SAHBNET, an Accessible Surface-Based Elastic Network: An Application to Membrane Protein

Abstract
:1. Introduction
2. Results and Discussion
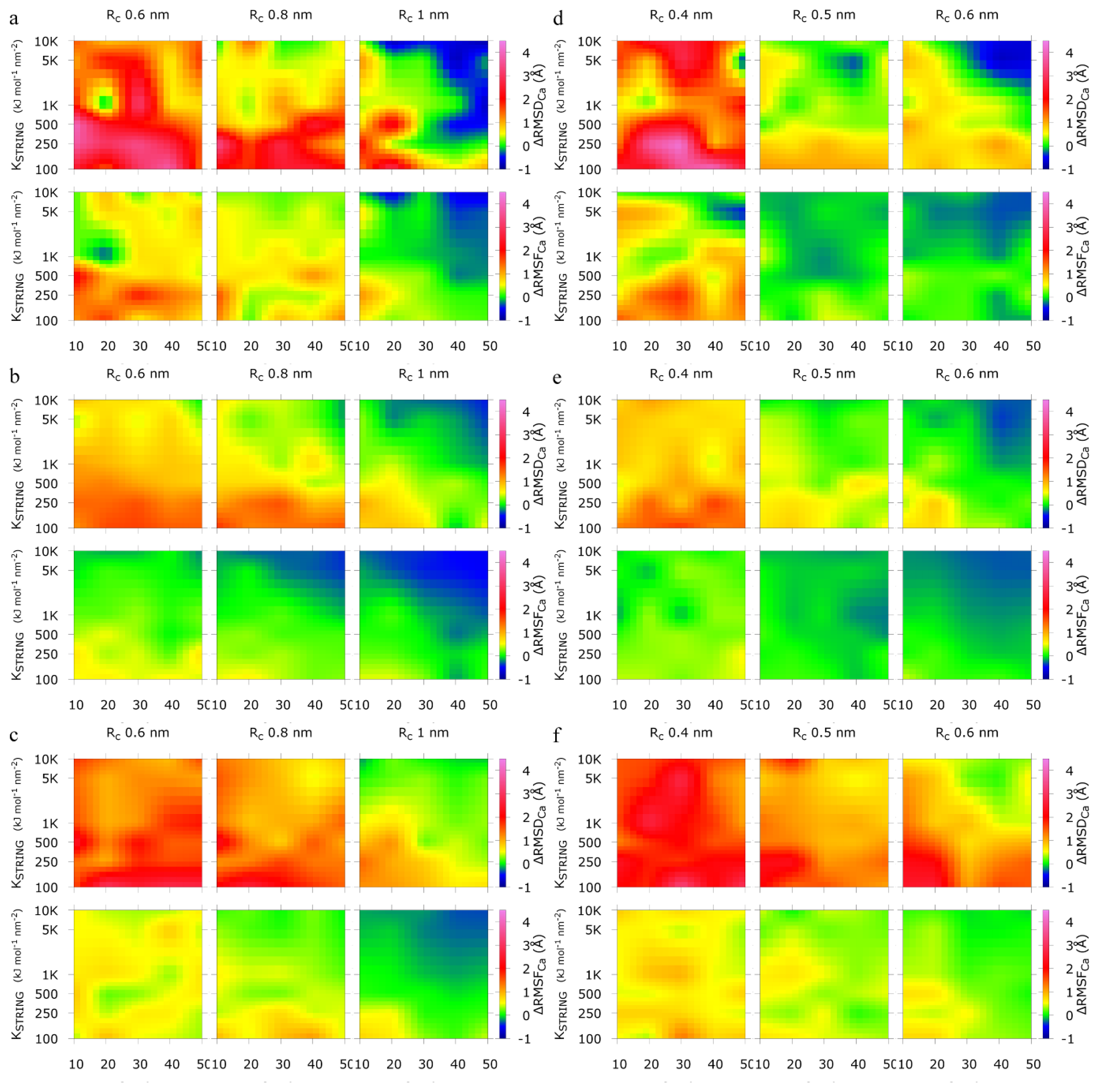
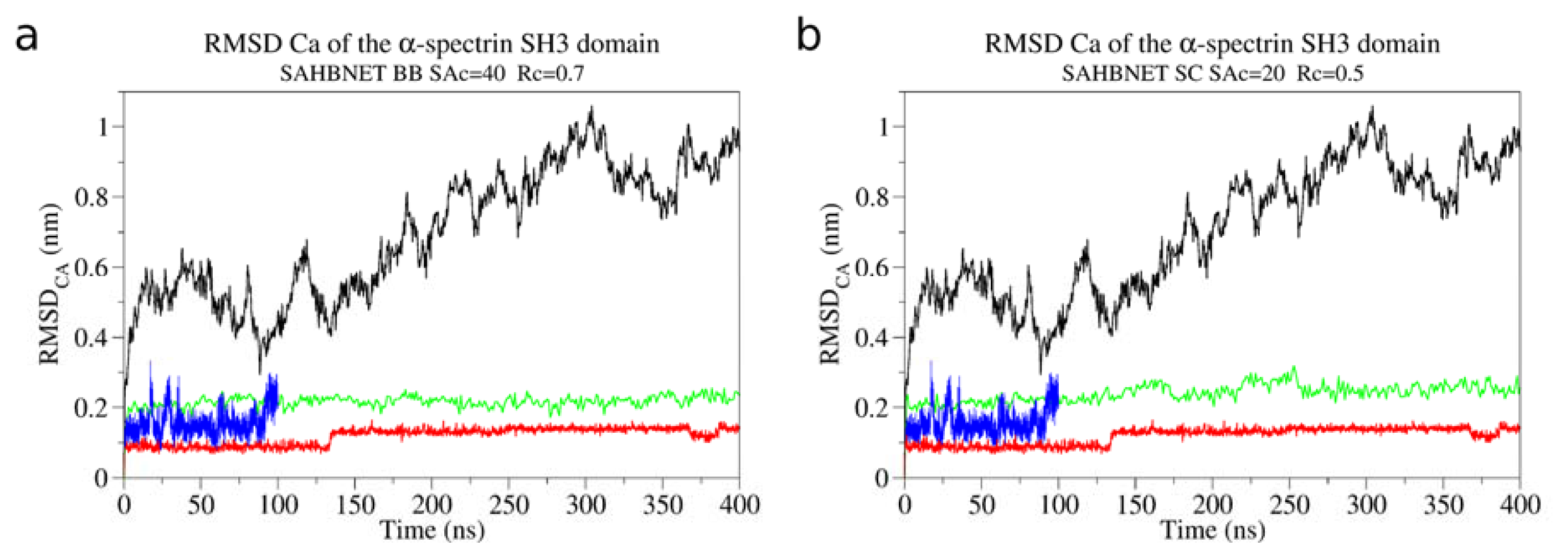
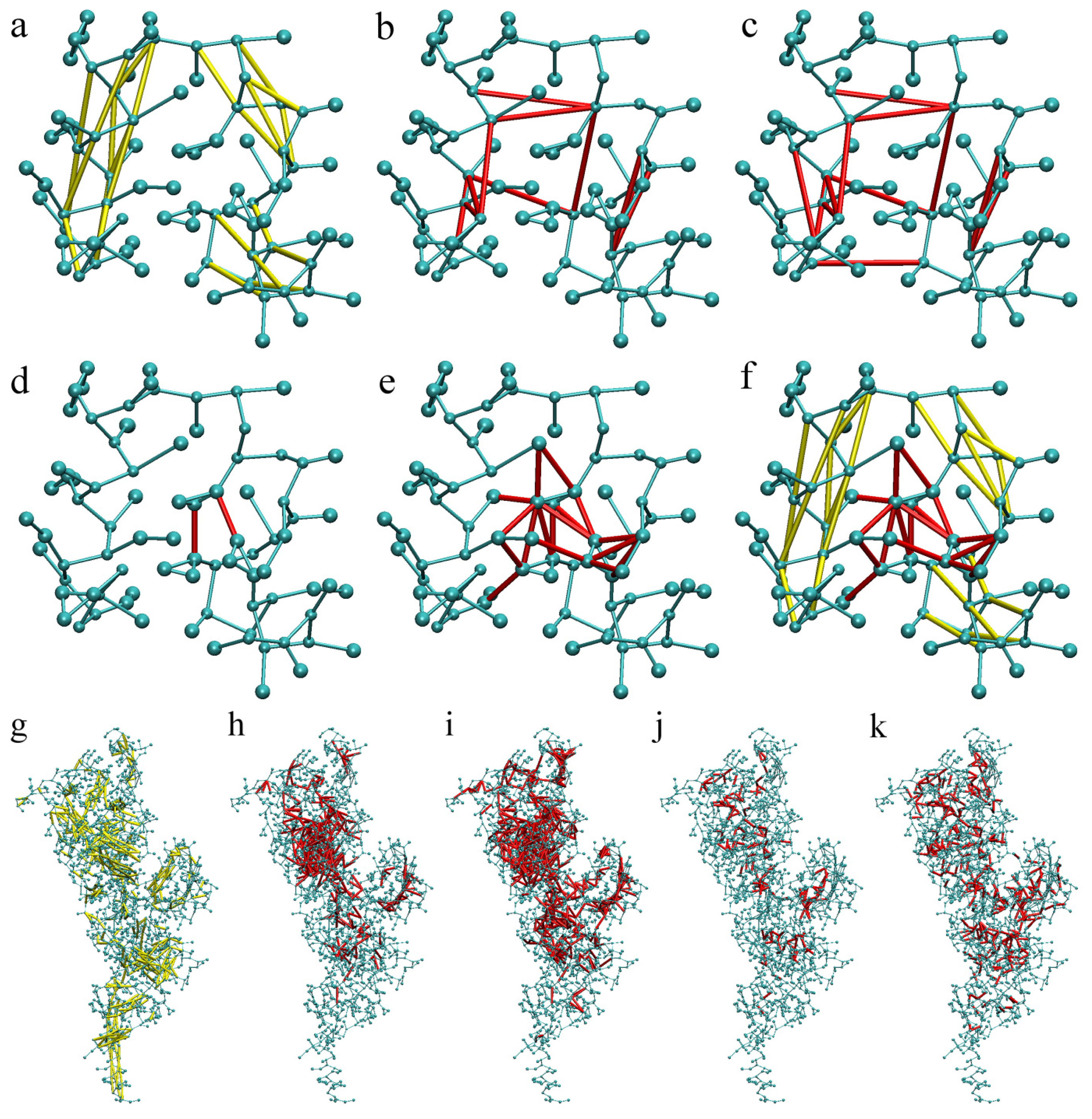
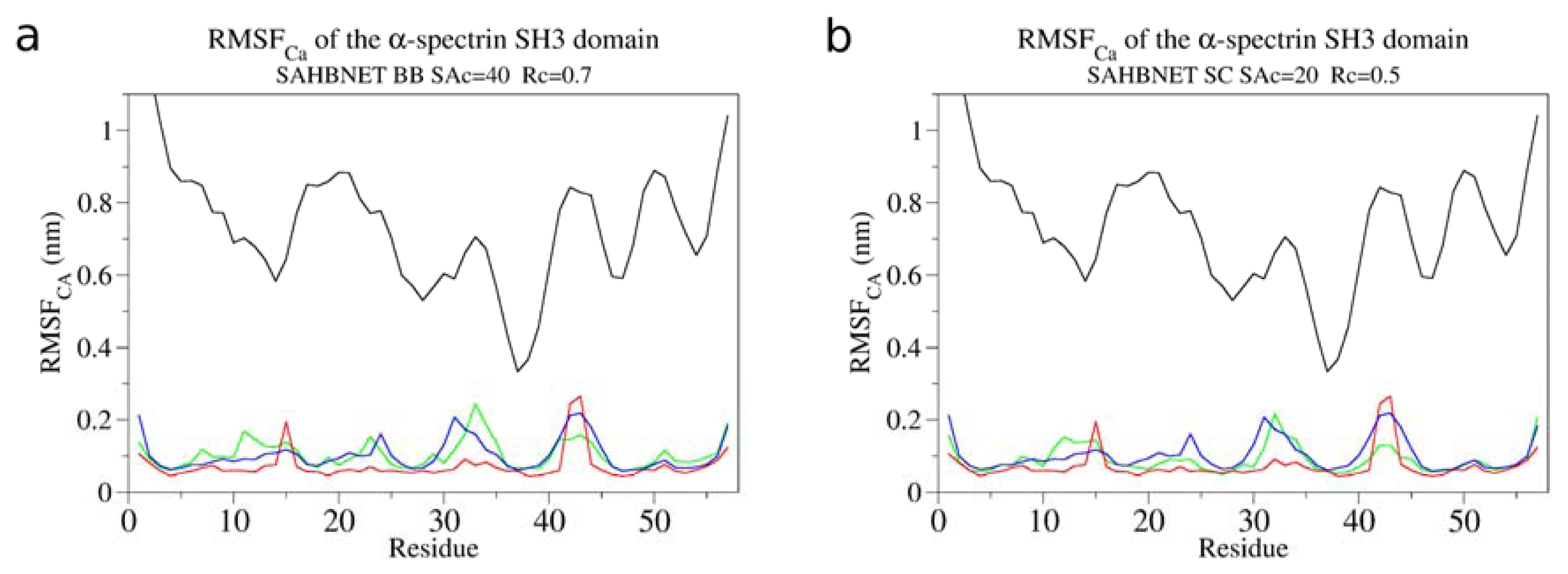
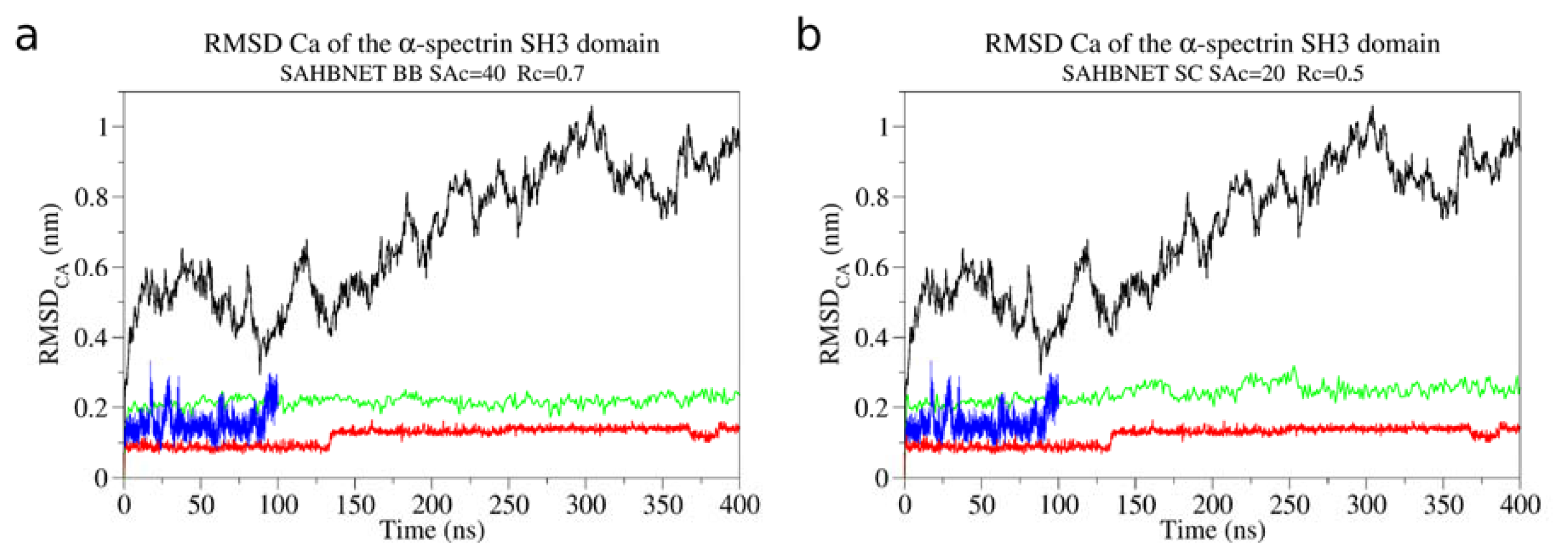
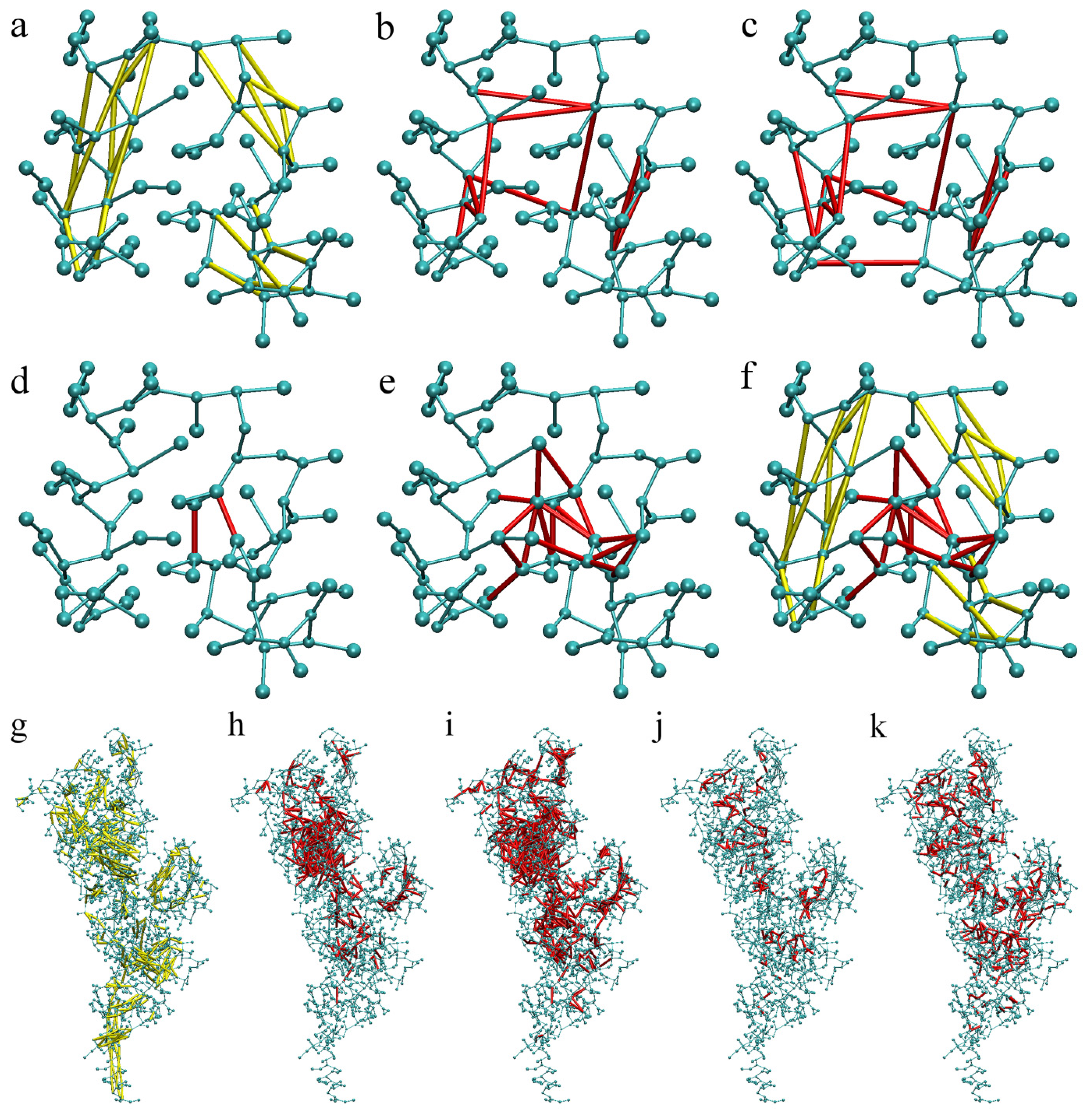
2.1. Calibration of the Surface Accessibility Hydrogen-Bonds Elastic NETwork (SAHBNET) against Small Soluble Proteins in a Protein/Water System
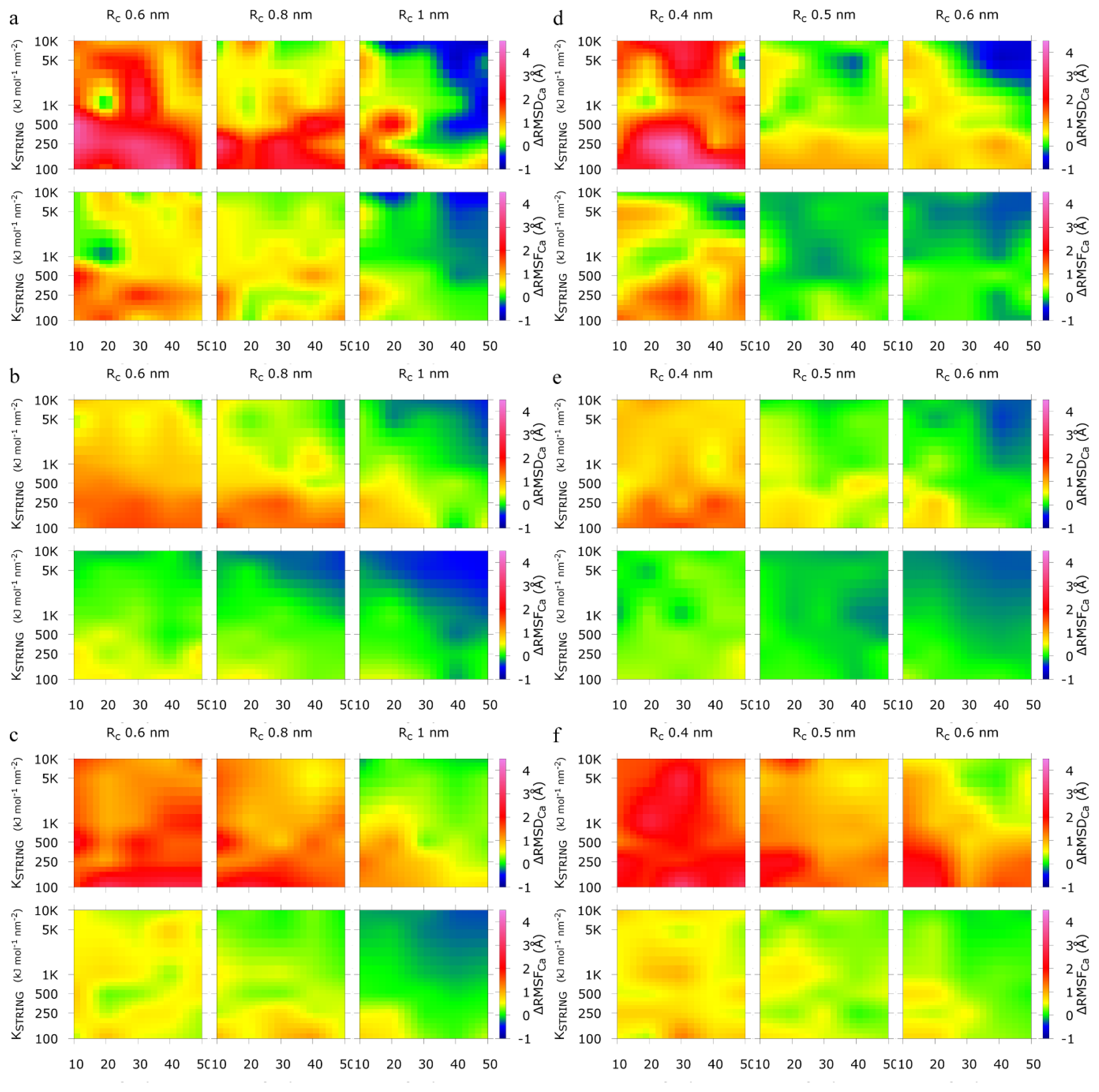
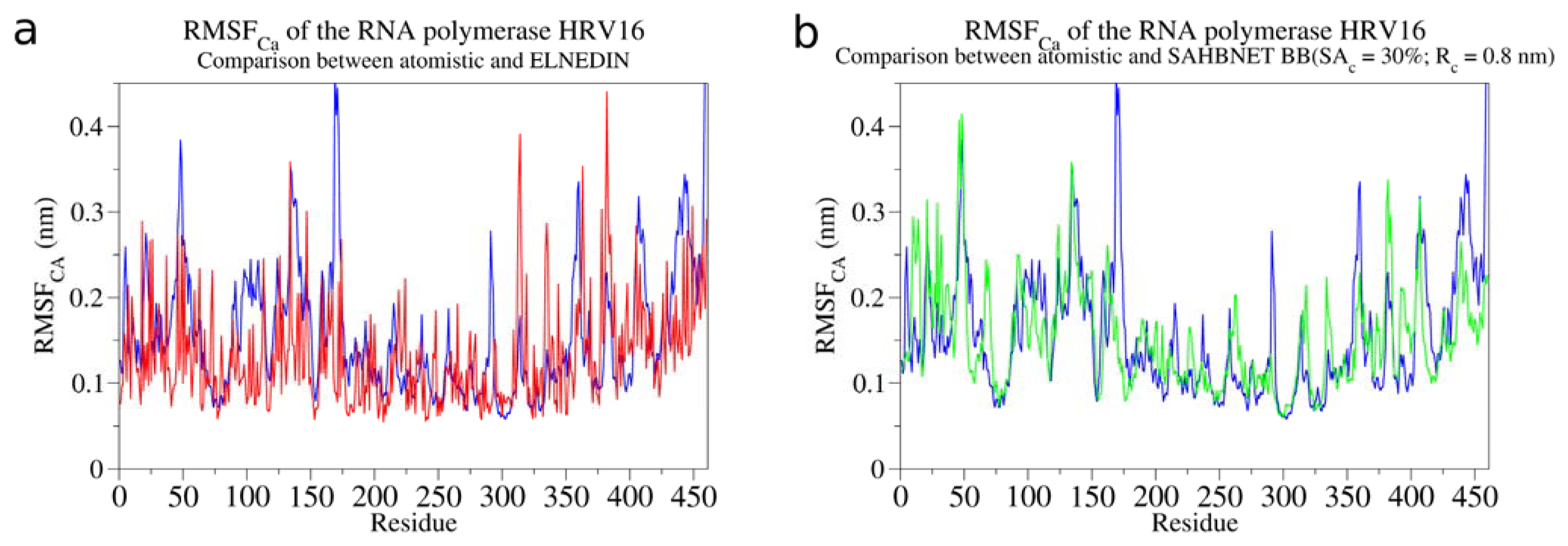
2.2. Comparison between the SAHBNET and ELNEDYN Networks
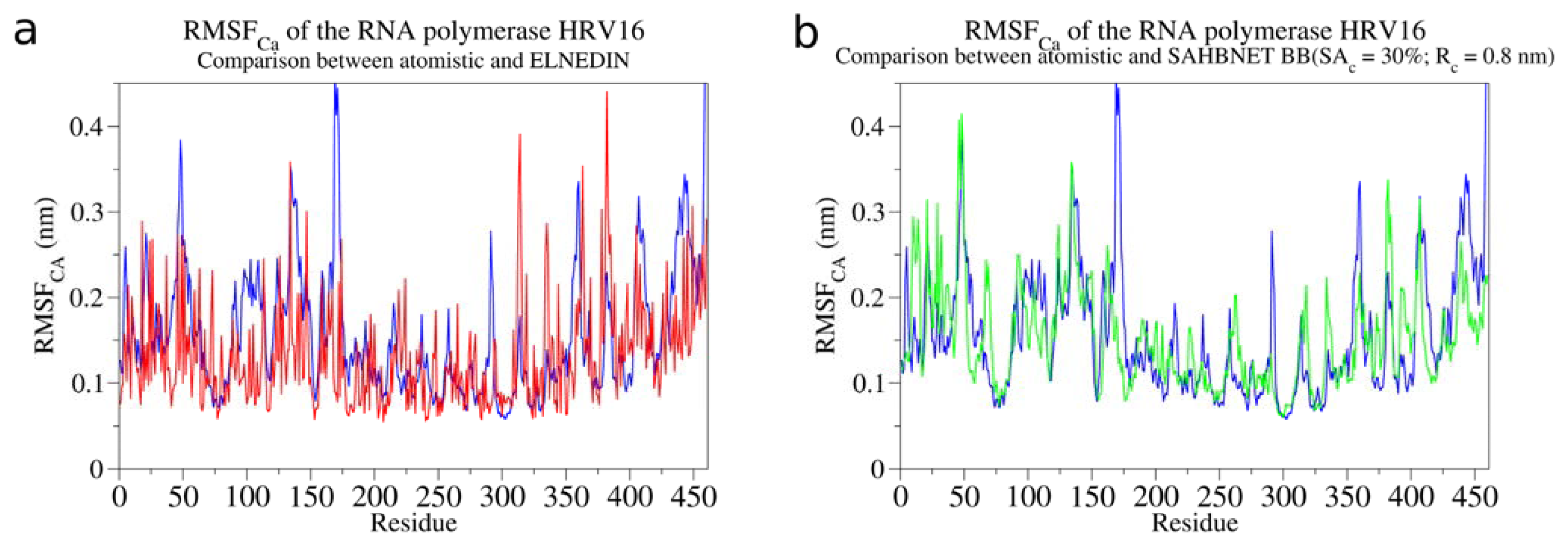
2.3. Testing SAHBNET on a Larger Soluble Protein: The RNA-Dependent RNA Polymerase from Human Rhinovirus 16
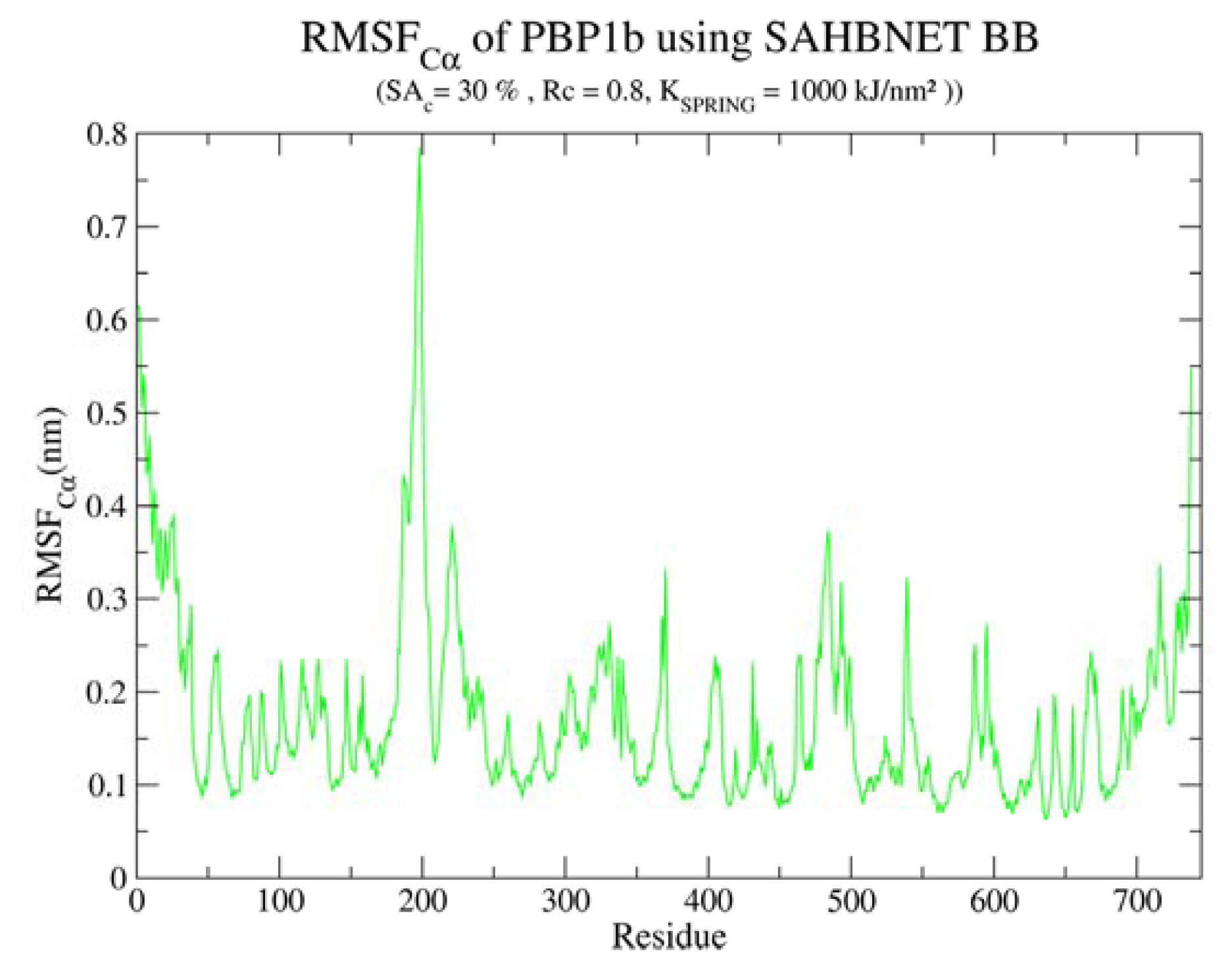
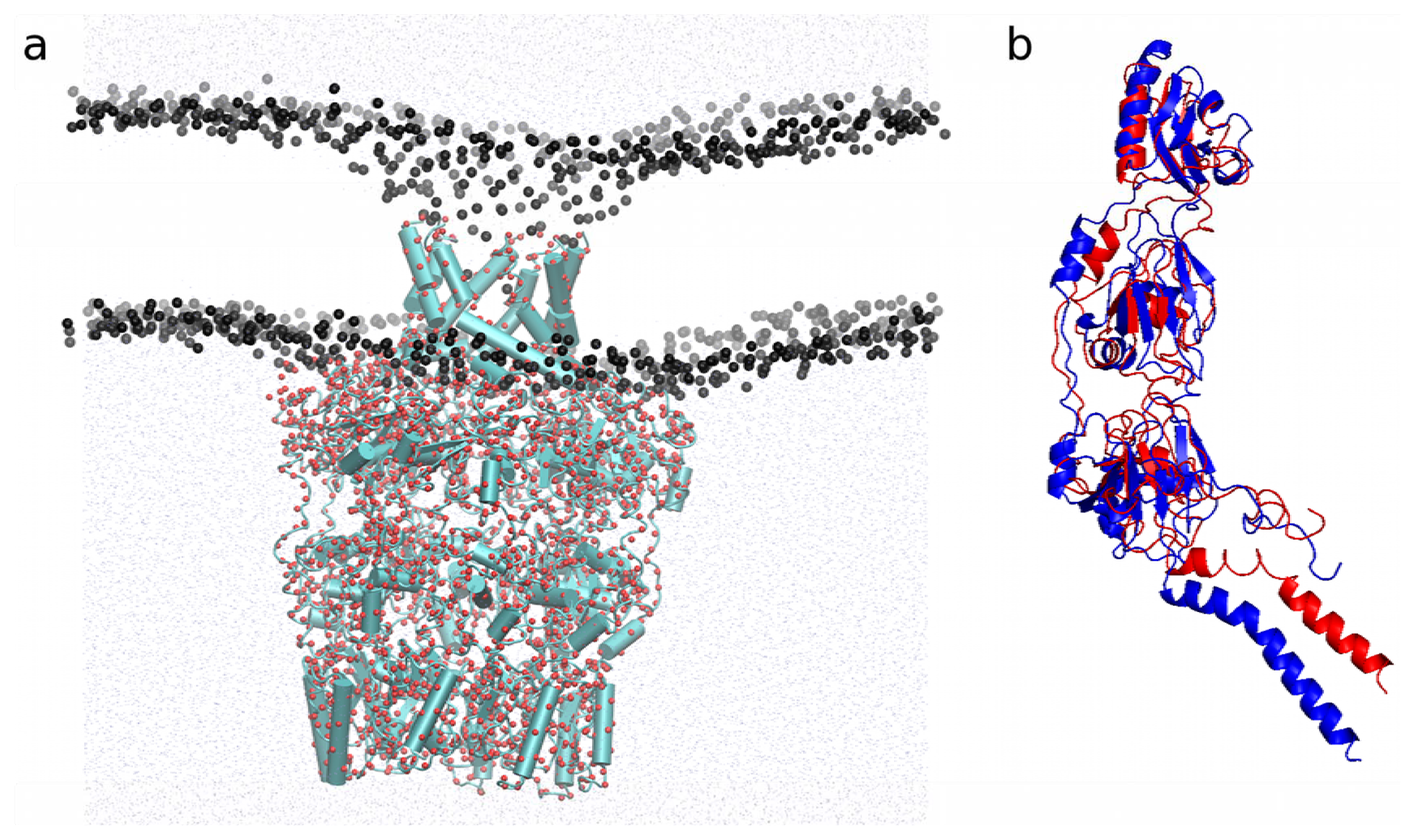
2.4. Application of SAHBNET to Insert Membrane Proteins
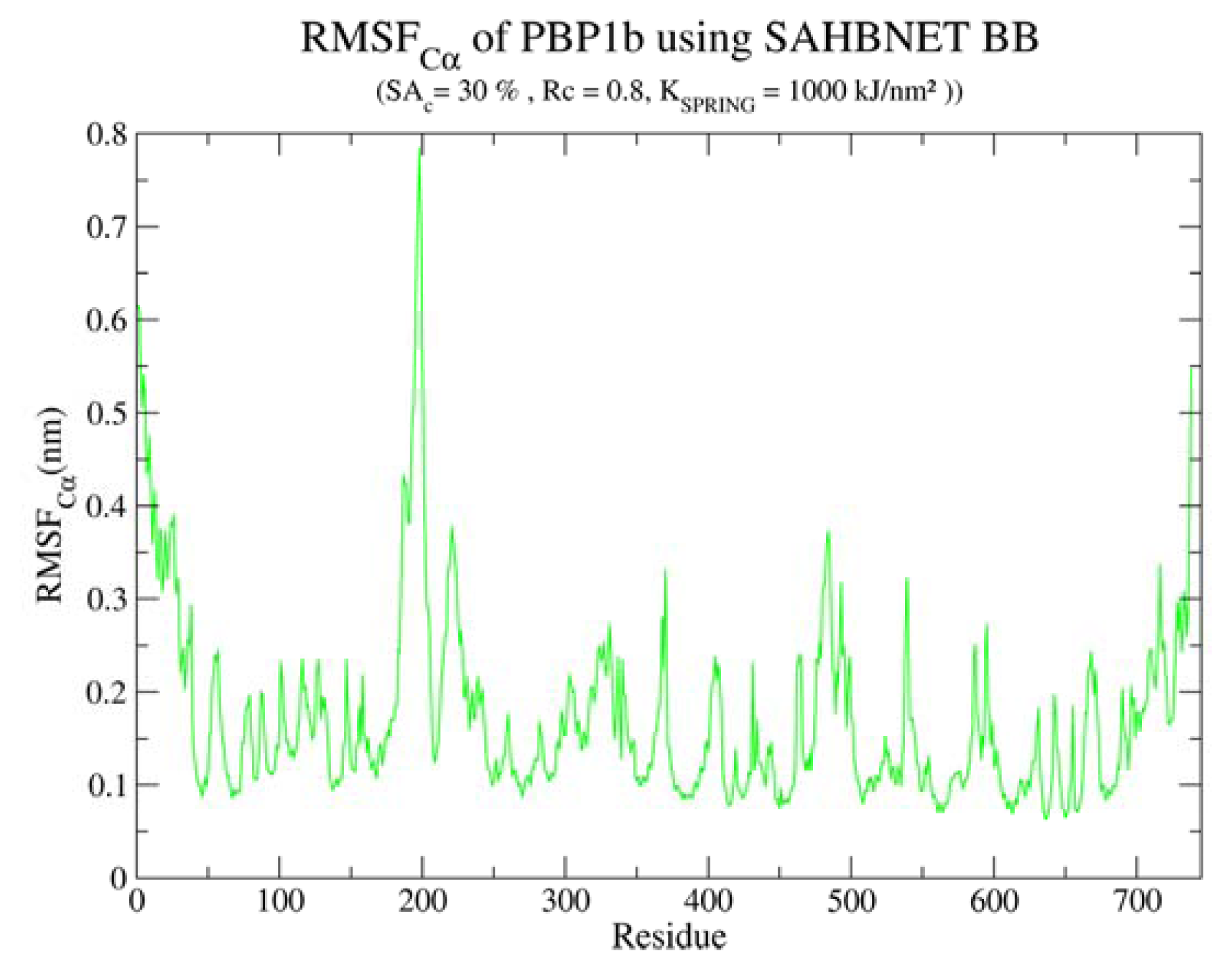
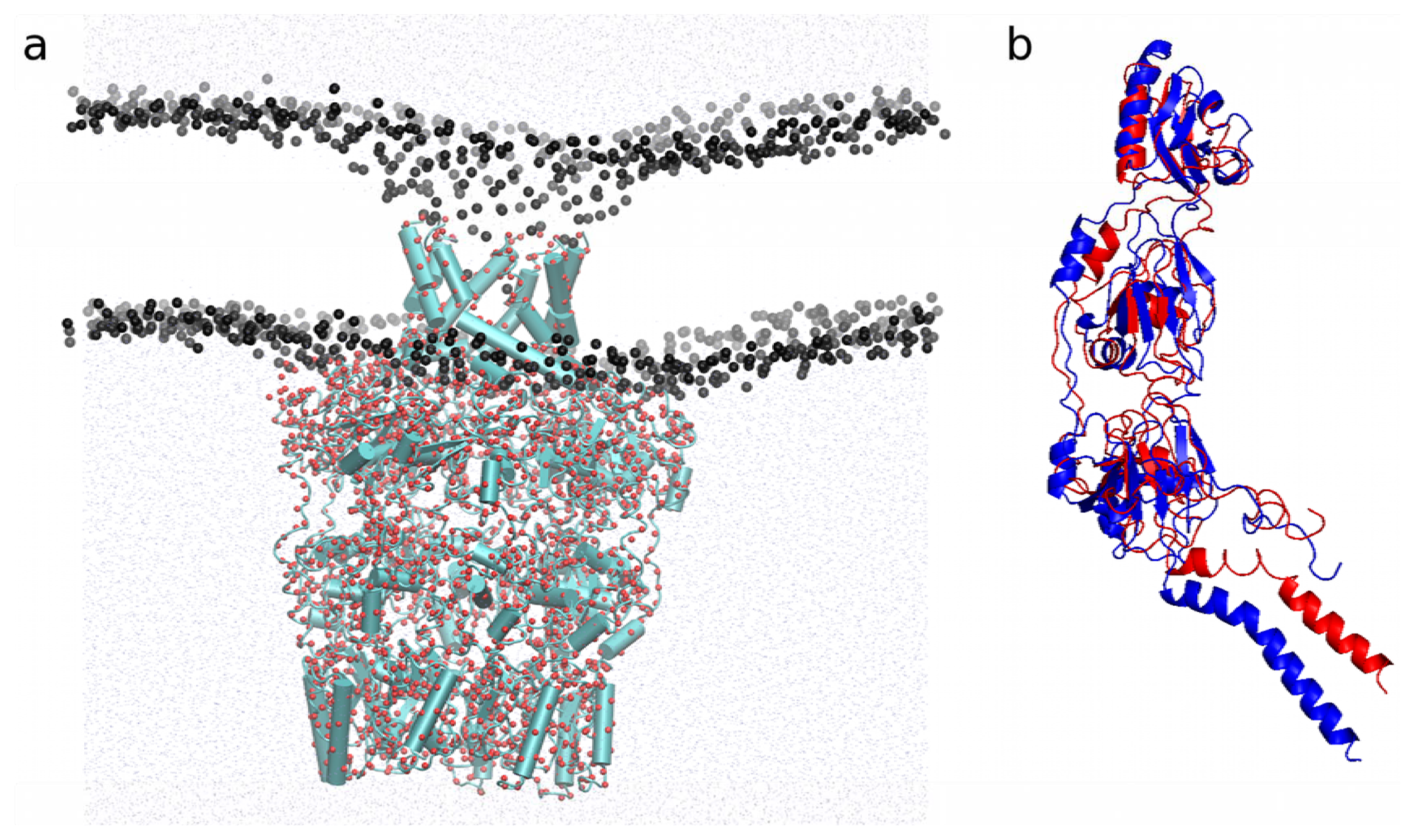
2.4.1. Insertion of PBP1b, a Monotopic Membrane Protein from Escherichia coli
2.4.2. Application to the Outer Membrane Lipoprotein WZA
3. Method
3.1. Molecular Systems
3.2. Coarse-Grained Simulations
3.3. Comparison between SAHBNET, ELNEDYN and Atomistic Simulations
3.4. Atomistic Simulations
3.5. Modifications of Genbox
4. Conclusions
Supplementary Information
