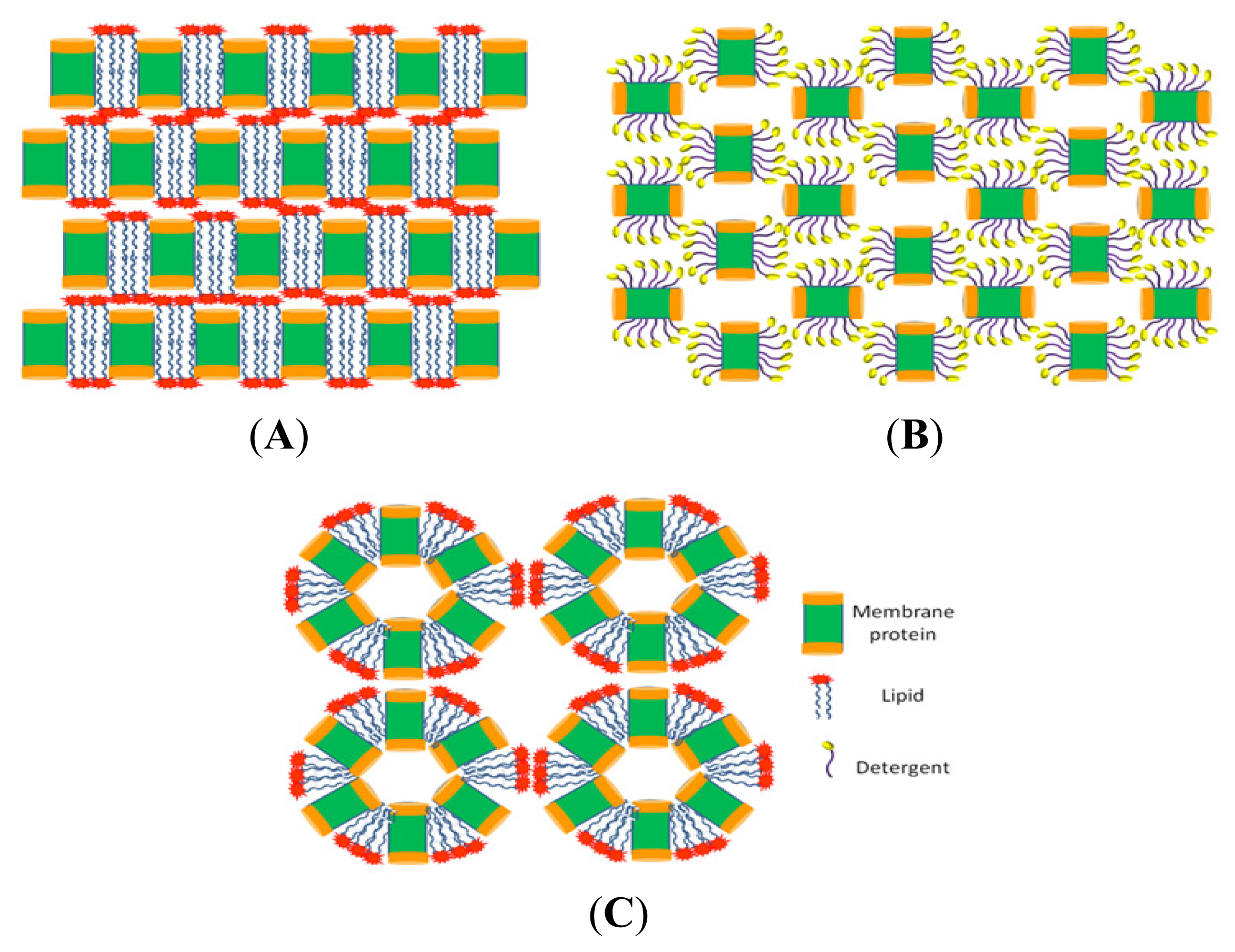
8. Membrane Proteins
8.1. Detergent-Based
8.2. Lipidic Cubic Phase
8.3. Lipidic Sponge Phase
8.4. Bilayered Micelles
8.5. Antibody-Fragment-Mediated
9. Real-Time Monitoring of Crystals
10. Improvement of Crystallizability and of X-Ray Diffraction Limits
11. Concluding Remarks
Acknowledgments
Conflict of Interest
References and Notes
- McPherson, A. Protein crystallization in the structural genomics era. J. Struct. Funct. Genomics 2004, 5, 3–12. [Google Scholar]
- Hoffman, I.D. Protein crystallization for structure-based drug design. Methods Mol. Biol 2012, 841, 67–91. [Google Scholar]
- Deschamps, J.R. The role of crystallography in drug design. AAPS J 2005, 7, E813–E819. [Google Scholar]
- Cachau, R.E.; Podjarny, A.D. High-resolution crystallography and drug design. J. Mol. Recognit 2005, 18, 196–202. [Google Scholar]
- Blundell, T.L.; Patel, S. High-throughput X-ray crystallography for drug discovery. Curr. Opin. Pharmacol 2004, 4, 490–496. [Google Scholar]
- Heinemann, U.; Bussow, K.; Mueller, U.; Umbach, P. Facilities and methods for the high-throughput crystal structural analysis of human proteins. Acc. Chem. Res 2003, 36, 157–163. [Google Scholar]
- Abola, E.; Kuhn, P.; Earnest, T.; Stevens, R.C. Automation of X-ray crystallography. Nat. Struct. Biol 2000, 7, 973–977. [Google Scholar]
- Jaskolski, M. From atomic resolution to molecular giants: An overview of crystallographic studies of biological macromolecules with synchrotron radiation. Synchrotron. Radiat. Nat. Sci 2010, 9, 1–2. [Google Scholar]
- Bergfors, T.M. Protein Crystallization: Techniques, Strategies and Tips; International University Line: La Jolla, CA, USA, 1999. [Google Scholar]
- Ducruix, A.; Giege, R. Crystallization of Nucleic Acids and Proteins, A Practical Approach, 2nd ed.; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Sauter, C.; Lorber, B.; McPherson, A.; Giege, R. Crystallization. General methods. In International Tables for Crystallography. Volume F: Crystallography of Biological Macromolecules, 2nd ed.; Arnold, E., Himmel, D.M., Rossmann, M.G., Eds.; Wiley: Chichester, NH, USA, 2012; pp. 99–121. [Google Scholar]
- Rupp, B. Biomolecular Crystallography: Principles, Practice, and Application to Structural Biology; Garland Publishing: New York, NY, USA, 2010. [Google Scholar]
- Amorphous precipitate is the result of a disordered aggregation of folded molecules, it is usually white and could still give crystals, differently from precipitate of denaturated proteins, which forms in conditions where the native state is not stable. Moreover some precipitates are actually formed by submicroscopic crystals (microcrystalline precipitate).
- McPherson, A. Crystallization of Biological Macromolecules; Cold Spring Harbor Laboratory Press: Cold Spring Harbor: New York, NY, USA, 1999. [Google Scholar]
- McPherson, A. Introduction to protein crystallization. Methods 2004, 34, 254–265. [Google Scholar]
- Kelly, S.M.; Price, N.C. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci 2000, 1, 349–384. [Google Scholar]
- George, A.; Chiang, Y.; Guo, B.; Arabshahi, A.; Cai, Z.; Wilson, W.W. Second virial coefficient as predictor in protein crystal growth. Methods Enzymol 1997, 276, 100–110. [Google Scholar]
- George, A.; Wilson, W.W. Predicting protein crystallization from a dilute solution property. Acta Crystallogr. D 1994, 50, 361–365. [Google Scholar]
- Bonnete, F.; Vivares, D. Interest of the normalized second virial coefficient and interaction potentials for crystallizing large macromolecules. Acta Crystallogr. D 2002, 58, 1571–1575. [Google Scholar]
- Deszczynski, M.; Harding, S.E.; Winzor, D.J. Negative second virial coefficients as predictors of protein crystal growth: Evidence from sedimentation equilibrium studies that refutes the designation of those light scattering parameters as osmotic virial coefficients. Biophys. Chem 2006, 120, 106–113. [Google Scholar]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering with Applications to Chemistry, Biology, and Physics; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Pecora, R. Dynamic Light Scattering: Applications of Photon Correlation Spectroscopy; Plenum Press: New York, NY, USA, 1985. [Google Scholar]
- Saridakis, E.; Dierks, K.; Moreno, A.; Dieckmann, M.W.; Chayen, N.E. Separating nucleation and growth in protein crystallization using dynamic light scattering. Acta Crystallogr. D 2002, 58, 1597–600. [Google Scholar]
- Rosenberger, F.; Howard, S.B.; Sowers, J.W.; Nyce, T.A. Temperature dependence of protein solubility: Determination and application to crystallization in X-ray capillaries. J. Cryst. Growth 1993, 129, 1–12. [Google Scholar]
- Chernov, A.A.; Komatsu, H. Principles of Crystal Growth in Protein Crystallization. In Science and Technology of Crystal Growth; van der Eerden, J.P., Bruinsma, O.S.L., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995. [Google Scholar]
- Dumetz, A.C.; Chockla, A.M.; Kaler, E.W.; Lenhoff, A.M. Effects of pH on protein-protein interactions and implications for protein phase behavior. Biochim. Biophys. Acta 2008, 1784, 600–610. [Google Scholar]
- Kantardjieff, K.A.; Rupp, B. Protein isoelectric point as a predictor for increased crystallization screening efficiency. Bioinformatics 2004, 20, 2162–2168. [Google Scholar]
- Dupeux, F.; Rower, M.; Seroul, G.; Blot, D.; Marquez, J.A. A thermal stability assay can help to estimate the crystallization likelihood of biological samples. Acta Crystallogr. D 2011, 67, 915–919. [Google Scholar]
- Malawski, G.A.; Hillig, R.C.; Monteclaro, F.; Eberspaecher, U.; Schmitz, A.A.; Crusius, K.; Huber, M.; Egner, U.; Donner, P.; Muller-Tiemann, B. Identifying protein construct variants with increased crystallization propensity-a case study. Protein Sci 2006, 15, 2718–2728. [Google Scholar]
- Ericsson, U.B.; Hallberg, B.M.; Detitta, G.T.; Dekker, N.; Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. BioChem 2006, 357, 289–298. [Google Scholar]
- Doerr, A. Widening the protein crystallization bottleneck. Nat. Methods 2006, 3, 961. [Google Scholar]
- Hofmeister, F. Zur Lehre von der Wirkung der Salze (About the science of the effect of salts). Arch. Exp. Pathol. Pharmakol 1888, 24, 247–260. [Google Scholar]
- McPherson, A. Crystallization of proteins from polyethylene glycol. J. Biol. Chem 1976, 251, 6300–6303. [Google Scholar]
