Silicon Era of Carbon-Based Life: Application of Genomics and Bioinformatics in Crop Stress Research



Abstract
:1. Introduction
2. General Bioinformatics Resources
2.1. Databases
2.2. Biological Ontologies Related to Crop Stress Research
3. Recent Advances and Challenges in Crop Genomics
3.1. Polyploidy as a Major Challenge in Crop Genome Assembly
3.2. Reduced Genetic Diversity of Modern Crops
3.3. Sequence and Structural Variations in Genomes Providing Clues for Stress Studies
3.4. Advances in Ultra-High-Density Genetic Mapping Using SNPs
3.5. Genomic Selections
3.6. Identification of Stress-Related Gene Families
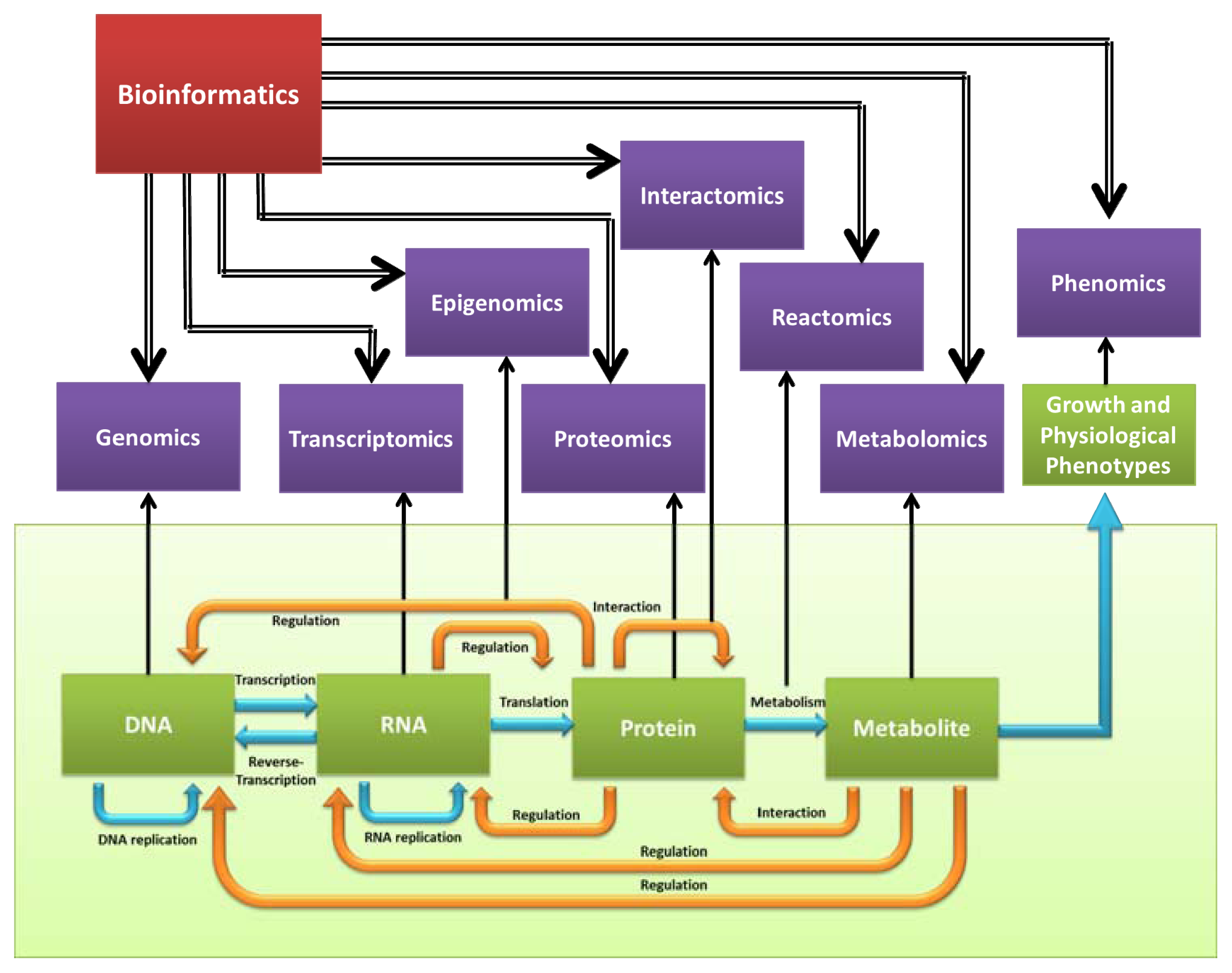
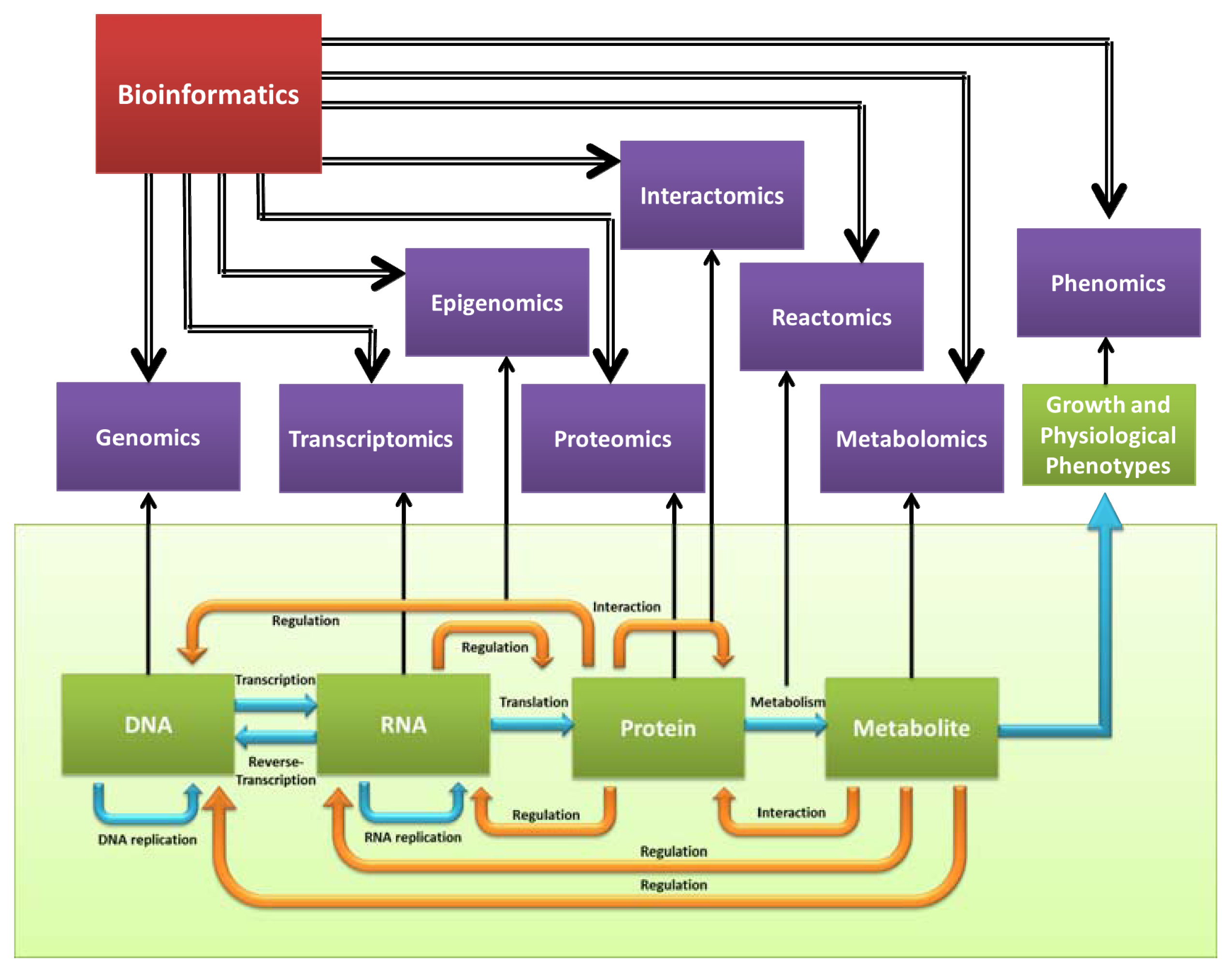
4. Functional Genomics
4.1. Transcriptome
4.2. Proteome
4.3. Interactome
4.4. Epigenome
4.5. Phenome
5. Future Perspectives
Supplementary Information
ijms-14-11444-s001.pdfAcknowledgments
Conflict of Interest
References
- FAO, How to Feed the World in 2050; FAO: Rome, Italy, 2009; p. 35.