ijms-14-11510-s001.pdfAcknowledgments
Conflict of Interest
References
- Terstappen, G.C.; Reggiani, A. In silico research in drug discovery. Trends Pharmacol. Sci 2001, 22, 23–26. [Google Scholar]
- Forrest, L.R.; Krämer, R.; Ziegler, C. The structural basis of secondary active transport mechanisms. Biochim. Biophys. Acta 2011, 1807, 167–188. [Google Scholar]
- Stroud, R.M.; Reiling, K.; Wiener, M.; Freymann, D. Ion-channel-forming colicins. Curr. Opin. Struct. Biol 1998, 8, 525–533. [Google Scholar]
- Cho, W.; Stahelin, R.V. Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 119–151. [Google Scholar]
- DeFelice, L.J. Transporter structure and mechanism. Trends Neurosci 2004, 27, 352–359. [Google Scholar]
- Tusnády, G.E.; Dosztányi, Z.; Simon, I. Transmembrane proteins in the Protein Data Bank: Identification and classification. Bioinformatics 2004, 20, 2964–2972. [Google Scholar]
- Kozma, D.; Simon, I.; Tusnády, G.E. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res 2013, 41, D524–D529. [Google Scholar]
- Dowhan, W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem 1997, 66, 199–232. [Google Scholar]
- Dowhan, W.; Bogdanov, M. Functional Roles of Lipids in Membranes. In Biochemistry of Lipids, Lipoproteins and Membranes, 5th ed.; Elsevier: San Diego, CA, USA, 2008; pp. 1–37. [Google Scholar]