- Majeed, S.; Ofek, G.; Belachew, A.; Huang, C.C.; Zhou, T.; Kwong, P.D. Enhancing protein crystallization through precipitant synergy. Structure 2003, 11, 1061–1070. [Google Scholar]
- McPherson, A.; Nguyen, C.; Cudney, R.; Larson, S.B. The role of small molecule additives and chemical modification in protein crystallization. Cryst. Growth Des 2011, 11, 1469–1474. [Google Scholar]
- McPherson, A. The Preparation and Analysis of Protein Crystals; John Wiley and Sons: New York, NY, USA, 1982. [Google Scholar]
- Piazza, R.; Pierno, M. Protein interactions near crystallization: A microscopic approach to the Hofmeister series. J. Phys. Condens. Matter 2000, 12, A443–A449. [Google Scholar]
- Zhang, Y.; Cremer, P.S. Interactions between macromolecules and ions: The Hofmeister series. Curr. Opin. Chem. Biol 2006, 10, 658–663. [Google Scholar]
- Hekmat, D.; Hebel, D.; Joswig, S.; Schmidt, M.; Weuster-Botz, D. Advanced protein crystallization using water-soluble ionic liquids as crystallization additives. Biotechnol. Lett 2007, 29, 1703–1711. [Google Scholar]
- Judge, R.A.; Takahashi, S.; Longenecker, K.L.; Fry, E.H.; Abad-Zapatero, C.; Chiu, M.L. The effect of ionic liquids on protein crystallization and X-ray diffraction resolution. Cryst. Growth Des 2009, 9, 3463–3469. [Google Scholar]
- Larson, S.B.; Day, J.S.; Cudney, R.; McPherson, A. A novel strategy for the crystallization of proteins: X-ray diffraction validation. Acta Crystallogr. D 2007, 63, 310–318. [Google Scholar]
- Sauter, C.; Lorber, B.; Kern, D.; Cavarelli, J.; Moras, D.; Giege, R. Crystallogenesis studies on yeast aspartyl-tRNA synthetase: Use of phase diagram to improve crystal quality. Acta Crystallogr. D 1999, 55, 149–156. [Google Scholar]
- Sauter, C.; Ng, J.D.; Lorber, B.; Keith, G.; Brion, P.; Hosseini, M.W.; Lehn, J.-M.; Giege, R. Additives for the crystallization of proteins and nucleic acids. J. Cryst. Growth 1999, 196, 365–376. [Google Scholar]
- Shim, J.U.; Cristobal, G.; Link, D.R.; Thorsen, T.; Fraden, S. Using microfluidics to decouple nucleation and growth of protein crystals. Cryst. Growth Des 2007, 7, 2192–2194. [Google Scholar]
- Tanaka, S.; Ataka, M.; Ito, K. Pattern formation and coarsening during metastable phase separation in lysozyme solutions. Phys. Rev. E 2002, 65, 051804. [Google Scholar]
- Sica, F.; Adinolfi, S.; Berisio, R.; de Lorenzo, C.; Mazzarella, L.; Piccoli, R.; Vitagliano, L.; Zagari, A. Crystallization of multiple forms of bovine seminal ribonuclease in the liganded and unliganded state. J. Cryst. Growth 1999, 196, 305–312. [Google Scholar]
- Sousa, R. Use of glycerol, polyols and other protein structure stabilizing agents in protein crystallization. Acta Crystallogr. D 1995, 51, 271–277. [Google Scholar]
- Tomčová, I.; Branca, R.M.; Bodo, G.; Bagyinka, C.; Smatanová, I.K. Cross-crystallization method used for the crystallization and preliminary diffraction analysis of a novel di-haem cytochrome c4. Acta Crystallogr. Sect. F 2006, 62, 820–824. [Google Scholar]
- Tomčová, I.; Smatanová, I.K. Copper co-crystallization and divalent metal salts cross-influence effect: A new optimization tool improving crystal morphology and diffraction quality. J. Cryst. Growth 2007, 306, 383–389. [Google Scholar]
- Kundrot, C.E.; Judge, R.A.; Pusey, M.L.; Snell, E.H. Microgravity and macromolecular crystallography. Cryst. Growth Des 2001, 1, 87–99. [Google Scholar]
- Otalora, F.; Novella, M.L.; Gavira, J.A.; Thomas, B.R.; Garcia Ruiz, J.M. Experimental evidence for the stability of the depletion zone around a growing protein crystal under microgravity. Acta Crystallogr. D 2001, 57, 412–417. [Google Scholar]
- Wakayama, N.I.; Yin, D.C.; Harata, K.; Kiyoshi, T.; Fujiwara, M.; Tanimoto, Y. Macromolecular crystallization in microgravity generated by a superconducting magnet. Ann. N. Y. Acad. Sci 2006, 1077, 184–193. [Google Scholar]
- Vergara, A.; Lorber, B.; Sauter, C.; Giege, R.; Zagari, A. Lessons from crystals grown in the advanced protein crystallisation facility for conventional crystallisation applied to structural biology. Biophys. Chem 2005, 118, 102–112. [Google Scholar]
- Sazaki, G. Crystal quality enhancement by magnetic fields. Prog. Biophys. Mol. Biol 2009, 101, 45–55. [Google Scholar]
- Yin, D.C.; Wakayama, N.I.; Harata, K.; Fujiwara, M.; Kiyoshi, T.; Wada, H.; Niimura, N.; Arai, S.; Huang, W.D.; Tanimoto, Y. Formation of protein crystals (orthorhombic lysozyme) in quasi-microgravity environment obtained by superconducting magnet. J. Cryst. Growth 2004, 270, 184–191. [Google Scholar]
- Okada, H.; Hirota, N.; Matsumoto, S.; Wada, H. Simulation of fluid flow during protein crystal growth in magnetic fields. J. Appl. Phys 2011, 110, 043903–043906. [Google Scholar]
- Ataka, M.; Katoh, E.; Wakayama, N.I. Magnetic orientation as a tool to study the initial stage of crystallization of lysozyme. J. Cryst. Growth 1997, 173, 592–596. [Google Scholar]
- Gavira, J.A.; Garcia-Ruiz, J.M. Effects of a magnetic field on lysozyme crystal nucleation and growth in a diffusive environment. Cryst. Growth Design 2009, 9, 2610–2615. [Google Scholar]
- Penkova, A.; Pan, W.; Hodjaoglu, F.; Vekilov, P.G. Nucleation of protein crystals under the influence of solution shear flow. Ann. N. Y. Acad. Sci 2006, 1077, 214–231. [Google Scholar]
- Koizumi, H.; Uda, S.; Fujiwara, K.; Nozawa, J. Control of effect on the nucleation rate for hen egg white lysozyme crystals under application of an external ac electric field. Langmuir 2011, 27, 8333–8338. [Google Scholar]
- Koizumi, H.; Fujiwara, K.; Uda, S. Control of nucleation rate for tetragonal hen-egg white lysozyme crystals by application of an electric field with variable frequencies. Cryst. Growth Des 2009, 9, 2420–2424. [Google Scholar]
- Taleb, M.; Didierjean, C.; Jelsch, C.; Mangeot, J.P.; Aubry, A. Equilibrium kinetics of lysozyme crystallization under an external electric field. J. Cryst. Growth 2001, 232, 250–255. [Google Scholar]
- Taleb, M.; Didierjean, C.; Jelsch, C.; Mangeot, J.P.; Capelle, B.; Aubry, A. Crystallization of proteins under an external electric field. J. Cryst. Growth 1999, 200, 575–582. [Google Scholar]
- Nanev, C.N.; Penkova, A. Nucleation of lysozyme crystals under external electric and ultrasonic fields. J. Cryst. Growth 2001, 232, 285–293. [Google Scholar]
- Charron, C.; Didierjean, C.; Mangeot, J.P.; Aubry, A. The “Octopus” plate for protein crystallization under an electric field. J. Appl. Crystallogr 2003, 36, 1482–1483. [Google Scholar]