- Arabidopsis Genome Initiative. The Arabidopsis genome initiative analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815.
- BGI Library of Digital Life: Plants. Available online: http://ldl.genomics.org.cn/page/pa-plant.jsp (accessed on 5 January 2013).
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2012, 40, D13–D25. [Google Scholar]
- Leinonen, R.; Akhtar, R.; Birney, E.; Bower, L.; Cerdeno-Tárraga, A.; Cheng, Y.; Cleland, I.; Faruque, N.; Goodgame, N.; Gibson, R.; et al. The European nucleotide archive. Nucleic Acids Res 2011, 39, D28–D31. [Google Scholar]
- Miyazaki, S.; Sugawara, H.; Ikeo, K.; Gojobori, T.; Tateno, Y. DDBJ in the stream of various biological data. Nucleic Acids Res 2004, 32, D31–D34. [Google Scholar]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res 2012, 40, D1178–D1186. [Google Scholar]
- Liang, C.; Jaiswal, P.; Hebbard, C.; Avraham, S.; Buckler, E.S.; Casstevens, T.; Hurwitz, B.; McCouch, S.; Ni, J.; Pujar, A.; et al. Gramene: A growing plant comparative genomics resource. Nucleic Acids Res 2008, 36, D947–D953. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008, 36, D480–D484. [Google Scholar]
- Duvick, J.; Fu, A.; Muppirala, U.; Sabharwal, M.; Wilkerson, M.D.; Lawrence, C.J.; Lushbough, C.; Brendel, V. PlantGDB: A resource for comparative plant genomics. Nucleic Acids Res 2008, 36, D959–D965. [Google Scholar]
- Kersey, P.J.; Staines, D.M.; Lawson, D.; Kulesha, E.; Derwent, P.; Humphrey, J.C.; Hughes, D.S.T.; Keenan, S.; Kerhornou, A.; Koscielny, G.; et al. Ensembl Genomes: An integrative resource for genome-scale data from non-vertebrate species. Nucleic Acids Res 2012, 40, D91–D97. [Google Scholar]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res 2004, 32, W273–W279. [Google Scholar]
- Proost, S.; van Bel, M.; Sterck, L.; Billiau, K.; van Parys, T.; van de Peer, Y.; Vandepoele, K. PLAZA: A comparative genomics resource to study gene and genome evolution in plants. Plant Cell 2009, 21, 3718–3731. [Google Scholar]
- Sneddon, T.P.; Li, P.; Edmunds, S.C. GigaDB: Announcing the GigaScience database. GigaScience 2012, 1, 1–2. [Google Scholar]
- Bombarely, A.; Menda, N.; Tecle, I.Y.; Buels, R.M.; Strickler, S.; Fischer-York, T.; Pujar, A.; Leto, J.; Gosselin, J.; Mueller, L.A. The Sol Genomics Network (solgenomics.net): Growing tomatoes using Perl. Nucleic Acids Res 2011, 39, D1149–D1155. [Google Scholar]
- Carollo, V.; Matthews, D.E.; Lazo, G.R.; Blake, T.K.; Hummel, D.D.; Lui, N.; Hane, D.L.; Anderson, O.D. GrainGenes 2.0. An improved resource for the small-grains community. Plant Physiol 2005, 139, 643–651. [Google Scholar]
- Gonzales, M.D.; Archuleta, E.; Farmer, A.; Gajendran, K.; Grant, D.; Shoemaker, R.; Beavis, W.D.; Waugh, M.E. The Legume Information System (LIS): An integrated information resource for comparative legume biology. Nucleic Acids Res 2005, 33, D660–D665. [Google Scholar]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar]
- The International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716.
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotech 2012, 30, 549–554. [Google Scholar]
- Messmer, R.; Fracheboud, Y.; Bänziger, M.; Vargas, M.; Stamp, P.; Ribaut, J.-M. Drought stress and tropical maize: QTL-by-environment interactions and stability of QTLs across environments for yield components and secondary traits. Theor. Appl. Genet 2009, 119, 913–930. [Google Scholar]
- Sanseverino, W.; Hermoso, A.; D’Alessandro, R.; Vlasova, A.; Andolfo, G.; Frusciante, L.; Lowy, E.; Roma, G.; Ercolano, M.R. PRGdb 2.0: Towards a community-based database model for the analysis of R-genes in plants. Nucleic Acids Res 2013, 41, D1167–D1171. [Google Scholar]
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.-C.; Iwamoto, M.; Abe, T.; et al. Rice annotation project database (RAP-DB): An integrative and interactive database for rice genomics. Plant Cell Physiol 2013, 54, e6. [Google Scholar]
- Schaeffer, M.L.; Harper, L.C.; Gardiner, J.M.; Andorf, C.M.; Campbell, D.A.; Cannon, E.K.S.; Sen, T.Z.; Lawrence, C.J. MaizeGDB: Curation and outreach go hand-in-hand. Database 2011, 2011, bar022. [Google Scholar]
- Grant, D.; Nelson, R.T.; Cannon, S.B.; Shoemaker, R.C. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res 2010, 38, D843–D846. [Google Scholar]
- Fei, Z.; Joung, J.-G.; Tang, X.; Zheng, Y.; Huang, M.; Lee, J.M.; McQuinn, R.; Tieman, D.M.; Alba, R.; Klee, H.J.; et al. Tomato functional genomics database: A comprehensive resource and analysis package for tomato functional genomics. Nucleic Acids Res 2011, 39, D1156–D1163. [Google Scholar]
- Gene Ontology Consortium. The Gene Ontology Website. Available online: http://www.geneontology.org/ (accessed on 10 January 2013).
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet 2000, 25, 25–29. [Google Scholar]
- Torto-Alalibo, T.; Collmer, C.; Gwinn-Giglio, M. The Plant-Associated Microbe Gene Ontology (PAMGO) Consortium: Community development of new Gene Ontology terms describing biological processes involved in microbe-host interactions. BMC Microbiol 2009, 9, 1–5. [Google Scholar]
- Avraham, S.; Tung, C.W.; Ilic, K.; Jaiswal, P.; Kellogg, E.A.; McCouch, S.; Pujar, A.; Reiser, L.; Rhee, S.Y.; Sachs, M.M.; et al. The Plant Ontology Database: A community resource for plant structure and developmental stages controlled vocabulary and annotations. Nucleic Acids Res 2008, 36, D449–D454. [Google Scholar]
- Eilbeck, K.; Lewis, S.E.; Mungall, C.J.; Yandell, M.; Stein, L.; Durbin, R.; Ashburner, M. The Sequence Ontology: A tool for the unification of genome annotations. Genome Biol 2005, 6, R44. [Google Scholar]
- The Catalogue of Life. Available online: http://www.catalogueoflife.org/ (accessed on 8 January 2013).