- Cullis, P.R.; Fenske, D.B.; Hope, M.J. Chapter 1 Physical properties and functional roles of lipids in membranes. New Compr. Biochem. 1996, 31, 1–33. [Google Scholar]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol 2008, 9, 112–124. [Google Scholar]
- Wang, X.; Bogdanov, M.; Dowhan, W. Topology of polytopic membrane protein subdomains is dictated by membrane phospholipid composition. EMBO J 2002, 21, 5673–5681. [Google Scholar]
- Cabiaux, V.; Quertenmont, P.; Conrath, K.; Brasseur, R.; Capiau, C.; Ruysschaert, J.M.; Triomphe, B.; Chemistry, P. Topology of diphtheria toxin B fragment inserted in lipid vesicles. Mol. Microbiol 1994, 11, 43–50. [Google Scholar]
- Knol, J.; Sjollema, K.; Poolman, B. Detergent-mediated reconstitution of membrane proteins. Biochemistry 1998, 37, 16410–16415. [Google Scholar]
- Montigny, C.; Penin, F.; Lethias, C.; Falson, P. Overcoming the toxicity of membrane peptide expression in bacteria by upstream insertion of Asp-Pro sequence. Biochim. Biophys. Acta 2004, 1660, 53–65. [Google Scholar]
- Lacapère, J.-J.; Pebay-Peyroula, E.; Neumann, J.-M.; Etchebest, C. Determining membrane protein structures: Still a challenge! Trends Biochem. Sci 2007, 32, 259–270. [Google Scholar]
- Rigaud, J.-L.; Lévy, D. Reconstitution of membrane proteins into liposomes. Methods Enzymol 2003, 372, 65–86. [Google Scholar]