- Mirkin, N.; Frontana-Uribe, B.A.; Rodrıguez-Romero, A.; Hernandez-Santoyo, A.; Moreno, A. The influence of an internal electric field upon protein crystallization using the gel-acupunture method. Acta Crystallogr. D 2003, D59, 1533–1538. [Google Scholar]
- Al-Haq, M.I.; Lebrasseur, E.; Choi, W.-K.; Tsuchiya, H.; Torii, T.; Yamazaki, H.; Shinohara, E. An apparatus for electric-field-induced protein crystallization. J. Appl. Crystallogr 2007, 40, 199–201. [Google Scholar]
- Sazaki, G.; Moreno, A.; Nakajima, K. Novel coupling effects of the magnetic and electric fields on protein crystallization. J. Cryst. Growth 2004, 262, 499–502. [Google Scholar]
- Hammadi, Z.; Veesler, S. New approaches on crystallization under electric fields. Progr. Biophys. Mol. Biol 2009, 101, 38–44. [Google Scholar]
- Gutiérrez-Quezada, A.E.; Arreguín-Espinosa, R.; Moreno, A.; Natarajan, S.; Kalkura, S.N.; Celestian, A.J.; Parise, J.B.; Clearfield, A.; Nikl, M.; Vedda, A.; et al. Special Topics in Crystal Growth. In Springer Handbook of Crystal Growth; Dhanaraj, G., Byrappa, K., Prasad, V., Dudley, M., Eds.; Springer: New York, NY, USA, 2010; Volume H, pp. 1583–1736. [Google Scholar]
- Adachi, H.; Takano, K.; Niino, A.; Matsumura, H.; Kinoshita, T.; Warizaya, M.; Inoue, T.; Mori, Y.; Sasaki, T. Solution stirring initiates nucleation and improves the quality of adenosine deaminase crystals. Acta Crystallogr. D 2005, 61, 759–762. [Google Scholar]
- Adachi, H.; Takano, K.; Matsumura, H.; Inoue, T.; Mori, Y.; Sasaki, T. Protein crystal growth with a two-liquid system and stirring solution. J. Synchrotron. Rad 2004, 11, 121–124. [Google Scholar]
- Yaoi, M.; Adachi, H.; Takano, K.; Matsumura, H.; Inoue, T.; Mori, Y.; Sasaki, T. The effects of solution stirring on protein crystal growth. Jpn. J. Appl. Phys 2004, 43, L686–L688. [Google Scholar]
- Adachi, H.; Niino, A.; Kinoshita, T.; Warizaya, M.; Maruki, R.; Takano, K.; Matsumura, H.; Inoue, T.; Murakami, S.; Mori, Y.; et al. Solution-stirring method improves crystal quality of human triosephosphate isomerase. J. BioSci. Bioeng 2006, 101, 83–86. [Google Scholar]
- Nagata, A.; Ohnishi, H.; Yoshimura, M.; Ogawa, A.; Ujita, S.; Adachi, H.; Okada, M.; Matozaki, T.; Nakagawa, A. Crystallization and preliminary X-ray analysis of rat SHPS-1. Acta Crystallogr. Sect. F 2006, 62, 189–191. [Google Scholar]
- Chayen, N.E. Comparative studies of protein crystallization by vapour-diffusion and microbatch techniques. Acta Crystallogr. D 1998, 1, 8–15. [Google Scholar]
- Benvenuti, M.; Mangani, S. Crystallization of soluble proteins in vapor diffusion for X-ray crystallography. Nat. Prot 2007, 2, 1633–1651. [Google Scholar]
- Korczynska, J.; Hu, T.C.; Smith, D.K.; Jenkins, J.; Lewis, R.; Edwards, T.; Brzozowski, A.M. Microscale vapour diffusion for protein crystallization. Acta Crystallogr. D 2007, 63, 1009–1015. [Google Scholar]
- Whon, T.W.; Lee, Y.H.; An, D.S.; Song, H.K.; Kim, S.G. A simple technique to convert sitting-drop vapor diffusion into hanging-drop vapor diffusion by solidifying the reservoir solution with agarose. J. Appl. Crystallogr 2009, 42, 975–976. [Google Scholar]
- Lu, Q.Q.; Yin, D.C.; Xie, S.X.; Liu, Y.M.; Chen, R.Q. The effect of diluting crystallization droplets on protein crystallization in vapor diffusion method. Cryst. Res. Technol 2011, 46, 917–925. [Google Scholar]
- Chayen, N.E. A novel technique to control the rate of vapor diffusion, giving larger protein crystals. J. Appl. Crystallogr 1997, 30, 198–201. [Google Scholar]
- Chayen, N.E. Crystallization with oils: A new dimension in macromolecular crystal growth. J. Cryst. Growth 1999, 196, 434–441. [Google Scholar]
- Chayen, N.E.; Shaw Stewart, P.D.; Maeder, D.L.; Blow, D.M. An automated system for micro-batch protein crystallization and screening. J. Appl. Crystallogr 1990, 23, 297–302. [Google Scholar]
- Brumshtein, B.; Greenblatt, H.M.; Futerman, A.H.; Silman, I.; Sussman, J.L. Control of the rate of evaporation in protein crystallization by the “microbatch under oil” method. J. Appl. Crystallogr 2008, 41, 969–971. [Google Scholar]
- D’Arcy, A.; Elmore, C.; Stihle, M.; Johnston, J.E. A novel approach to crystallising proteins under oil. J. Cryst. Growth 1996, 168, 175–180. [Google Scholar]
- D’Arcy, A.; Mac Sweeney, A.; Stihle, M.; Haber, A. The advantages of using a modified microbatch method for rapid screening of protein crystallization conditions. Acta Crystallogr. D 2003, 59, 396–399. [Google Scholar]
- Merlino, A.; Russo Krauss, I.; Albino, A.; Pica, A.; Vergara, A.; Masullo, M.; de Vendittis, E.; Sica, F. Improving protein crystal quality by the without-oil microbatch method: Crystallization and preliminary X-ray diffraction analysis of glutathione synthetase from Pseudoalteromonas haloplanktis. Int. J. Mol. Sci 2011, 12, 6312–6319. [Google Scholar]
- Reid, B.R.; Koch, G.L.; Boulanger, Y.; Hartley, B.S.; Blow, D.M. Letter: Crystallization and preliminary X-ray diffraction studies on tyrosyl-transfer RNA synthetase from Bacillus stearothermophilus. J. Mol. Biol 1973, 80, 199–201. [Google Scholar]
- Thomas, D.H.; Rob, A.; Rice, D.W. A novel dialysis procedure for the crystallization of proteins. Protein Eng 1989, 2, 489–491. [Google Scholar]
- Salemme, F.R. A free interface diffusion technique for the crystallization of proteins for X-ray crystallography. Arch. BioChem. Biophys 1972, 151, 533–539. [Google Scholar]
- Berisio, R.; Lamzin, V.S.; Sica, F.; Wilson, K.S.; Zagari, A.; Mazzarella, L. Protein titration in the crystal state. J. Mol. Biol 1999, 292, 845–854. [Google Scholar]
- Vitagliano, L.; Merlino, A.; Zagari, A.; Mazzarella, L. Productive and nonproductive binding to ribonuclease A: X-ray structure of two complexes with uridylyl(2′,5′)guanosine. Protein Sci 2000, 9, 1217–1225. [Google Scholar]
- Garcia-Ruiz, J.M. Counter-diffusion methods for macromolecular crystallization. Methods Enzymol 2003, 368, 130–154. [Google Scholar]
- Li, L.; Du, W.; Ismagilov, R.F. Multiparameter screening on SlipChip used for nanoliter protein crystallization combining free interface diffusion and microbatch methods. J. Am. Chem. Soc 2010, 132, 112–119. [Google Scholar]
- McPherson, A. Effects of a microgravity environment on the crystallization of biological macromolecules; Presented at VIII European Symposium on Materials and Fluid Sciences in Microgravity, Free University of Brussels, Brussels, Belgium, 12–16 April 1992, Legros, J.C., Ed.; Volume II, pp. 619–626.