- Integrated Taxonomic Information System. Available online: http://www.itis.gov/ (accessed on 8 January 2013).
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet 2012, 44, 1098–1103. [Google Scholar]
- Shulaev, V.; Korban, S.S.; Sosinski, B.; Abbott, A.G.; Aldwinckle, H.S.; Folta, K.M.; Iezzoni, A.; Main, D.; Arús, P.; Dandekar, A.M.; et al. Multiple models for rosaceae genomics. Plant Physiol 2008, 147, 985–1003. [Google Scholar]
- Shulaev, V.; Sargent, D.J.; Crowhurst, R.N.; Mockler, T.C.; Folkerts, O.; Delcher, A.L.; Jaiswal, P.; Mockaitis, K.; Liston, A.; Mane, S.P.; et al. The genome of woodland strawberry (Fragaria vesca). Nat. Genet 2011, 43, 109–116. [Google Scholar]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.A.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar]
- Jia, J.; Zhao, S.; Kong, X.; Li, Y.; Zhao, G.; He, W.; Appels, R.; Pfeifer, M.; Tao, Y.; Zhang, X.; et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 2013, 496, 91–95. [Google Scholar]
- Ling, H.-Q.; Zhao, S.; Liu, D.; Wang, J.; Sun, H.; Zhang, C.; Fan, H.; Li, D.; Dong, L.; Tao, Y.; et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 2013, 496, 87–90. [Google Scholar]
- Lam, H.M.; Xu, X.; Liu, X.; Chen, W.B.; Yang, G.H.; Wong, F.L.; Li, M.W.; He, W.M.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet 2010, 42, 1053–1059. [Google Scholar]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol 2012, 30, 105–111. [Google Scholar]
- Hufford, M.B.; Xu, X.; van Heerwaarden, J.; Pyhajarvi, T.; Chia, J.-M.; Cartwright, R.A.; Elshire, R.J.; Glaubitz, J.C.; Guill, K.E.; Kaeppler, S.M.; et al. Comparative population genomics of maize domestication and improvement. Nat. Genet 2012, 44, 808–811. [Google Scholar]
- Kim, M.Y.; Lee, S.; Van, K.; Kim, T.-H.; Jeong, S.-C.; Choi, I.-Y.; Kim, D.-S.; Lee, Y.-S.; Park, D.; Ma, J.; et al. Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proc. Natl. Acad. Sci. USA 2010, 107, 22032–22037. [Google Scholar]
- Lai, J.; Li, R.; Xu, X.; Jin, W.; Xu, M.; Zhao, H.; Xiang, Z.; Song, W.; Ying, K.; Zhang, M.; et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet 2010, 42, 1027–1030. [Google Scholar]
- Garcia-Mas, J.; Benjak, A.; Sanseverino, W.; Bourgeois, M.; Mir, G.; Gonzalez, V.M.; Henaff, E.; Camara, F.; Cozzuto, L.; Lowy, E.; et al. The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. USA 2012, 109, 11872–11877. [Google Scholar]
- Dangl, J.L.; Jones, J.D.G. Plant pathogens and integrated defence responses to infection. Nature 2001, 411, 826–833. [Google Scholar]
- Shanmugam, V. Role of extracytoplasmic leucine rich repeat proteins in plant defence mechanisms. Microbiol. Res 2005, 160, 83–94. [Google Scholar]
- Torii, K.U. Leucine-rich repeat receptor kinases in plants: Structure, function, and signal transduction pathways. Int. Rev. Cytol 2004, 234, 1–46. [Google Scholar]
- Van Ooijen, G.; Mayr, G.; Kasiem, M.M.A.; Albrecht, M.; Cornelissen, B.J.C.; Takken, F.L.W. Structure—Function analysis of the NB-ARC domain of plant disease resistance proteins. J. Exp. Bot 2008, 59, 1383–1397. [Google Scholar]
- McNally, K.L.; Childs, K.L.; Bohnert, R.; Davidson, R.M.; Zhao, K.; Ulat, V.J.; Zeller, G.; Clark, R.M.; Hoen, D.R.; Bureau, T.E.; et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. USA 2009, 106, 12273–12278. [Google Scholar]
- Zheng, L.-Y.; Guo, X.-S.; He, B.; Sun, L.-J.; Peng, Y.; Dong, S.-S.; Liu, T.-F.; Jiang, S.; Ramachandran, S.; Liu, C.-M.; et al. Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor). Genome Biol 2011, 12, R114. [Google Scholar]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet 2010, 11, 539–548. [Google Scholar]
- D’Hont, A.; Denoeud, F.; Aury, J.-M.; Baurens, F.-C.; Carreel, F.; Garsmeur, O.; Noel, B.; Bocs, S.; Droc, G.; Rouard, M.; et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012, 488, 213–217. [Google Scholar]
- Singh, K.B.; Foley, R.C.; Oñate-Sánchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol 2002, 5, 430–436. [Google Scholar]
- Collard, B.C.Y.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar]
- Rafalski, J.A. Association genetics in crop improvement. Curr. Opin. Plant Biol 2010, 13, 174–180. [Google Scholar]
- Huang, S.; Spielmeyer, W.; Lagudah, E.S.; James, R.A.; Platten, J.D.; Dennis, E.S.; Munns, R. A sodium transporter (HKT7) is a candidate for Nax1, a gene for salt tolerance in durum wheat. Plant Physiol 2006, 142, 1718–1727. [Google Scholar]
- Ren, Z.-H.; Gao, J.-P.; Li, L.-G.; Cai, X.-L.; Huang, W.; Chao, D.-Y.; Zhu, M.-Z.; Wang, Z.-Y.; Luan, S.; Lin, H.-X. A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat. Genet 2005, 37, 1141–1146. [Google Scholar]
- Sutton, T.; Baumann, U.; Hayes, J.; Collins, N.C.; Shi, B.-J.; Schnurbusch, T.; Hay, A.; Mayo, G.; Pallotta, M.; Tester, M.; et al. Boron-toxicity tolerance in barley arising from efflux transporter amplification. Science 2007, 318, 1446–1449. [Google Scholar]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res 2009, 19, 1068–1076. [Google Scholar]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet 2010, 42, U961–U976. [Google Scholar]
- Yu, H.; Xie, W.; Wang, J.; Xing, Y.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 2011, 6, e17595. [Google Scholar]
- Zou, G.; Zhai, G.; Feng, Q.; Yan, S.; Wang, A.; Zhao, Q.; Shao, J.; Zhang, Z.; Zou, J.; Han, B.; et al. Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J. Exp. Bot 2012, 63, 5451–5462. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar]
- Wetterstrand, K. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). Available online: http://www.genome.gov/sequencingcosts (accessed on 3 January 2013).