- Periole, X.; Knepp, A.M.; Sakmar, T.P.; Marrink, S.J.; Huber, T. Structural determinants of the supramolecular organization of G protein-coupled receptors in bilayers. J. Am. Chem. Soc 2012, 134, 10959–10965. [Google Scholar]
- Van denBogaart, G.; Meyenberg, K.; Risselada, H.J.; Amin, H.; Willig, K.I.; Hubrich, B.E.; Dier, M.; Hell, S.W.; Grubmüller, H.; Diederichsen, U.; et al. Membrane protein sequestering by ionic protein-lipid interactions. Nature 2011, 479, 552–555. [Google Scholar]
- Lins, L.; Ducarme, P.; Breukink, E.; Brasseur, R. Computational study of nisin interaction with model membrane. Biochim. Biophys. Acta 1999, 1420, 111–120. [Google Scholar]
- Pillot, T.; Goethals, M.; Vanloo, B.; Talussot, C.; Brasseur, R.; Vandekerckhove, J.; Rosseneu, M.; Lins, L. Fusogenic properties of the C-terminal domain of the Alzheimer beta-amyloid peptide. J. Biol. Chem 1996, 271, 28757–28765. [Google Scholar]
- Lins, L.; Decaffmeyer, M.; Thomas, A.; Brasseur, R. Relationships between the orientation and the structural properties of peptides and their membrane interactions. Biochim. Biophys. Acta 2008, 1778, 1537–1544. [Google Scholar]
- Mingeot-Leclercq, M.P.; Lins, L.; Bensliman, M.; van Bambeke, F.; van der Smissen, P.; Peuvot, J.; Schanck, A.; Brasseur, R. Membrane destabilization induced by beta-amyloid peptide 29–42: Importance of the amino-terminus. Chem. Phys. Lipids 2002, 120, 57–74. [Google Scholar]
- Stansfeld, P.; Sansom, M. Molecular simulation approaches to membrane proteins. Structure 2011, 19, 1562–1572. [Google Scholar]