- Bernard, Y.; Degoy, S.; Lefaucheux, F.; Robert, M.C. A gel-mediated feeding technique for protein crystal growth from hanging drops. Acta Crystallogr. D 1994, 50, 504–507. [Google Scholar]
- Lorber, B.; Sauter, C.; Theobald-Dietrich, A.; Moreno, A.; Schellenberger, P.; Robert, M.C.; Capelle, B.; Sanglier, S.; Potier, N.; Giege, R. Crystal growth of proteins, nucleic acids, and viruses in gels. Prog. Biophys. Mol. Biol 2009, 101, 13–25. [Google Scholar]
- Robert, M.C.; Lefaucheux, F. Crystal growth in gels: Principle and applications. J. Cryst. Growth 1988, 90, 358–367. [Google Scholar]
- Garcia-Ruiz, J.M.; Otálora, F.; Novella, M.L.; Gavira, J.A.; Sauter, C.; Vidal, O. A supersaturation wave of protein crystallization. J. Cryst. Growth 2001, 232, 165–172. [Google Scholar]
- Lorber, B.; Giege, R. Nucleation and growth of thaumatin crystals within a gel under microgravity on STS-95 mission vs. under Earth’s gravity. J. Cryst. Growth 2001, 231, 252–261. [Google Scholar]
- Sauter, C.; Lorber, B.; Giege, R. Towards atomic resolution with crystals grown in gel: The case of thaumatin seen at room temperature. Proteins 2002, 48, 146–150. [Google Scholar]
- Zhu, D.W.; Lorber, B.; Sauter, C.; Ng, J.D.; Benas, P.; Le Grimellec, C.; Giege, R. Growth kinetics, diffraction properties and effect of agarose on the stability of a novel crystal form of Thermus thermophilus aspartyl-tRNA synthetase-1. Acta Crystallogr. D 2001, 57, 552–558. [Google Scholar]
- Moreno, A.; Yokaichiya, F.; Dimasi, E.; Stojanoff, V. Growth and characterization of high-quality protein crystals for X-ray crystallography. Ann. N. Y. Acad. Sci 2009, 1161, 429–436. [Google Scholar]
- Chayen, N.E. Turning protein crystallisation from an art into a science. Curr. Opin. Struct. Biol 2004, 14, 577–583. [Google Scholar]
- Biertumpfel, C.; Basquin, J.; Suck, D.; Sauter, C. Crystallization of biological macromolecules using agarose gel. Acta Crystallogr. D 2002, 58, 1657–1659. [Google Scholar]
- Sugiyama, S.; Maruyama, M.; Sazaki, G.; Hirose, M.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; Mori, Y.; Matsumura, H. Growth of protein crystals in hydrogels prevents osmotic shock. J. Am. Chem. Soc 2012, 134, 5786–5789. [Google Scholar]
- Hasenaka, H.; Sugiyama, S.; Hirose, M.; Shimizu, N.; Kitatani, T.; Takahashi, Y.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; et al. Femtosecond laser processing of protein crystals grown in agarose gel. J. Cryst. Growth 2010, 312, 73–78. [Google Scholar]
- Van Driessche, A.E. S.; Otalora, F.; Gavira, J.A.; Sazaki, G. Is agarose an impurity or an impurity filter? In situ observation of the joint gel/impurity effect on protein crystal growth kinetics. Cryst. Growth Design 2008, 8, 3623–3629. [Google Scholar]
- Garcia Ruiz, J.M. The uses of crystal growth in gels and other diffusing-reacting systems. Key Eng. Mater 1991, 58, 87–106. [Google Scholar]
- Otalora, F.; Gavira, J.A.; Ng, J.D.; Garcia-Ruiz, J.M. Counterdiffusion methods applied to protein crystallization. Prog. Biophys. Mol. Biol 2009, 101, 26–37. [Google Scholar]
- Garcia Ruiz, J.M.; Moreno, A.; Viedma, C.; Coll, M. Crystal quality of lysozyme single crystals grown by the gel acupuncture method. Mater. Res. Bull 1993, 28, 541–546. [Google Scholar]
- Oberthuer, D.; Melero-Garcia, E.; Dierks, K.; Meyer, A.; Betzel, C.; Garcia-Caballero, A.; Gavira, J.A. Monitoring and scoring counter-diffusion protein crystallization experiments in capillaries by In situ dynamic light scattering. PLoS One 2012, 7, e33545. [Google Scholar]
- Ng, J.D.; Gavira, J.A.; Garcia-Ruiz, J.M. Protein crystallization by capillary counter-diffusion for applied crystallographic structure determination. J. Struct. Biol 2003, 142, 218–231. [Google Scholar]
- Garcia Ruiz, J.M.; Gonzales-Ramirez, J.A.; Gavira, J.A.; Otalora, F. Granada Crystallisation Box: A new device for protein crystallisation by counter-diffusion techniques. Acta Crystallogr. D 2002, D58, 1638–1642. [Google Scholar]
- Vergara, A.; Castagnolo, D.; Carotenuto, L.; Vitagliano, L.; Berisio, R.; Sorrentino, G.; Gonzalez-Ramirez, L.; Garcia-Ruiz, J.; Zagari, A. Phase behavior and crystallogenesis under counter-diffusion conditions of the collagen-model peptide (Pro-Pro-Gly)(10). J. Cryst. Growth 2009, 311, 304–309. [Google Scholar]
- Malecki, P.H.; Rypniewski, W.; Szymanski, M.; Barciszewski, J.; Meyer, A. Binding of the plant hormone kinetin in the active site of Mistletoe Lectin I from Viscum album. Biochim. Biophys. Acta 2012, 1824, 334–338. [Google Scholar]
- Meyer, A.; Rypniewski, W.; Szymanski, M.; Voelter, W.; Barciszewski, J.; Betzel, C. Structure of mistletoe lectin I from Viscum album in complex with the phytohormone zeatin. Biochim. Biophys. Acta 2008, 1784, 1590–1595. [Google Scholar]
- Shieh, H.S.; Stallings, W.C.; Stevens, A.M.; Stegeman, R.A. Using sampling techniques in protein crystallization. Acta Crystallogr. D 1995, 51, 305–310. [Google Scholar]
- Kingston, R.L.; Baker, H.M.; Baker, E.N. Search designs for protein crystallization based on orthogonal arrays. Acta Crystallogr. D 1994, 50, 429–440. [Google Scholar]
- Jancarik, J.; Kim, S.-H. Sparse matrix sampling: A screening method for crystallization of proteins. J. Appl. Crystallogr 1991, 24, 409–411. [Google Scholar]
- Carter, C.W., Jr; Carter, C.W. Protein crystallization using incomplete factorial experiments. J. Biol. Chem. 1979, 254, 12219–12223. [Google Scholar]
- Cudney, R.; Patel, S.; Weisgraber, K.; Newhouse, Y.; McPherson, A. Screening and optimization strategies for macromolecular crystal growth. Acta Crystallogr. D 1994, 50, 414–423. [Google Scholar]
- Stura, E.A.; Wilson, I.A. Applications of the streak seeding technique in protein crystallization. J. Cryst. Growth 1991, 110, 270–282. [Google Scholar]
- Stewart, P.D. S.; Kolek, S.A.; Briggs, R.A.; Chayen, N.E.; Baldock, P.F.M. Random microseeding: A theoretical and practical exploration of seed stability and seeding techniques for successful protein crystallization. Cryst. Growth Design 2011, 11, 3432–3441. [Google Scholar]
- Khurshid, S.; Haire, L.F.; Chayen, N.E. Automated seeding for the optimization of crystal quality. J. Appl. Crystallogr 2010, 43, 752–756. [Google Scholar]
- Georgiev, A.; Vorobiev, S.; Edstrom, W.; Song, T.; Laine, A.; Hunt, J.; Allen, P. Automated streak-seeding with micromachined silicon tools. Acta Crystallogr. D 2006, D62, 1039–1045. [Google Scholar]
- McPherson, A.; Shlichta, P. Heterogeneous and epitaxial nucleation of protein crystals on mineral surfaces. Science 1988, 239, 385–387. [Google Scholar]
- McPherson, A.; Shlichta, P.J. Facilitation of the growth of protein crystals by heterogeneous/epitaxial nucleation. J. Cryst. Growth 1987, 85, 206–214. [Google Scholar]
- Falini, G.; Fermani, S.; Conforti, G.; Ripamonti, A. Protein crystallisation on chemically modified mica surfaces. Acta Crystallogr. D 2002, 58, 1649–1652. [Google Scholar]
- Tang, L.; Huang, Y.B.; Liu, D.Q.; Li, J.L.; Mao, K.; Liu, L.; Cheng, Z.J.; Gong, W.M.; Hu, J.; He, J.H. Effects of the silanized mica surface on protein crystallization. Acta Crystallogr. D 2005, 61, 53–59. [Google Scholar]
- Tosi, G.; Fermani, S.; Falini, G.; Gavira Gallardo, J.A.; Garcia Ruiz, J.M. Crystallization of proteins on functionalized surfaces. Acta Crystallogr. D 2008, 64, 1054–1061. [Google Scholar]
- Rong, L.; Komatsu, H.; Natsuisaka, M.; Yoda, S. Epitaxial nucleation of protein crystal on poly-L-lysine modified surface. Jpn. J. Appl. Phys 2001, 40, 6677–6678. [Google Scholar]
- Rong, L.; Komatsu, H.; Yoda, S. Control of heterogeneous nucleation of lysozyme crystals by using Poly-L-Lysine modified substrate. J. Cryst. Growth 2002, 235, 489–493. [Google Scholar]
- Fermani, S.; Falini, G.; Minnucci, M.; Ripamonti, A. Protein crystallization on polymeric film surfaces. J. Cryst. Growth 2001, 224, 327–334. [Google Scholar]
- Sanjoh, A.; Tsukihara, T. Spatiotemporal protein crystal growth studies using microfluidic silicon devices. J. Cryst. Growth 1999, 196, 691–702. [Google Scholar]
- Sanjoh, A.; Tsukihara, T.; Gorti, S. Surface-potential controlled Si-microarray devices for heterogeneous protein crystallization screening. J. Cryst. Growth 2001, 232, 618–628. [Google Scholar]
- Di Profio, G.; Curcio, E.; Drioli, E. Trypsin crystallization by membrane-based techniques. J. Struct. Biol 2005, 150, 41–49. [Google Scholar]