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.-L. Genomic selection for crop improvement. Crop Sci 2009, 49, 1–12. [Google Scholar]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Zhao, Y.; Gowda, M.; Liu, W.; Würschum, T.; Maurer, H.P.; Longin, F.H.; Ranc, N.; Reif, J.C. Accuracy of genomic selection in European maize elite breeding populations. Theor. Appl. Genet 2012, 124, 769–776. [Google Scholar]
- Heffner, E.L.; Jannink, J.-L.; Iwata, H.; Souza, E.; Sorrells, M.E. Genomic selection accuracy for grain quality traits in biparental wheat populations. Crop Sci 2011, 51, 2597–2606. [Google Scholar]
- Heffner, E.L.; Jannink, J.-L.; Sorrells, M.E. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Gen 2011, 4, 65–75. [Google Scholar]
- Zhong, S.Q.; Dekkers, J.C.M.; Fernando, R.L.; Jannink, J.L. Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines: A barley case study. Genetics 2009, 182, 355–364. [Google Scholar]
- Oliveira, E.; Resende, M.; Silva Santos, V.; Ferreira, C.; Oliveira, G.; Silva, M.; Oliveira, L.; Aguilar-Vildoso, C. Genome-wide selection in cassava. Euphytica 2012, 187, 263–276. [Google Scholar]
- Xu, S. Estimating polygenic effects using markers of the entire genome. Genetics 2003, 163, 789–801. [Google Scholar]
- Nakaya, A.; Isobe, S.N. Will genomic selection be a practical method for plant breeding? Ann. Bot 2012, 110, 1303–1316. [Google Scholar]
- Jannink, J.-L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genomics 2010, 9, 166–177. [Google Scholar]
- Rutkoski, J.; Heffner, E.; Sorrells, M. Genomic selection for durable stem rust resistance in wheat. Euphytica 2011, 179, 161–173. [Google Scholar]
- Salse, J. In silico archeogenomics unveils modern plant genome organisation, regulation and evolution. Curr. Opin. Plant Biol 2012, 15, 122–130. [Google Scholar]
- Salamov, A.A.; Solovyev, V.V. Ab initio gene finding in drosophila genomic DNA. Genome Res 2000, 10, 516–522. [Google Scholar]
- Howe, K.L.; Chothia, T.; Durbin, R. GAZE: A generic framework for the integration of gene-prediction data by dynamic programming. Genome Res 2002, 12, 1418–1427. [Google Scholar]
- Allen, J.E.; Salzberg, S.L. JIGSAW: Integration of multiple sources of evidence for gene prediction. Bioinformatics 2005, 21, 3596–3603. [Google Scholar]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res 2010, 38, D211–D222. [Google Scholar]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New developments in the family and domain prediction database. Nucleic Acids Res 2012, 40, D306–D312. [Google Scholar]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2012, 40, D109–D114. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res 2012, 40, D302–D305. [Google Scholar]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res 2011, 39, W29–W37. [Google Scholar]
- Sharoni, A.M.; Nuruzzaman, M.; Satoh, K.; Shimizu, T.; Kondoh, H.; Sasaya, T.; Choi, I.-R.; Omura, T.; Kikuchi, S. Gene structures, classification and expression models of the AP2/EREBP transcription factor family in rice. Plant Cell Physiol 2011, 52, 344–360. [Google Scholar]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet 2011, 12, 87–98. [Google Scholar]
- Rustici, G.; Kolesnikov, N.; Brandizi, M.; Burdett, T.; Dylag, M.; Emam, I.; Farne, A.; Hastings, E.; Ison, J.; Keays, M.; et al. ArrayExpress update—Trends in database growth and links to data analysis tools. Nucleic Acids Res 2013, 41, D987–D990. [Google Scholar]
- Lou, S.-K.; Ni, B.; Lo, L.-Y.; Tsui, S.K.-W.; Chan, T.-F.; Leung, K.-S. ABMapper: A suffix array-based tool for multi-location searching and splice-junction mapping. Bioinformatics 2011, 27, 421–422. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Meth 2012, 9, 357–359. [Google Scholar]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 2012, 7, 562–578. [Google Scholar]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Meth 2010, 7, 909–912. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol 2011, 29, 644–652. [Google Scholar]
- Sekhon, R.S.; Lin, H.; Childs, K.L.; Hansey, C.N.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. Genome-wide atlas of transcription during maize development. Plant J 2011, 66, 553–563. [Google Scholar]
- Severin, A.; Woody, J.; Bolon, Y.-T.; Joseph, B.; Diers, B.; Farmer, A.; Muehlbauer, G.; Nelson, R.; Grant, D.; Specht, J.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol 2010, 10, 160. [Google Scholar]
- Ge, Y.; Li, Y.; Zhu, Y.-M.; Bai, X.; Lv, D.-K.; Guo, D.; Ji, W.; Cai, H. Global transcriptome profiling of wild soybean (Glycine soja) roots under NaHCO3 treatment. BMC Plant Biol 2010, 10, 153. [Google Scholar]
- Ma, T.-L.; Wu, W.-H.; Wang, Y. Transcriptome analysis of rice root responses to potassium deficiency. BMC Plant Biol 2012, 12, 161. [Google Scholar]
- An, D.; Yang, J.; Zhang, P. Transcriptome profiling of low temperature-treated cassava apical shoots showed dynamic responses of tropical plant to cold stress. BMC Genomics 2012, 13, 64. [Google Scholar]
- Zabala, G.; Zou, J.; Tuteja, J.; Gonzalez, D.; Clough, S.; Vodkin, L. Transcriptome changes in the phenylpropanoid pathway of Glycine max in response to Pseudomonas syringae infection. BMC Plant Biol 2006, 6, 26. [Google Scholar]
- Wang, C.; Chen, H.; Hao, Q.; Sha, A.; Shan, Z.; Chen, L.; Zhou, R.; Zhi, H.; Zhou, X. Transcript profile of the response of two soybean genotypes to potassium deficiency. PLoS One 2012, 7, e39856. [Google Scholar]
- Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Bansal, K.C. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol. J 2011, 9, 315–327. [Google Scholar]
- Zhang, T.; Zhao, X.; Wang, W.; Pan, Y.; Huang, L.; Liu, X.; Zong, Y.; Zhu, L.; Yang, D.; Fu, B. Comparative transcriptome profiling of chilling stress responsiveness in two contrasting rice genotypes. PLoS One 2012, 7, e43274. [Google Scholar]
- Li, Y.C.; Meng, F.R.; Zhang, C.Y.; Zhang, N.; Sun, M.S.; Ren, J.P.; Niu, H.B.; Wang, X.; Yin, J. Comparative analysis of water stress-responsive transcriptomes in drought-susceptible and -tolerant wheat (Triticum aestivum L.). J. Plant Biol 2012, 55, 349–360. [Google Scholar]
- Zahaf, O.; Blanchet, S.; de Zelicourt, A.; Alunni, B.; Plet, J.; Laffont, C.; de Lorenzo, L.; Imbeaud, S.; Ichante, J.-L.; Diet, A.; et al. Comparative transcriptomic analysis of salt adaptation in roots of contrasting Medicago truncatula genotypes. Mol. Plant 2012, 5, 1068–1081. [Google Scholar]
- Puranik, S.; Jha, S.; Srivastava, P.S.; Sreenivasulu, N.; Prasad, M. Comparative transcriptome analysis of contrasting foxtail millet cultivars in response to short-term salinity stress. J. Plant Physiol 2011, 168, 280–287. [Google Scholar]