- Leekumjorn, S.; Sum, A.K. Molecular simulation study of structural and dynamic properties of mixed DPPC/DPPE bilayers. Biophys. J 2006, 90, 3951–3965. [Google Scholar]
- Kawamoto, S.; Takasu, M.; Miyakawa, T.; Morikawa, R.; Oda, T.; Futaki, S.; Nagao, H. Inverted micelle formation of cell-penetrating peptide studied by coarse-grained simulation: Importance of attractive force between cell-penetrating peptides and lipid head group. J. Chem. Phys 2011, 134, 095103. [Google Scholar]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; De Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem 2007, 111, 7812–7824. [Google Scholar]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.-J. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput 2008, 4, 819–834. [Google Scholar]
- Nasica-Labouze, J.; Meli, M.; Derreumaux, P.; Colombo, G.; Mousseau, N. A multiscale approach to characterize the early aggregation steps of the amyloid-forming peptide GNNQQNY from the yeast prion sup-35. PLoS Comput. Biol 2011, 7, e1002051. [Google Scholar]
- Nishizawa, M.; Nishizawa, K. Curvature-driven lipid sorting: Coarse-grained dynamics simulations of a membrane mimicking a hemifusion intermediate. J. Biophys. Chem 2010, 1, 86–95. [Google Scholar]