- Curcio, E.; Fontananova, E.; di Profio, G.; Drioli, E. Influence of the structural properties of poly(vinylidene fluoride) membranes on the heterogeneous nucleation rate of protein crystals. J. Phys. Chem. B 2006, 110, 12438–12445. [Google Scholar]
- Chayen, N.E.; Saridakis, E.; El-Bahar, R.; Nemirovsky, Y. Porous silicon: An effective nucleation-inducing material for protein crystallization. J. Mol. Biol 2001, 312, 591–595. [Google Scholar]
- Stolyarova, S.; Saridakis, E.; Chayen, N.E.; Nemirovsky, Y. A model for enhanced nucleation of protein crystals on a fractal porous substrate. Biophys. J 2006, 91, 3857–3863. [Google Scholar]
- Rong, L.; Komatsu, H.; Yoshizaki, I.; Kadowaki, A.; Yoda, S. Protein crystallization by using porous glass substrate. J. Synchrotron. Rad 2004, 11, 27–29. [Google Scholar]
- Chayen, N.E.; Saridakis, E.; Sear, R.P. Experiment and theory for heterogeneous nucleation of protein crystals in a porous medium. Proc. Natl. Acad. Sci. USA 2006, 103, 597–601. [Google Scholar]
- Asanithi, P.; Saridakis, E.; Govada, L.; Jurewicz, I.; Brunner, E.W.; Ponnusamy, R.; Cleaver, J.A.; Dalton, A.B.; Chayen, N.E.; Sear, R.P. Carbon-nanotube-based materials for protein crystallization. ACS Appl. Mater. Interfaces 2009, 1, 1203–1210. [Google Scholar]
- Sugahara, M.; Asada, Y.; Morikawa, Y.; Kageyama, Y.; Kunishima, N. Nucleant-mediated protein crystallization with the application of microporous synthetic zeolites. Acta Crystallogr. D 2008, 64, 686–695. [Google Scholar]
- Georgieva, D.G.; Kuil, M.E.; Oosterkamp, T.H.; Zandbergen, H.W.; Abrahams, J.P. Heterogeneous nucleation of three-dimensional protein nanocrystals. Acta Crystallogr. D 2007, 63, 564–570. [Google Scholar]
- Takehara, M.; Ino, K.; Takakusagi, Y.; Oshikane, H.; Nureki, O.; Ebina, T.; Mizukami, F.; Sakaguchi, K. Use of layer silicate for protein crystallization: Effects of micromica and chlorite powders in hanging drops. Anal. BioChem 2008, 373, 322–329. [Google Scholar]
- Saridakis, E.; Khurshid, S.; Govada, L.; Phan, Q.; Hawkins, D.; Crichlow, G.V.; Lolis, E.; Reddy, S.M.; Chayen, N.E. Protein crystallization facilitated by molecularly imprinted polymers. Proc. Natl. Acad. Sci. USA 2011, 108, 11081–11086. [Google Scholar] [Green Version]
- Thakur, A.S.; Robin, G.; Guncar, G.; Saunders, N.F.; Newman, J.; Martin, J.L.; Kobe, B. Improved success of sparse matrix protein crystallization screening with heterogeneous nucleating agents. PLoS One 2007, 2, e1091. [Google Scholar]
- Nederlof, I.; Hosseini, R.; Georgieva, D.; Luo, J.; Li, D.; Abrahams, J.P. A straightforward and robust method for introducing human hair as a nucleant into high throughput crystallization trials. Cryst. Growth Des 2011, 11, 1170–1176. [Google Scholar]
- Munshi, P.; Chung, S.L.; Blakeley, M.P.; Weiss, K.L.; Myles, D.A.; Meilleur, F. Rapid visualization of hydrogen positions in protein neutron crystallographic structures. Acta Crystallogr. D 2012, 68, 35–41. [Google Scholar]
- Budayova-Spano, M.; Dauvergne, F.; Audiffren, M.; Bactivelane, T.; Cusack, S. A methodology and an instrument for the temperature-controlled optimization of crystal growth. Acta Crystallogr. D 2007, 63, 339–347. [Google Scholar]
- Matsumura, H.; Kakiniuchi, K.; Nakamura, T.; Sugiyama, S.; Maruyama, M.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; Mori, Y. Growth of protein crystals by syringe-type top-seeded solution growth. Cryst. Growth Des 2011, 11, 1486–1492. [Google Scholar]
- Matsumura, H.; Sugiyama, S.; Hirose, M.; Kakinouchi, K.; Maruyama, M.; Murai, R.; Adachi, H.; Takano, K.; Murakami, S.; Mori, Y.; et al. Approach for growth of high-quality and large protein crystals. J. Synchrotron. Rad 2011, 18, 16–19. [Google Scholar]
- Sugiyama, S.; Hasenaka, H.; Hirose, M.; Shimizu, N.; Kitatani, T.; Takahashi, Y.; Adachi, H.; Takano, K.; Murakami, S.; Inoue, T.; et al. Femtosecond laser processing of agarose gel surrounding protein crystals for development of an automated crystal capturing system. Jpn. J. Appl. Phys 2009, 48, 105502–105506. [Google Scholar]
- Murai, R.; Yoshikawa, H.Y.; Takahashi, Y.; Maruyama, M.; Sugiyama, S.; Sazaki, G.; Adachi, H.; Takano, K.; Matsumura, H.; Murakami, S.; et al. Enhancement of femtosecond laser-induced nucleation of protein in a gel solution. Appl. Phys. Lett 2010, 96, 043702–043704. [Google Scholar]
- Sherlin, L.D.; Bullock, T.L.; Nissan, T.A.; Perona, J.J.; Lariviere, F.J.; Uhlenbeck, O.C.; Scaringe, S.A. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA 2001, 7, 1671–1678. [Google Scholar]
- Choi, J.; Majima, T. Conformational changes of non-B DNA. Chem. Soc. Rev 2011, 40, 5893–5909. [Google Scholar]
- Mooers, B.H. Crystallographic studies of DNA and RNA. Methods 2009, 47, 168–176. [Google Scholar]
- Batey, R.T.; Rambo, R.P.; Lucast, L.; Rha, B.; Doudna, J.A. Crystal structure of the ribonucleoprotein core of the signal recognition particle. Science 2000, 287, 1232–1239. [Google Scholar]
- Ferre-D’Amare, A.R.; Zhou, K.; Doudna, J.A. A general module for RNA crystallization. J. Mol. Biol 1998, 279, 621–631. [Google Scholar]
- Rupert, P.B.; Ferre-D’Amare, A.R. Crystallization of the hairpin ribozyme: Illustrative protocols. Methods Mol. Biol 2004, 252, 303–311. [Google Scholar]
- Ferre-D’Amare, A.R. Use of the spliceosomal protein U1A to facilitate crystallization and structure determination of complex RNAs. Methods 2010, 52, 159–167. [Google Scholar]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5′-UTR G-quadruplexes: Diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res 2007, 35, 7429–7455. [Google Scholar]
- Lech, C.J.; Heddi, B.; Phan, A.T. Guanine base stacking in G-quadruplex nucleic acids. Nucleic Acids Res 2013, 41, 2034–2046. [Google Scholar]
- Xu, Y.; Komiyama, M. Structure, function and targeting of human telomere RNA. Methods 2012, 57, 100–105. [Google Scholar]
- Neidle, S. Human telomeric G-quadruplex: The current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J 2010, 277, 1118–1125. [Google Scholar]
- Haider, S.M.; Neidle, S.; Parkinson, G.N. A structural analysis of G-quadruplex/ligand interactions. Biochimie 2011, 93, 1239–1251. [Google Scholar]
- Lane, A.N.; Chaires, J.B.; Gray, R.D.; Trent, J.O. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res 2008, 36, 5482–5515. [Google Scholar]
- Bugaut, A.; Balasubramanian, S. A sequence-independent study of the influence of short loop lengths on the stability and topology of intramolecular DNA G-quadruplexes. BioChemistry 2008, 47, 689–697. [Google Scholar]
- Hud, N.V.; Smith, F.W.; Anet, F.A.; Feigon, J. The selectivity for K+ versus Na+ in DNA quadruplexes is dominated by relative free energies of hydration: A thermodynamic analysis by 1H NMR. BioChemistry 1996, 35, 15383–15390. [Google Scholar]
- Campbell, N.H.; Parkinson, G.N. Crystallographic studies of quadruplex nucleic acids. Methods 2007, 43, 252–263. [Google Scholar]
- Perederina, A.; Krasilnikov, A.S. Crystallization of RNA-protein complexes: From synthesis and purification of individual components to crystals. Methods Mol. Biol 2012, 905, 123–143. [Google Scholar]
- Tan, S.; Hunziker, Y.; Pellegrini, L.; Richmond, T.J. Crystallization of the yeast MATalpha2/MCM1/DNA ternary complex: General methods and principles for protein/DNA cocrystallization. J. Mol. Biol 2000, 297, 947–959. [Google Scholar]
- Jordan, S.R.; Whitcombe, T.V.; Berg, J.M.; Pabo, C.O. Systematic variation in DNA length yields highly ordered repressor-operator cocrystals. Science 1985, 230, 1383–1385. [Google Scholar]
- Ke, A.; Doudna, J.A. Crystallization of RNA and RNA-protein complexes. Methods 2004, 34, 408–414. [Google Scholar]
- Brown, D.G.; Freemont, P.S. Crystallography in the study of protein-DNA interactions. Methods Mol. Biol 1996, 56, 293–318. [Google Scholar]
- Hollis, T. Crystallization of Protein-DNA Complexes. In Macromolecular Crystallography Protocols: Volume 1, Preparation and Crystallization of Macromolecules; Springer: New York, NY, USA, 2007; Volume 363. [Google Scholar]
- Conlin, R.M.; Brown, R.S. Reconstitution of Protein-DNA Complexes for Crystallization. In DNA-Protein Interactions: Principles and Protocols; Springer: New York, NY, USA, 1994; Volume 30. [Google Scholar]