- Delker, C.; Quint, M. Expression level polymorphisms: Heritable traits shaping natural variation. Trends Plant Sci 2011, 16, 481–488. [Google Scholar]
- Holloway, B.; Luck, S.; Beatty, M.; Rafalski, J.-A.; Li, B. Genome-wide expression quantitative trait loci (eQTL) analysis in maize. BMC Genomics 2011, 12, 336. [Google Scholar]
- Wang, J.; Yu, H.; Xie, W.; Xing, Y.; Yu, S.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A global analysis of QTLs for expression variations in rice shoots at the early seedling stage. Plant J 2010, 63, 1063–1074. [Google Scholar]
- Chen, X.; Hackett, C.A.; Niks, R.E.; Hedley, P.E.; Booth, C.; Druka, A.; Marcel, T.C.; Vels, A.; Bayer, M.; Milne, I.; et al. An eQTL Analysis of partial resistance to Puccinia hordei in barley. PLoS One 2010, 5, e8598. [Google Scholar]
- Mann, M. Can proteomics retire the western blot? J. Proteome Res 2008, 7, 3065–3065. [Google Scholar]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol 1999, 17, 994–999. [Google Scholar]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar]
- Wiese, S.; Reidegeld, K.A.; Meyer, H.E.; Warscheid, B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7, 340–350. [Google Scholar]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–1372. [Google Scholar]
- Old, W.M.; Meyer-Arendt, K.; Aveline-Wolf, L.; Pierce, K.G.; Mendoza, A.; Sevinsky, J.R.; Resing, K.A.; Ahn, N.G. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 2005, 4, 1487–1502. [Google Scholar]
- Jorrín, J.V.; Maldonado, A.M.; Castillejo, M.A. Plant proteome analysis: A 2006 update. Proteomics 2007, 7, 2947–2962. [Google Scholar]
- Jorrin-Novo, J.V.; Maldonado, A.M.; Echevarria-Zomeno, S.; Valledor, L.; Castillejo, M.A.; Curto, M.; Valero, J.; Sghaier, B.; Donoso, G.; Redondo, I. Plant proteomics update (2007–2008): Second-generation proteomic techniques, an appropriate experimental design, and data analysis to fulfill MIAPE standards, increase plant proteome coverage and expand biological knowledge. J. Proteomics 2009, 72, 285–314. [Google Scholar]
- Kamal, A.H.M.; Cho, K.; Kim, D.-E.; Uozumi, N.; Chung, K.-Y.; Lee, S.Y.; Choi, J.-S.; Cho, S.-W.; Shin, C.-S.; Woo, S.H. Changes in physiology and protein abundance in salt-stressed wheat chloroplasts. Mol. Biol. Rep 2012, 39, 9059–9074. [Google Scholar]
- Ahsan, N.; Nakamura, T.; Komatsu, S. Differential responses of microsomal proteins and metabolites in two contrasting cadmium (Cd)-accumulating soybean cultivars under Cd stress. Amino Acids 2012, 42, 317–327. [Google Scholar]
- Wang, H.; Wang, S.; Xia, Y. Identification and verification of redox-sensitive proteins in Arabidopsis thaliana. Methods Mol. Biol 2012, 876, 83–94. [Google Scholar]
- Galant, A.; Koester, R.P.; Ainsworth, E.A.; Hicks, L.M.; Jez, J.M. From climate change to molecular response: Redox proteomics of ozone-induced responses in soybean. New Phytol 2012, 194, 220–229. [Google Scholar]
- Nakagami, H.; Sugiyama, N.; Mochida, K.; Daudi, A.; Yoshida, Y.; Toyoda, T.; Tomita, M.; Ishihama, Y.; Shirasu, K. Large-scale comparative phosphoproteomics identifies conserved phosphorylation sites in plants. Plant Physiol 2010, 153, 1161–1174. [Google Scholar]
- Agrawal, G.K.; Jwa, N.-S.; Lebrun, M.-H.; Job, D.; Rakwal, R. Plant secretome: Unlocking secrets of the secreted proteins. Proteomics 2010, 10, 799–827. [Google Scholar]
- Alexandersson, E.; Ashfaq, A.; Resjö, S.; Andreasson, E. Plant secretome proteomics. Front. Plant Sci 2013, 4, 9. [Google Scholar]
- Catalá, C.; Howe, K.J.; Hucko, S.; Rose, J.K.C.; Thannhauser, T.W. Towards characterization of the glycoproteome of tomato (Solanum lycopersicum) fruit using Concanavalin A lectin affinity chromatography and LC-MALDI-MS/MS analysis. Proteomics 2011, 11, 1530–1544. [Google Scholar]
- Ruiz-May, E.; Kim, S.J.; Brandizzi, F.; Rose, J.K.C. The secreted plant n-glycoproteome and associated secretory pathways. Front. Plant Sci 2012, 3, 117. [Google Scholar]
- Pawson, T.; Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev 2000, 14, 1027–1047. [Google Scholar]
- Zhang, Y.; Gao, P.; Yuan, J.S. Plant protein-protein interaction network and interactome. Curr. Genomics 2010, 11, 40–46. [Google Scholar]
- Wittig, I.; Braun, H.-P.; Schagger, H. Blue native PAGE. Nat. Protoc 2006, 1, 418–428. [Google Scholar]
- Hue, M.; Riffle, M.; Vert, J.-P.; Noble, W. Large-scale prediction of protein-protein interactions from structures. BMC Bioinforma 2010, 11, 144. [Google Scholar] [Green Version]
- Moal, I.H.; Agius, R.; Bates, P.A. Protein-protein binding affinity prediction on a diverse set of structures. Bioinformatics 2011, 27, 3002–3009. [Google Scholar]
- Cusick, M.E.; Yu, H.; Smolyar, A.; Venkatesan, K.; Carvunis, A.-R.; Simonis, N.; Rual, J.-F.; Borick, H.; Braun, P.; Dreze, M.; et al. Literature-curated protein interaction datasets. Nat. Meth 2009, 6, 39–46. [Google Scholar]
- Ho, C.-L.; Wu, Y.; Shen, H.-B.; Provart, N.; Geisler, M. A predicted protein interactome for rice. Rice 2012, 5, 15. [Google Scholar]
- Cui, J.; Li, P.; Li, G.; Xu, F.; Zhao, C.; Li, Y.H.; Yang, Z.N.; Wang, G.; Yu, Q.B.; Li, Y.X.; et al. AtPID: Arabidopsis thaliana protein interactome database—An integrative platform for plant systems biology. Nucleic Acids Res 2008, 36, D999–D1008. [Google Scholar]
- Arabidopsis interactome mapping consortium. Evidence for network evolution in an Arabidopsis interactome map. Science 2011, 333, 601–607.
- Seo, Y.-S.; Chern, M.; Bartley, L.E.; Han, M.; Jung, K.-H.; Lee, I.; Walia, H.; Richter, T.; Xu, X.; Cao, P.; et al. Towards establishment of a rice stress response interactome. PLoS Genet 2011, 7, e1002020. [Google Scholar]
- Gu, H.; Zhu, P.; Jiao, Y.; Meng, Y.; Chen, M. PRIN: A predicted rice interactome network. BMC Bioinforma 2011, 12, 161. [Google Scholar]
- Chatr-aryamontri, A.; Breitkreutz, B.-J.; Heinicke, S.; Boucher, L.; Winter, A.; Stark, C.; Nixon, J.; Ramage, L.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res 2013, 41, D816–D823. [Google Scholar]
- Xenarios, I.; Salwínski, L.; Duan, X.J.; Higney, P.; Kim, S.-M.; Eisenberg, D. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res 2002, 30, 303–305. [Google Scholar]
- Mingwei, M.; Haoyang, C.; Wen, Z.; Zhirui, Y.; Xiao, L.; Xinjian, F.; Quansheng, F. PlaPID: A Database of Protein-Protein Interactions in Plants. Proceedings of the 4th International Conference on Bioinformatics and Biomedical Engineering (iCBBE), Chengdu, China, 18–20 June 2010; pp. 1–4.