- Vaidya, N.; Huang, H.; Takagi, S. Coarse grained molecular dynamics simulation of interaction between hemagglutinin fusion peptides and lipid bilayer membranes. Adv. Appl. Math. Mech 2010, 2, 430–450. [Google Scholar]
- Wu, C.; Shea, J.-E. Coarse-grained models for protein aggregation. Curr. Opin. Struct. Biol 2011, 21, 209–220. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 2008, 4, 435–447. [Google Scholar]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved parameters for the martini coarse-grained protein force field. J. Chem. Theory Comput 2013, 9, 687–697. [Google Scholar]
- Periole, X.; Cavalli, M.; Marrink, S.; Ceruso, M.A. Combining an elastic network with a coarse-grained molecular force field: Structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput 2009, 5, 2531–2543. [Google Scholar]
- Shen, H.; Moustafa, I.M.; Cameron, C.E.; Colina, C.M. Exploring the dynamics of four RNA-dependent RNA polymerases by a coarse-grained model. J. Phys. Chem. B 2012, 116, 14515–14524. [Google Scholar]
- Globisch, C.; Krishnamani, V.; Deserno, M.; Peter, C. Optimization of an elastic network augmented coarse grained model to study CCMV capsid deformation. PLoS One 2013, 8, e60582. [Google Scholar]
- Lins, L.; Thomas, A.; Brasseur, R. Analysis of accessible surface of residues in proteins. Protein Sci 2003, 12, 1406–1417. [Google Scholar]