- Nadassy, K.; Wodak, S.J.; Janin, J. Structural features of protein-nucleic acid recognition sites. BioChemistry 1999, 38, 1999–2017. [Google Scholar]
- Yang, X.L.; Otero, F.J.; Ewalt, K.L.; Liu, J.; Swairjo, M.A.; Kohrer, C.; RajBhandary, U.L.; Skene, R.J.; McRee, D.E.; Schimmel, P. Two conformations of a crystalline human tRNA synthetase-tRNA complex: Implications for protein synthesis. EMBO J 2006, 25, 2919–2929. [Google Scholar]
- Rice, P.A.; Yang, S.; Mizuuchi, K.; Nash, H.A. Crystal structure of an IHF-DNA complex: A protein-induced DNA U-turn. Cell 1996, 87, 1295–1306. [Google Scholar]
- Wedekind, J.E.; McKay, D.B. Crystal structure of a lead-dependent ribozyme revealing metal binding sites relevant to catalysis. Nat. Struct. Biol 1999, 6, 261–268. [Google Scholar]
- Russo Krauss, I.; Merlino, A.; Randazzo, A.; Mazzarella, L.; Sica, F. Crystallization and preliminary X-ray analysis of the complex of human alpha-thrombin with a modified thrombin-binding aptamer. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 2010, 66, 961–963. [Google Scholar]
- Russo Krauss, I.; Merlino, A.; Giancola, C.; Randazzo, A.; Mazzarella, L.; Sica, F. Thrombin-aptamer recognition: A revealed ambiguity. Nucleic Acids Res 2011, 39, 7858–7867. [Google Scholar]
- Russo Krauss, I.; Merlino, A.; Randazzo, A.; Novellino, E.; Mazzarella, L.; Sica, F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res 2012, 40, 8119–8128. [Google Scholar]
- Garavito, R.M.; Rosenbusch, J.P. Three-dimensional crystals of an integral membrane protein: An initial x-ray analysis. J. Cell Biol 1980, 86, 327–329. [Google Scholar]
- Bill, R.M.; Henderson, P.J.; Iwata, S.; Kunji, E.R.; Michel, H.; Neutze, R.; Newstead, S.; Poolman, B.; Tate, C.G.; Vogel, H. Overcoming barriers to membrane protein structure determination. Nat. Biotechnol 2011, 29, 335–340. [Google Scholar]
- Prive, G.G. Detergents for the stabilization and crystallization of membrane proteins. Methods 2007, 41, 388–397. [Google Scholar]
- Newstead, S.; Ferrandon, S.; Iwata, S. Rationalizing alpha-helical membrane protein crystallization. Protein Sci 2008, 17, 466–472. [Google Scholar]
- Hunte, C.; Michel, H. Crystallisation of membrane proteins mediated by antibody fragments. Curr. Opin. Struct. Biol 2002, 12, 503–508. [Google Scholar]
- Landau, E.M.; Rosenbusch, J.P. Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 14532–14535. [Google Scholar]
- Wadsten, P.; Wohri, A.B.; Snijder, A.; Katona, G.; Gardiner, A.T.; Cogdell, R.J.; Neutze, R.; Engstrom, S. Lipidic sponge phase crystallization of membrane proteins. J. Mol. Biol 2006, 364, 44–53. [Google Scholar]
- Faham, S.; Bowie, J.U. Bicelle crystallization: A new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J. Mol. Biol 2002, 316, 1–6. [Google Scholar]
- Wiener, M.C. A pedestrian guide to membrane protein crystallization. Methods 2004, 34, 364–372. [Google Scholar]
- Michel, H. Crystallization of membrane proteins. Trends BioChem. Sci 1983, 8, 56–59. [Google Scholar]
- Ujwal, R.; Bowie, J.U. Crystallizing membrane proteins using lipidic bicelles. Methods 2011, 55, 337–341. [Google Scholar]
- Luecke, H.; Schobert, B.; Richter, H.T.; Cartailler, J.P.; Lanyi, J.K. Structural changes in bacteriorhodopsin during ion transport at 2 angstrom resolution. Science 1999, 286, 255–261. [Google Scholar]
- Katona, G.; Snijder, A.; Gourdon, P.; Andreasson, U.; Hansson, O.; Andreasson, L.E.; Neutze, R. Conformational regulation of charge recombination reactions in a photosynthetic bacterial reaction center. Nat. Struct. Mol. Biol 2005, 12, 630–631. [Google Scholar]
- Caffrey, M.; Cherezov, V. Crystallizing membrane proteins using lipidic mesophases. Nat. Protocols 2009, 4, 706–731. [Google Scholar]
- Caffrey, M. Crystallizing membrane proteins for structure-function studies using lipidic mesophases. BioChem. Soc. Trans 2011, 39, 725–732. [Google Scholar]
- Cherezov, V. Lipidic cubic phase technologies for membrane protein structural studies. Curr. Opin. Struct. Biol 2011, 21, 559–566. [Google Scholar]
- Li, D.; Caffrey, M. Lipid cubic phase as a membrane mimetic for integral membrane protein enzymes. Proc. Natl. Acad. Sci. USA 2011, 108, 8639–8644. [Google Scholar]
- Aherne, M.; Lyons, J.A.; Caffrey, M. A fast, simple and robust protocol for growing crystals in the lipidic cubic phase. J. Appl. Crystallogr 2012, 45, 1330–1333. [Google Scholar]
- Pebay-Peyroula, E.; Rummel, G.; Rosenbusch, J.P.; Landau, E.M. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 1997, 277, 1676–1681. [Google Scholar]
- Royant, A.; Nollert, P.; Edman, K.; Neutze, R.; Landau, E.M.; Pebay-Peyroula, E.; Navarro, J. X-ray structure of sensory rhodopsin II at 2.1-A resolution. Proc. Natl. Acad. Sci. USA 2001, 98, 10131–10136. [Google Scholar]
- Gordeliy, V.I.; Labahn, J.; Moukhametzianov, R.; Efremov, R.; Granzin, J.; Schlesinger, R.; Buldt, G.; Savopol, T.; Scheidig, A.J.; Klare, J.P.; et al. Molecular basis of transmembrane signalling by sensory rhodopsin II-transducer complex. Nature 2002, 419, 484–487. [Google Scholar]
- Vogeley, L.; Sineshchekov, O.A.; Trivedi, V.D.; Sasaki, J.; Spudich, J.L.; Luecke, H. Anabaena sensory rhodopsin: A photochromic color sensor at 2.0 A. Science 2004, 306, 1390–1393. [Google Scholar]
- Kato, H.E.; Zhang, F.; Yizhar, O.; Ramakrishnan, C.; Nishizawa, T.; Hirata, K.; Ito, J.; Aita, Y.; Tsukazaki, T.; Hayashi, S.; et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature 2012, 482, 369–374. [Google Scholar]
- Katona, G.; Andreasson, U.; Landau, E.M.; Andreasson, L.E.; Neutze, R. Lipidic cubic phase crystal structure of the photosynthetic reaction centre from Rhodobacter sphaeroides at 2.35A resolution. J. Mol. Biol 2003, 331, 681–692. [Google Scholar]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar]
- Shimamura, T.; Shiroishi, M.; Weyand, S.; Tsujimoto, H.; Winter, G.; Katritch, V.; Abagyan, R.; Cherezov, V.; Liu, W.; Han, G.W.; et al. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. [Google Scholar] [Green Version]
- Haga, K.; Kruse, A.C.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; Weis, W.I.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [Google Scholar]
- Liu, W.; Cherezov, V. Crystallization of membrane proteins in lipidic mesophases. J. Visual. Exp. JoVE 2011, 49, 1259–1270. [Google Scholar]
- Li, D.; Boland, C.; Walsh, K.; Caffrey, M. Use of a robot for high-throughput crystallization of membrane proteins in lipidic mesophases. J. Visual. Exp. JoVE 2012, 67, e4000. [Google Scholar]
- Xu, F.; Liu, W.; Hanson, M.A.; Stevens, R.C.; Cherezov, V. Development of an automated high throughput LCP-FRAP assay to guide membrane protein crystallization in lipid mesophases. Cryst. Growth Des 2011, 11, 1193–1201. [Google Scholar]
- Kubicek, J.; Schlesinger, R.; Baeken, C.; Buldt, G.; Schafer, F.; Labahn, J. Controlled in meso phase crystallization–a method for the structural investigation of membrane proteins. PLoS One 2012, 7, e35458. [Google Scholar]
- Li, D.; Boland, C.; Aragao, D.; Walsh, K.; Caffrey, M. Harvesting and cryo-cooling crystals of membrane proteins grown in lipidic mesophases for structure determination by macromolecular crystallography. J. Visual. Exp. JoVE 2012, 67, e4001. [Google Scholar]
- Nollert, P.; Navarro, J.; Landau, E.M. Crystallization of membrane proteins in cubo. Methods Enzymol 2002, 343, 183–199. [Google Scholar]
- Johansson, L.C.; Wohri, A.B.; Katona, G.; Engstrom, S.; Neutze, R. Membrane protein crystallization from lipidic phases. Curr. Opin. Struct. Biol 2009, 19, 372–378. [Google Scholar]
- Fellouse, F.A.; Esaki, K.; Birtalan, S.; Raptis, D.; Cancasci, V.J.; Koide, A.; Jhurani, P.; Vasser, M.; Wiesmann, C.; Kossiakoff, A.A.; et al. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol 2007, 373, 924–940. [Google Scholar]
- Lim, H.H.; Fang, Y.; Williams, C. High-efficiency screening of monoclonal antibodies for membrane protein crystallography. PLoS One 2011, 6, e24653. [Google Scholar]
- Sennhauser, G.; Grutter, M.G. Chaperone-assisted crystallography with DARPins. Structure 2008, 16, 1443–1453. [Google Scholar]
- Ostermeier, C.; Michel, H. Crystallization of membrane proteins. Curr. Opin. Struct. Biol 1997, 7, 697–701. [Google Scholar]