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res 2012, 40, D841–D846. [Google Scholar]
- Cooper, B.; Clarke, J.D.; Budworth, P.; Kreps, J.; Hutchison, D.; Park, S.; Guimil, S.; Dunn, M.; Luginbühl, P.; Ellero, C.; et al. A network of rice genes associated with stress response and seed development. Proc. Natl. Acad. Sci. USA 2003, 100, 4945–4950. [Google Scholar]
- Tardif, G.; Kane, N.; Adam, H.; Labrie, L.; Major, G.; Gulick, P.; Sarhan, F.; Laliberté, J.-F. Interaction network of proteins associated with abiotic stress response and development in wheat. Plant Mol. Biol 2007, 63, 703–718. [Google Scholar]
- Afzal, A.J.; Natarajan, A.; Saini, N.; Iqbal, M.J.; Geisler, M.; El Shemy, H.A.; Mungur, R.; Willmitzer, L.; Lightfoot, D.A. The nematode resistance allele at the rhg1 locus alters the proteome and primary metabolism of soybean roots. Plant Physiol 2009, 151, 1264–1280. [Google Scholar]
- Gendler, K.; Paulsen, T.; Napoli, C. ChromDB: The chromatin database. Nucleic Acids Res 2008, 36, D298–D302. [Google Scholar]
- Morison, I.M.; Ramsay, J.P.; Spencer, H.G. A census of mammalian imprinting. Trends Genet 2005, 21, 457–465. [Google Scholar]
- Tsukahara, S.; Kobayashi, A.; Kawabe, A.; Mathieu, O.; Miura, A.; Kakutani, T. Bursts of retrotransposition reproduced in Arabidopsis. Nature 2009, 461, 423–426. [Google Scholar]
- Zhang, X.Y.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.L.; Chen, H.M.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet 2007, 39, 61–69. [Google Scholar]
- Seifert, M.; Cortijo, S.; Colome-Tatche, M.; Johannes, F.; Roudier, F.; Colot, V. MeDIP-HMM: Genome-wide identification of distinct DNA methylation states from high-density tiling arrays. Bioinformatics 2012, 28, 2930–2939. [Google Scholar]
- Cokus, S.J.; Feng, S.H.; Zhang, X.Y.; Chen, Z.G.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 2005, 33, 5868–5877. [Google Scholar]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar]
- Wang, W.S.; Pan, Y.J.; Zhao, X.Q.; Dwivedi, D.; Zhu, L.H.; Ali, J.; Fu, B.Y.; Li, Z.K. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot 2011, 62, 1951–1960. [Google Scholar]
- Zhong, L.; Xu, Y.H.; Wang, J.B. DNA-methylation changes induced by salt stress in wheat Triticum aestivum. Afr. J. Biotechnol 2009, 8, 6201–6207. [Google Scholar]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.; van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijo, J.A.; Becker, J.D.; et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 2012, 151, 194–205. [Google Scholar]
- Holeski, L.M.; Jander, G.; Agrawal, A.A. Transgenerational defense induction and epigenetic inheritance in plants. Trends Ecol. Evol 2012, 27, 618–626. [Google Scholar]
- Kou, H.P.; Li, Y.; Song, X.X.; Ou, X.F.; Xing, S.C.; Ma, J.; von Wettstein, D.; Liu, B. Heritable alteration in DNA methylation induced by nitrogen-deficiency stress accompanies enhanced tolerance by progenies to the stress in rice (Oryza sativa L.). J. Plant Physiol 2011, 168, 1685–1693. [Google Scholar]
- Lutsik, P.; Feuerbach, L.; Arand, J.; Lengauer, T.; Walter, J.; Bock, C. BiQ Analyzer HT: Locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Res 2011, 39, W551–W556. [Google Scholar]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar]
- Harris, E.Y.; Ponts, N.; Le Roch, K.G.; Lonardi, S. BRAT-BW: Efficient and accurate mapping of bisulfite-treated reads. Bioinformatics 2012, 28, 1795–1796. [Google Scholar]
- Hansen, K.D.; Langmead, B.; Irizarry, R.A. BSmooth: From whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol 2012, 13, R83. [Google Scholar]
- Chen, P.Y.; Cokus, S.J.; Pellegrini, M. BS Seeker: Precise mapping for bisulfite sequencing. BMC Bioinforma 2010, 11, 203. [Google Scholar]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinforma 2009, 10, 232. [Google Scholar]
- Su, J.Z.; Yan, H.D.; Wei, Y.J.; Liu, H.B.; Liu, H.; Wang, F.; Lv, J.; Wu, Q.; Zhang, Y. CpG_MPs: Identification of CpG methylation patterns of genomic regions from high-throughput bisulfite sequencing data. Nucleic Acids Res 2013, 41, e4. [Google Scholar]
- Benoukraf, T.; Wongphayak, S.; Hadi, L.H.; Wu, M.; Soong, R. GBSA: A comprehensive software for analysing whole genome bisulfite sequencing data. Nucleic Acids Res. 2012. [Google Scholar] [CrossRef]
- Gruntman, E.; Qi, Y.J.; Slotkin, R.K.; Roeder, T.; Martienssen, R.A.; Sachidanandam, R. Kismeth: Analyzer of plant methylation states through bisulfite sequencing. BMC Bioinforma 2008, 9, 371. [Google Scholar]
- Kumaki, Y.; Oda, M.; Okano, M. QUMA: Quantification tool for methylation analysis. Nucleic Acids Res 2008, 36, W170–W175. [Google Scholar]
- Xi, Y.X.; Bock, C.; Muller, F.; Sun, D.Q.; Meissner, A.; Li, W. RRBSMAP: A fast, accurate and user-friendly alignment tool for reduced representation bisulfite sequencing. Bioinformatics 2012, 28, 430–432. [Google Scholar]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar]
- Marino-Ramirez, L.; Levine, K.M.; Morales, M.; Zhang, S.Y.; Moreland, R.T.; Baxevanis, A.D.; Landsman, D. The histone database: An integrated resource for histones and histone fold-containing proteins. Database-Oxford 2011. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol 2007, 8, 284–295. [Google Scholar]
- Barski, A.; Cuddapah, S.; Cui, K.R.; Roh, T.Y.; Schones, D.E.; Wang, Z.B.; Wei, G.; Chepelev, I.; Zhao, K.J. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar]
- Oki, M.; Aihara, H.; Ito, T. Role Of Histone Phosphorylation In Chromatin Dynamics And Its Implications in Diseases. In Chromatin and Disease; Kundu, T., Bittman, R., Dasgupta, D., Engelhardt, H., Flohe, L., Herrmann, H., Holzenburg, A., Nasheuer, H.P., Rottem, S., Wyss, M., Zwickl, P., Eds.; Springer: London, UK, 2007; Volume 41, pp. 323–340. [Google Scholar]
- Shivaswamy, S.; Iyer, V.R. Genome-wide analysis of chromatin status using tiling microarrays. Methods 2007, 41, 304–311. [Google Scholar]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar]
- Papaefthimiou, D.; Tsaftaris, A.S. Characterization of a drought inducible trithorax-like H3K4 methyltransferase from barley. Biol. Plant 2012, 56, 683–692. [Google Scholar]
- Ding, B.; Bellizzi, M.D.R.; Ning, Y.; Meyers, B.C.; Wang, G.-L. HDT701, a Histone H4 deacetylase, negatively regulates plant innate immunity by modulating histone H4 acetylation of defense-related genes in rice. Plant Cell 2012, 24, 3783–3794. [Google Scholar]
- Song, Y.G.; Ji, D.D.; Li, S.; Wang, P.; Li, Q.; Xiang, F.N. The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS One 2012, 7, e41274. [Google Scholar]
- Zong, W.; Zhong, X.; You, J.; Xiong, L. Genome-wide profiling of histone H3K4-tri-methylation and gene expression in rice under drought stress. Plant Mol. Biol 2013, 81, 175–188. [Google Scholar]