- Sung, M.-T.; Lai, Y.-T.; Huang, C.-Y.; Chou, L.-Y.; Shih, H.-W.; Cheng, W.-C.; Wong, C.-H.; Ma, C. Crystal structure of the membrane-bound bifunctional transglycosylase PBP1b from Escherichia coli. Proc. Natl. Acad. Sci. USA 2009, 106, 8824–8829. [Google Scholar]
- Rzepiela, A.J.; Schäfer, L.V.; Goga, N.; Risselada, H.J.; de Vries, A.H.; Marrink, S.J. Reconstruction of atomistic details from coarse-grained structures. J. Comput. Chem 2010, 31, 1333–1343. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar]
- Chiu, T.K.; Kubelka, J.; Herbst-Irmer, R.; Eaton, W.A.; Hofrichter, J.; Davies, D.R. High-resolution X-ray crystal structures of the villin headpiece subdomain, an ultrafast folding protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7517–7522. [Google Scholar]
- Martinez, J.C.; Pisabarro, M.T.; Serrano, L. Obligatory steps in protein folding and the conformational diversity of the transition state. Nat. Struct. Biol 1998, 5, 721–729. [Google Scholar]
- Gallagher, T.; Alexander, P.; Bryan, P.; Gilliland, G.L. Two crystal structures of the B1 immunoglobulin-binding domain of streptococcal protein G and comparison with NMR. Biochemistry 1994, 33, 4721–4729. [Google Scholar]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys 1984, 81, 3684. [Google Scholar]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem 1999, 103, 3596–3607. [Google Scholar]
- Tironi, I.G.; Sperb, R.; Smith, P.E.; van Gunsteren, W.F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys 1995, 102, 5451. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem 1992, 13, 952–962. [Google Scholar]