- Liu, Z.; Yan, H.; Wang, K.; Kuang, T.; Zhang, J.; Gui, L.; An, X.; Chang, W. Crystal structure of spinach major light-harvesting complex at 2.72 Å resolution. Nature 2004, 428, 287–292. [Google Scholar]
- Newman, J.; Bolton, E.E.; Muller-Dieckmann, J.; Fazio, V.J.; Gallagher, D.T.; Lovell, D.; Luft, J.R.; Peat, T.S.; Ratcliffe, D.; Sayle, R.A.; et al. On the need for an international effort to capture, share and use crystallization screening data. Acta Crystallogr. Sect. F 2012, 68, 253–258. [Google Scholar]
- Sazaki, G.; Van Driessche, A.E.; Dai, G.; Okada, M.; Matsui, T.; Otalora, F.; Tsukamoto, K.; Nakajima, K. In situ observation of elementary growth processes of protein crystals by advanced optical microscopy. Protein Peptide Lett 2012, 19, 743–760. [Google Scholar]
- Wiencek, J.M. New strategies for protein crystal growth. Ann. Rev. Biomed. Eng 1999, 1, 505–534. [Google Scholar]
- Qutub, Y.; Reviakine, I.; Maxwell, C.; Navarro, J.; Landau, E.M.; Vekilov, P.G. Crystallization of transmembrane proteins in cubo: Mechanisms of crystal growth and defect formation. J. Mol. Biol 2004, 343, 1243–1254. [Google Scholar]
- Glassford, S.E.; Govada, L.; Chayen, N.E.; Byrne, B.; Kazarian, S.G. Micro ATR FTIR imaging of hanging drop protein crystallisation. Vib. Spectrosc 2012, 63, 492–498. [Google Scholar]
- Echalier, A.; Glazer, R.L.; Fulop, V.; Geday, M.A. Assessing crystallization droplets using birefringence. Acta Crystallogr. D 2004, 60, 696–702. [Google Scholar]
- Wilson, W.W. Light scattering as a diagnostic for protein crystal growth—A practical approach. J. Struct. Biol 2003, 142, 56–65. [Google Scholar]
- Gliko, O.; Pan, W.; Katsonis, P.; Neumaier, N.; Galkin, O.; Weinkauf, S.; Vekilov, P.G. Metastable liquid clusters in super- and undersaturated protein solutions. J. Phys. Chem. B 2007, 111, 3106–3114. [Google Scholar]
- Dierks, K.; Meyer, A.; Einspahr, H.; Betzel, C. Dynamic light scattering in protein crystallization droplets: Adaptations for analysis and optimization of crystallization processes. Cryst. Growth Des 2008, 8, 1628–1634. [Google Scholar]
- Watts, D.; Muller-Dieckmann, J.; Tsakanova, G.; Lamzin, V.S.; Groves, M.R. Quantitive evaluation of macromolecular crystallization experiments using 1,8-ANS fluorescence. Acta Crystallogr. D 2010, 66, 901–908. [Google Scholar]
- Fiedler, S.; Müller-Dieckmann, J.; Watts, D.; Lamzin, V.S.; Groves, M.R. In situ Protein Crystal Diffraction Screening’. In Crystallography, Research Technology and Applications; Nova Science Publishers: Hamburg, Germany, 2012; pp. 31–44. [Google Scholar]
- Trache, A.; Meininger, G.A. Total internal reflection fluorescence (TIRF) microscopy. Curr. Protocols Microbiol. 2008. [Google Scholar] [CrossRef]
- Boudjemline, A.; Saridakis, E.; Swann, M.J.; Govada, L.; Mavridis, I.M.; Chayen, N.E. Use of dual polarization interferometry as a diagnostic tool for protein crystallization. Anal. Chem 2011, 83, 7881–7887. [Google Scholar]
- Merlino, A.; Vitagliano, L.; Balsamo, A.; Nicoletti, F.P.; Howes, B.D.; Giordano, D.; Coppola, D.; di Prisco, G.; Verde, C.; Smulevich, G.; et al. Crystallization, preliminary X-ray diffraction studies and Raman microscopy of the major haemoglobin from the sub-Antarctic fish Eleginops maclovinus in the carbomonoxy form. Acta Crystallogr. Sect. F 2011, 66, 1536–1540. [Google Scholar]
- Vergara, A.; Merlino, A.; Pizzo, E.; D’Alessio, G.; Mazzarella, L. A novel method for detection of selenomethionine incorporation in protein crystals via Raman microscopy. Acta Crystallogr. D 2008, 64, 167–171. [Google Scholar]
- Merlino, A.; Sica, F.; Vergara, A. Monitoring Preparation of Derivative Protein Crystals via Raman Microscopy. In Current Trends in X-Ray Crystallography; Chandrasekaran, D.A., Ed.; InTech: Rijeka, Croatia, 2011; pp. 393–408. [Google Scholar]
- DeWalt, E.L.; Begue, V.J.; Ronau, J.A.; Sullivan, S.Z.; Das, C.; Simpson, G.J. Polarization-resolved second-harmonic generation microscopy as a method to visualize protein-crystal domains. Acta Crystallogr. D 2013, 69, 74–81. [Google Scholar]
- Wampler, R.D.; Kissick, D.J.; Dehen, C.J.; Gualtieri, E.J.; Grey, J.L.; Wang, H.F.; Thompson, D.H.; Cheng, J.X.; Simpson, G.J. Selective detection of protein crystals by second harmonic microscopy. J. Am. Chem. Soc 2008, 130, 14076–14077. [Google Scholar]
- Stradner, A.; Sedgwick, H.; Cardinaux, F.; Poon, W.C.; Egelhaaf, S.U.; Schurtenberger, P. Equilibrium cluster formation in concentrated protein solutions and colloids. Nature 2004, 432, 492–495. [Google Scholar]
- Cardinaux, F.; Zaccarelli, E.; Stradner, A.; Bucciarelli, S.; Farago, B.; Egelhaaf, S.U.; Sciortino, F.; Schurtenberger, P. Cluster-driven dynamical arrest in concentrated lysozyme solutions. J. Phys. Chem. B 2011, 115, 7227–7237. [Google Scholar]
- Devaud, G.; Furcinitti, P.S.; Fleming, J.C.; Lyon, M.K.; Douglas, K. Direct observation of defect structure in protein crystals by atomic force and transmission electron microscopy. Biophys. J 1992, 63, 630–638. [Google Scholar]
- Gomery, K.; Humphrey, E.C.; Herring, R. Imaging and diffraction of protein crystallization using TEM. J. Electron. Microsc. 2012. [Google Scholar] [CrossRef]
- Desbois, S.; Seabrook, S.A.; Newman, J. Some practical guidelines for UV imaging in the protein crystallization laboratory. Acta Crystallogr. Sect. F 2013, 69, 201–208. [Google Scholar]
- Hu, Z.W.; Thomas, B.R.; Chernov, A.A. Laboratory multiple-crystal X-ray topography and reciprocal-space mapping of protein crystals: Influence of impurities on crystal perfection. Acta Crystallogr. D 2001, 57, 840–846. [Google Scholar]
- Capelle, B.; Epelboin, Y.; Hartwig, J.; Moraleda, A.B.; Otalora, F.; Stojanoff, V. Characterization of dislocations in protein crystals by means of synchrotron double-crystal topography. J. Appl. Cryst 2004, 37, 67–71. [Google Scholar]
- Gill, H.S. Evaluating the efficacy of tryptophan fluorescence and absorbance as a selection tool for identifying protein crystals. Acta Crystallogr. Sect. F 2010, 66, 364–372. [Google Scholar]
- Madden, J.T.; DeWalt, E.L.; Simpson, G.J. Two-photon excited UV fluorescence for protein crystal detection. Acta Crystallogr. D 2011, 67, 839–846. [Google Scholar]
- Carpentier, P.; Royant, A.; Ohana, J.; Bourgeois, D. Advances in spectroscopic methods for biological crystals. 2. Raman spectroscopy. J. Appl. Crystallogr 2007, 40, 1113–1122. [Google Scholar]
- Vergara, A.; Vitagliano, L.; Merlino, A.; Sica, F.; Marino, K.; Verde, C.; di Prisco, G.; Mazzarella, L. An order-disorder transition plays a role in switching off the Root effect in fish hemoglobins. J. Biol. Chem 2010, 285, 32568–32575. [Google Scholar]
- Katona, G.; Carpentier, P.; Niviere, V.; Amara, P.; Adam, V.; Ohana, J.; Tsanov, N.; Bourgeois, D. Raman-assisted crystallography reveals end-on peroxide intermediates in a nonheme iron enzyme. Science 2007, 316, 449–453. [Google Scholar]
- Derewenda, Z.S. Rational protein crystallization by mutational surface engineering. Structure 2004, 12, 529–535. [Google Scholar]
- Kobayashi, M.; Kubota, M.; Matsuura, Y. Crystallization and improvement of crystal quality for x-ray diffraction of maltooligosyl trehalose synthase by reductive methylation of lysine residues. Acta Crystallogr. D 1999, 55, 931–933. [Google Scholar]
- Walter, T.S.; Meier, C.; Assenberg, R.; Au, K.F.; Ren, J.; Verma, A.; Nettleship, J.E.; Owens, R.J.; Stuart, D.I.; Grimes, J.M. Lysine methylation as a routine rescue strategy for protein crystallization. Structure 2006, 14, 1617–1622. [Google Scholar]
- Heras, B.; Martin, J.L. Post-crystallization treatments for improving diffraction quality of protein crystals. Acta Crystallogr. D 2005, 61, 1173–1180. [Google Scholar]
- Derewenda, Z.S.; Vekilov, P.G. Entropy and surface engineering in protein crystallization. Acta Crystallogr. D 2006, 62, 116–124. [Google Scholar]
- Harp, J.M.; Hanson, B.L.; Timm, D.E.; Bunick, G.J. Macromolecular crystal annealing: Evaluation of techniques and variables. Acta Crystallogr. D 1999, 55, 1329–1334. [Google Scholar]
- Kriminski, S.; Caylor, C.L.; Nonato, M.C.; Finkelstein, K.D.; Thorne, R.E. Flash-cooling and annealing of protein crystals. Acta Crystallogr. D 2002, 58, 459–471. [Google Scholar]
- Russo Krauss, I.; Sica, F.; Mattia, C.A.; Merlino, A. Increasing the X-ray diffraction power of protein crystals by dehydration: The case of bovine serum albumin and a survey of literature data. Int. J. Mol. Sci 2012, 13, 3782–3800. [Google Scholar]