- Munns, R.; James, R.A.; Sirault, X.R.R.; Furbank, R.T.; Jones, H.G. New phenotyping methods for screening wheat and barley for beneficial responses to water deficit. J. Exp. Bot 2010, 61, 3499–3507. [Google Scholar]
- Golzarian, M.; Frick, R.; Rajendran, K.; Berger, B.; Roy, S.; Tester, M.; Lun, D. Accurate inference of shoot biomass from high-throughput images of cereal plants. Plant Methods 2011, 7, 2. [Google Scholar]
- Paproki, A.; Sirault, X.; Berry, S.; Furbank, R.; Fripp, J. A novel mesh processing based technique for 3D plant analysis. BMC Plant Biol 2012, 12, 63. [Google Scholar]
- Spielbauer, G.; Armstrong, P.; Baier, J.W.; Allen, W.B.; Richardson, K.; Shen, B.; Settles, A.M. High-throughput near-infrared reflectance spectroscopy for predicting quantitative and qualitative composition phenotypes of individual maize kernels. Cereal Chem 2009, 86, 556–564. [Google Scholar]
- Baker, N.R. Chlorophyll fluorescence: A probe of photosynthesis in vivo. Annu. Rev. Plant Biol 2008, 59, 89–113. [Google Scholar]
- Condon, A.G.; Richards, R.A.; Rebetzke, G.J.; Farquhar, G.D. Breeding for high water-use efficiency. J. Exp. Bot 2004, 55, 2447–2460. [Google Scholar]
- Hargreaves, C.; Gregory, P.; Bengough, A.G. Measuring root traits in barley (Hordeum vulgare ssp. vulgare and ssp. spontaneum) seedlings using gel chambers, soil sacs and X-ray microtomography. Plant Soil 2009, 316, 285–297. [Google Scholar]
- Rajendran, K.; Tester, M.; Roy, S.J. Quantifying the three main components of salinity tolerance in cereals. Plant Cell Environ 2009, 32, 237–249. [Google Scholar]
- Jones, H.G.; Serraj, R.; Loveys, B.R.; Xiong, L.; Wheaton, A.; Price, A.H. Thermal infrared imaging of crop canopies for the remote diagnosis and quantification of plant responses to water stress in the field. Funct. Plant Biol 2009, 36, 978–989. [Google Scholar]
- Moshou, D.; Bravo, C.; Oberti, R.; West, J.; Bodria, L.; McCartney, A.; Ramon, H. Plant disease detection based on data fusion of hyper-spectral and multi-spectral fluorescence imaging using Kohonen maps. Real-Time Imaging 2005, 11, 75–83. [Google Scholar]
- Genga, A.; Mattana, M.; Coraggio, I.; Locatelli, F.; Piffanelli, P.; Consonni, R. Plant Metabolomics: A Characterisation of Plant Responses to Abiotic Stresses. In Abiotic Stress in Plants—Mechanisms and Adaptations; Shanker, A., Venkateswarlu, B., Eds.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Kooke, R.; Keurentjes, J.J.B. Multi-dimensional regulation of metabolic networks shaping plant development and performance. J. Exp. Bot. 2011. [Google Scholar] [CrossRef]
- Cramer, G.R.; Urano, K.; Delrot, S.; Pezzotti, M.; Shinozaki, K. Effects of abiotic stress on plants: A systems biology perspective. BMC Plant Biol 2011, 11, 163. [Google Scholar]
- Huang, G.-T.; Ma, S.-L.; Bai, L.-P.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z.-F. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep 2012, 39, 969–987. [Google Scholar]
- Liland, K.H. Multivariate methods in metabolomics—From pre-processing to dimension reduction and statistical analysis. Trends Anal. Chem 2011, 30, 827–841. [Google Scholar]
- Stenlund, H.; Gorzsas, A.; Persson, P.; Sundberg, B.; Trygg, J. Orthogonal projections to latent structures discriminant analysis modeling on in situ FT-IR spectral imaging of liver tissue for identifying sources of variability. Anal. Chem 2008, 80, 6898–6906. [Google Scholar]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemom 2002, 16, 119–128. [Google Scholar]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmüller, E.; Dörmann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. [email protected]: The golm metabolome database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res 2007, 35, W606–W612. [Google Scholar]
- Cui, Q.; Lewis, I.A.; Hegeman, A.D.; Anderson, M.E.; Li, J.; Schulte, C.F.; Westler, W.M.; Eghbalnia, H.R.; Sussman, M.R.; Markley, J.L. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol 2008, 26, 162–164. [Google Scholar]
- Brown, M.; Dunn, W.B.; Dobson, P.; Patel, Y.; Winder, C.L.; Francis-McIntyre, S.; Begley, P.; Carroll, K.; Broadhurst, D.; Tseng, A.; et al. Mass spectrometry tools and metabolite-specific databases for molecular identification in metabolomics. Analyst 2009, 134, 1322–1332. [Google Scholar]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom 2010, 45, 703–714. [Google Scholar]
- Carroll, A.; Badger, M.; Harvey Millar, A. The MetabolomeExpress Project: Enabling web-based processing, analysis and transparent dissemination of GC/MS metabolomics datasets. BMC Bioinforma 2010, 11, 376. [Google Scholar]
- Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol 2012, 30, 826–828. [Google Scholar]
- Zhang, F.; Robinette, S.L.; Bruschweiler-Li, L.; Brüschweiler, R. Web server suite for complex mixture analysis by covariance NMR. Magn. Reson. Chem 2009, 47, S118–S122. [Google Scholar]
- Biswas, A.; Mynampati, K.C.; Umashankar, S.; Reuben, S.; Parab, G.; Rao, R.; Kannan, V.S.; Swarup, S. MetDAT: A modular and workflow-based free online pipeline for mass spectrometry data processing, analysis and interpretation. Bioinformatics 2010, 26, 2639–2640. [Google Scholar]
- Zhou, B.; Wang, J.; Ressom, H.W. MetaboSearch: Tool for mass-based metabolite identification using multiple databases. PLoS One 2012, 7, e40096. [Google Scholar]
- Gavaghan, C.L.; Li, J.V.; Hadfield, S.T.; Hole, S.; Nicholson, J.K.; Wilson, I.D.; Howe, P.W.A.; Stanley, P.D.; Holmes, E. Application of NMR-based Metabolomics to the Investigation of Salt Stress in Maize (Zea mays). Phytochem. Anal 2011, 22, 214–224. [Google Scholar]
- Widodo Patterson, J.H.; Newbigin, E.; Tester, M.; Bacic, A.; Roessner, U. Metabolic responses to salt stress of barley (Hordeum vulgare L.) cultivars, Sahara and Clipper, which differ in salinity tolerance. J. Exp. Bot. 2009, 60, 4089–4103. [Google Scholar]
- Wu, J.; Yu, H.; Dai, H.; Mei, W.; Huang, X.; Zhu, S.; Peng, M. Metabolite profiles of rice cultivars containing bacterial blight-resistant genes are distinctive from susceptible rice. Acta Biochim. Biophys. Sin 2012, 44, 650–659. [Google Scholar]
- Levi, A.; Paterson, A.H.; Cakmak, I.; Saranga, Y. Metabolite and mineral analyses of cotton near-isogenic lines introgressed with QTLs for productivity and drought-related traits. Physiol. Plant 2011, 141, 265–275. [Google Scholar]
- Semel, Y.; Schauer, N.; Roessner, U.; Zamir, D.; Fernie, A.R. Metabolite analysis for the comparison of irrigated and non-irrigated field grown tomato of varying genotype. Metabolomics 2007, 3, 289–295. [Google Scholar]
- Silvente, S.; Sobolev, A.P.; Lara, M. Metabolite adjustments in drought tolerant and sensitive soybean genotypes in response to water stress. PLoS One 2012, 7, e38554. [Google Scholar]