- Schrödinger, L.L.C. The PyMOL molecular graphics system, version~1.3r1.2010.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Mean solvent accessible surface area |
|---|---|
| A | 103.8 |
| R | 231.1 |
| N | 157.6 |
| D | 156.7 |
| C | 130.8 |
| E | 195.0 |
| Q | 195.7 |
| G | 80.8 |
| H | 180.4 |
| I | 168.5 |
| L | 171.5 |
| K | 206.4 |
| M | 190.6 |
| F | 198.2 |
| P | 121.6 |
| S | 123.5 |
| T | 138.6 |
| W | 229.6 |
| Y | 219.7 |
| V | 144.7 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dony, N.; Crowet, J.M.; Joris, B.; Brasseur, R.; Lins, L. SAHBNET, an Accessible Surface-Based Elastic Network: An Application to Membrane Protein. Int. J. Mol. Sci. 2013, 14, 11510-11526. https://doi.org/10.3390/ijms140611510
Dony N, Crowet JM, Joris B, Brasseur R, Lins L. SAHBNET, an Accessible Surface-Based Elastic Network: An Application to Membrane Protein. International Journal of Molecular Sciences. 2013; 14(6):11510-11526. https://doi.org/10.3390/ijms140611510
Chicago/Turabian StyleDony, Nicolas, Jean Marc Crowet, Bernard Joris, Robert Brasseur, and Laurence Lins. 2013. "SAHBNET, an Accessible Surface-Based Elastic Network: An Application to Membrane Protein" International Journal of Molecular Sciences 14, no. 6: 11510-11526. https://doi.org/10.3390/ijms140611510