- Newman, J. A review of techniques for maximizing diffraction from a protein crystal in stilla. Acta Crystallogr. D 2006, 62, 27–31. [Google Scholar]
- Wine, Y.; Cohen-Hadar, N.; Freeman, A.; Frolow, F. Elucidation of the mechanism and end products of glutaraldehyde crosslinking reaction by X-ray structure analysis. Biotechnol. Bioeng 2007, 98, 711–718. [Google Scholar]
- Harp, J.M.; Timm, D.E.; Bunick, G.J. Macromolecular crystal annealing: Overcoming increased mosaicity associated with cryocrystallography. Acta Crystallogr. D 1998, 54, 622–628. [Google Scholar]
- Einstein, J.R. Humidity control device for the Buerger precession camera. J. Sci. Instrum 1961, 38, 449. [Google Scholar]
- Huxley, H.E.; Kendrew, J.C. Discontinuous lattice changes in haemoglobin crystals. Acta Cryst 1953, 6, 76–80. [Google Scholar]
- Pickford, M.G.; Garman, E.F.; Jones, E.Y.; Stuart, D.I. A design of crystal mounting cell that allows the controlled variation of humidity at the protein crystal during X-ray diffraction. J. Appl. Crystallogr 1993, 26, 465–466. [Google Scholar]
- Sjögren, T.; Carlsson, G.; Larsson, G.; Hajdu, A.; Andersson, C.; Pettersson, H.; Hajdu, J. Protein crystallography in a vapour stream: Data collection, reaction initiation and intermediate trapping in naked hydrated protein crystals. J. Appl. Crystallogr 2002, 35, 113–116. [Google Scholar]
- Kiefersauer, R.; Stetefeld, J.; Gomis-Rüth, F.X.; Romão, M.J.; Lottspeich, F.; Huber, R. Protein-crystal density by volume measurement and amino-acid analysis. J. Appl. Crystallogr 1996, 29, 311–317. [Google Scholar]
- Kiefersauer, R.; Than, M.E.; Dobbek, H.; Gremer, L.; Melero, M.; Strobl, S.; Dias, J.M.; Soulimane, T.; Huber, R. A novel free-mounting system for protein crystals: Transformation and improvement of diffraction power by accurately controlled humidity changes. J. Appl. Crystallogr 2000, 33, 1223–1230. [Google Scholar]
- AA.VV. Transformation and improvement of macromolecular crystal diffraction through accurately controlled humidity changes: Proteros Free Mounting System. Rigaku J. 2005, 22, 46–49. [Google Scholar]
- Collins, M.D.; Kim, C.U.; Gruner, S.M. High-pressure protein crystallography and NMR to explore protein conformations. Ann. Rev. Biophys 2011, 40, 81–98. [Google Scholar]
- Suzuki, Y.; Sazaki, G.; Miyashita, S.; Sawada, T.; Tamura, K.; Komatsu, H. Protein crystallization under high pressure. Biochim. Biophys. Acta 2002, 1595, 345–356. [Google Scholar]
- Suzuki, Y. Protein Crystal Growth under High Pressure. In Modern Aspects of Bulk Crystal and Thin Film Preparation; InTech: Rijeka, Croatia, 2012; pp. 439–462. [Google Scholar]
- Li, L.; Ismagilov, R.F. Protein crystallization using microfluidic technologies based on valves, droplets, and SlipChip. Ann. Rev. Biophys 2010, 39, 139–518. [Google Scholar]
- Yamaguchi, H.; Maeki, M.; Yamashita, K.; Nakamura, H.; Miyazaki, M.; Maeda, H. Controlling one protein crystal growth by droplet-based microfluidic system. J. BioChem 2013, 153, 339–346. [Google Scholar]
- Guha, S.; Perry, S.L.; Pawate, A.S.; Kenis, P.J.A. Fabrication of X-ray compatible microfluidic platforms for protein crystallization. Sensor Actuat. B 2012, 174, 1–9. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physical factors | Chemical factors | Biochemical factors |
|---|---|---|
| Temperature | Precipitant type | Sample purity |
| Pressure | Precipitant concentration | Sample homogeneity |
| Gravity | pH | Sequence modifications |
| Magnetic fields | Buffer type | Posttranslational modifications |
| Electric fields | Ionic strength | Chemical modifications |
| Dielectric properties | Sample concentration | Aggregation |
| Viscosity | Metal ions | Proteolysis |
| Vibrations and sound | Polyions | Sample pI |
| Time | Detergents | Ligands, co-factors, inhibitors |
| Equilibration rate | Heavy metals | |
| Nucleants | Small molecule impurities | |
| Methodology | Crosslinkers | |
| Surface of crystallization device | Reagent source | |
| Sample handling | Reagent formulation |
| Salts | Organic compounds | Polymers |
|---|---|---|
| Ammonium sulphate | 2-methyl-2,4-pentanediol | Polyethylene glycol 1000 |
| Lithium sulphate | Isopropanol | Polyethylene glycol 1500 |
| Ammonium acetate | Ethanol | Polyethylene glycol 2000 |
| Sodium chloride | 1,3-propanediol | Polyethylene glycol 3350 |
| Ammonium citrate | Dioxane | Polyethylene glycol 4000 |
| Ammonium formate | Acetone | Polyethylene glycol 6000 |
| Sodium citrate | Butanol | Polyethylene glycol 8000 |
| Sodium formate | Acetonitrile | Polyethylene glycol 10000 |
| Ammonium phosphate | Dimethyl sulfoxide | Polyethylene glycol 20000 |
| Sodium phosphate | 2,5-hexanediol | Polyethylene glycol 35000 |
| Potassium phosphate | Methanol | Polyethylene glycol monomethyl 2000 |
| Sodium/potassium phosphate | 1,3-butyrolactone | Polyethylene glycol monomethyl 5000 |
| Ammonium nitrate | Polyethylene glycol 200 | Polyvinylpyrrolidone K 15 |
| Potassium thiocyanate | Polyethylene glycol 400 | Pentaerythritolpropoxylate |
| Sodium/potassium tartrate | Malonic acid | Jeffamine |
| Ammonium tartrate | Malic acid | Polyacrylate |
| Magnesium sulphate | Succinic acid | Polypropylene glycol 400 |
| Sodium nitrate | Glycerol | Modified polycarboxylates |
| Sodium acetate | Imidazole | |
| Magnesium acetate | Ethylene glycol |
| Molecule | Category |
|---|---|
| Natural additives | Physiologically or biochemically relevant small molecules, such as coenzymes, substrate analogues, inhibitors, metal cofactors, or prosthetic groups |
| Chemical protectants | Molecules that assure protein integrity such as reductants and metal atoms scavengers |
| Solubilizing agents and detergents | Mild non-detergent molecules, such as sulfobetaines, low concentrations of chaotropic agents and surfactants and, in the case of membrane proteins, stronger solubilizing agents such as detergents |
| Poisons | Agents that partially inhibit nucleation thus facilitating the growth of few crystals of high quality, such as DMSO, DMF, low weigh alcohols and sugars |
| Osmolytes | Natural occurring molecules that help the protein in the adaptation to osmotic stress while maintaining native structure and function, such as TMAO, sarcosine and betaine |
| Non-covalent cross-linkers | Molecules able to stabilize the crystal lattice by mediating sample aggregation through reversible intermolecular interactions, electrostatic or hydrophobic, among surface groups on neighboring protein molecules |
| Covalent cross-linkers | Crosslinking reagents that may both reduce the conformational protein mobility and the stability of a protein-ligand complex |
| Method | Nucleation monitoring | Control of crystallization results | Crystal defect analysis | Checking of diffraction quality |
|---|---|---|---|---|
| Advanced optical microscopy | [227] | |||
| Atomic force microscopy | [228] | [229] | ||
| Attentuated total reflectance Fourier transform infrared spectroscopy | [230] | |||
| Birefringence | [231] | |||
| Dynamic Light Scattering | [232–234] | |||
| Fluorescence | [235] | |||
| In situ X-ray analysis | [236] | |||
| Internal reflection fluorescent microscopy | [237] | |||
| Polarization interferometry | [238] | |||
| Raman | [239–241] | |||
| Second-harmonic generation microscopy | [242] | [243] | ||
| Small-angle neutron scattering | [244] | |||
| Small-angle X-ray scattering | [244,245] | |||
| Transmission electron microscopy | [246,247] | |||
| Visible-UV | [248] | |||
| X-ray topography | [249,250] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Russo Krauss, I.; Merlino, A.; Vergara, A.; Sica, F. An Overview of Biological Macromolecule Crystallization. Int. J. Mol. Sci. 2013, 14, 11643-11691. https://doi.org/10.3390/ijms140611643
Russo Krauss I, Merlino A, Vergara A, Sica F. An Overview of Biological Macromolecule Crystallization. International Journal of Molecular Sciences. 2013; 14(6):11643-11691. https://doi.org/10.3390/ijms140611643
Chicago/Turabian StyleRusso Krauss, Irene, Antonello Merlino, Alessandro Vergara, and Filomena Sica. 2013. "An Overview of Biological Macromolecule Crystallization" International Journal of Molecular Sciences 14, no. 6: 11643-11691. https://doi.org/10.3390/ijms140611643