- Witt, S.; Galicia, L.; Lisec, J.; Cairns, J.; Tiessen, A.; Luis Araus, J.; Palacios-Rojas, N.; Fernie, A.R. Metabolic and phenotypic responses of greenhouse-grown maize hybrids to experimentally controlled drought stress. Mol. Plant 2012, 5, 401–417. [Google Scholar]
- Komatsu, S.; Yamamoto, A.; Nakamura, T.; Nouri, M.-Z.; Nanjo, Y.; Nishizawa, K.; Furukawa, K. Comprehensive analysis of mitochondria in roots and hypocotyls of soybean under flooding stress using proteomics and metabolomics techniques. J. Proteome Res 2011, 10, 3993–4004. [Google Scholar]
- Cho, K.; Shibato, J.; Agrawal, G.K.; Jung, Y.-H.; Kubo, A.; Jwa, N.-S.; Tamogami, S.; Satoh, K.; Kikuchi, S.; Higashi, T.; et al. Integrated transcriptomics, proteomics, and metabolomics analyses to survey ozone responses in the leaves of rice seedling. J. Proteome Res 2008, 7, 2980–2998. [Google Scholar]
- Aliferis, K.A.; Jabaji, S. FT-ICR/MS and GC-EI/MS metabolomics networking unravels global potato sprout’s responses to Rhizoctonia solani infection. PLoS One 2012, 7, e42576. [Google Scholar]
- Figueiredo, A.; Fortes, A.M.; Ferreira, S.; Sebastiana, M.; Choi, Y.H.; Sousa, L.; Acioli-Santos, B.; Pessoa, F.; Verpoorte, R.; Pais, M.S. Transcriptional and metabolic profiling of grape (Vitis vinifera L.) leaves unravel possible innate resistance against pathogenic fungi. J. Exp. Bot 2008, 59, 3371–3381. [Google Scholar]
- Hong, Y.-S.; Martinez, A.; Liger-Belair, G.; Jeandet, P.; Nuzillard, J.-M.; Cilindre, C. Metabolomics reveals simultaneous influences of plant defence system and fungal growth in Botrytis cinerea-infected Vitis vinifera cv. Chardonnay berries. J. Exp. Bot 2012, 63, 5773–5785. [Google Scholar]
- Cevallos-Cevallos, J.M.; Futch, D.B.; Shilts, T.; Folimonova, S.Y.; Reyes-De-Corcuera, J.I. GC-MS metabolomic differentiation of selected citrus varieties with different sensitivity to citrus huanglongbing. Plant Physiol. Biochem 2012, 53, 69–76. [Google Scholar]
- Ali, K.; Maltese, F.; Figueiredo, A.; Rex, M.; Fortes, A.M.; Zyprian, E.; Pais, M.S.; Verpoorte, R.; Choi, Y.H. Alterations in grapevine leaf metabolism upon inoculation with Plasmopara viticola in different time-points. Plant Sci 2012, 191, 100–107. [Google Scholar]
- Fumagalli, E.; Baldoni, E.; Abbruscato, P.; Piffanelli, P.; Genga, A.; Lamanna, R.; Consonni, R. NMR techniques coupled with multivariate statistical analysis: Tools to analyse Oryza sativa metabolic content under stress conditions. J. Agron. Crop Sci 2009, 195, 77–88. [Google Scholar]
- Rose, M.T.; Rose, T.J.; Pariasca-Tanaka, J.; Yoshihashi, T.; Neuweger, H.; Goesmann, A.; Frei, M.; Wissuwa, M. Root metabolic response of rice (Oryza sativa L.) genotypes with contrasting tolerance to zinc deficiency and bicarbonate excess. Planta 2012, 236, 959–973. [Google Scholar]
- Shulaev, V. Metabolomics technology and bioinformatics. Brief. Bioinforma 2006, 7, 128–139. [Google Scholar]
- Wu, W.; Zhang, Q.; Zhu, Y.; Lam, H.-M.; Cai, Z.; Guo, D. Comparative metabolic profiling reveals secondary metabolites correlated with soybean salt tolerance. J. Agric. Food Chem 2008, 56, 11132–11138. [Google Scholar]
- Johnson, H.E.; Broadhurst, D.; Goodacre, R.; Smith, A.R. Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 2003, 62, 919–928. [Google Scholar]
- Liu, L.; Li, Y.H.; Li, S.L.; Hu, N.; He, Y.M.; Pong, R.; Lin, D.N.; Lu, L.H.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012. [Google Scholar] [CrossRef]
- Kao, H.-L.; Gunsalus, K.C. Browsing Multidimensional Molecular Networks with the Generic Network Browser (N-Browse). In Current Protocols in Bioinformatics; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2002. [Google Scholar]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar]
- Katari, M.S.; Nowicki, S.D.; Aceituno, F.F.; Nero, D.; Kelfer, J.; Thompson, L.P.; Cabello, J.M.; Davidson, R.S.; Goldberg, A.P.; Shasha, D.E.; et al. VirtualPlant: A software platform to support systems biology research. Plant Physiol 2010, 152, 500–515. [Google Scholar]
- Jami, S.K.; Clark, G.B.; Ayele, B.T.; Ashe, P.; Kirti, P.B. Genome-wide comparative analysis of annexin superfamily in plants. PLoS One 2012, 7, e47801. [Google Scholar]
- Wan, H.; Yuan, W.; Bo, K.; Shen, J.; Pang, X.; Chen, J. Genome-wide analysis of NBS-encoding disease resistance genes in Cucumis sativus and phylogenetic study of NBS-encoding genes in Cucurbitaceae crops. BMC Genomics 2013, 14, 109. [Google Scholar]
- Hu, L.; Liu, S. Genome-wide identification and phylogenetic analysis of the ERF gene family in cucumbers. Genet. Mol. Biol 2011, 34, 624–633. [Google Scholar]
- Li, Q.; Zhang, C.; Li, J.; Wang, L.; Ren, Z. Genome-wide identification and characterization of R2R3MYB family in Cucumis sativus. PLoS One 2012, 7, e47576. [Google Scholar]
- Ling, J.; Jiang, W.; Zhang, Y.; Yu, H.; Mao, Z.; Gu, X.; Huang, S.; Xie, B. Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genomics 2011, 12, 471. [Google Scholar]
- Kang, Y.; Kim, K.; Shim, S.; Yoon, M.; Sun, S.; Kim, M.; Van, K.; Lee, S.-H. Genome-wide mapping of NBS-LRR genes and their association with disease resistance in soybean. BMC Plant Biol 2012, 12, 139. [Google Scholar]
- Du, H.; Yang, S.-S.; Liang, Z.; Feng, B.-R.; Liu, L.; Huang, Y.-B.; Tang, Y.-X. Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biol 2012, 12, 106. [Google Scholar]
- Dung Tien, L.; Nishiyama, R.; Watanabe, Y.; Mochida, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Lam-Son Phan, T. Genome-wide survey and expression analysis of the plant-specific NAC transcription factor family in soybean during development and dehydration stress. DNA Res 2011, 18, 263–276. [Google Scholar]
- Osorio, M.B.; Buecker-Neto, L.; Castilhos, G.; Turchetto-Zolet, A.C.; Wiebke-Strohm, B.; Bodanese-Zanettini, M.H.; Margis-Pinheiro, M. Identification and in silico characterization of soybean trihelix-GT and bHLH transcription factors involved in stress responses. Genet. Mol. Biol 2012, 35, 233–246. [Google Scholar]
- Tran, L.-S.P.; Quach, T.N.; Guttikonda, S.K.; Aldrich, D.L.; Kumar, R.; Neelakandan, A.; Valliyodan, B.; Nguyen, H.T. Molecular characterization of stress-inducible GmNAC genes in soybean. Mol. Genet. Genomics 2009, 281, 647–664. [Google Scholar]
- Zhou, Q.-Y.; Tian, A.-G.; Zou, H.-F.; Xie, Z.-M.; Lei, G.; Huang, J.; Wang, C.-M.; Wang, H.-W.; Zhang, J.-S.; Chen, S.-Y. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol. J 2008, 6, 486–503. [Google Scholar]
- Liang, D.; Xia, H.; Wu, S.; Ma, F. Genome-wide identification and expression profiling of dehydrin gene family in Malus domestica. Mol. Biol. Rep 2012, 39, 10759–10768. [Google Scholar]
- Zhao, T.; Liang, D.; Wang, P.; Liu, J.; Ma, F. Genome-wide analysis and expression profiling of the DREB transcription factor gene family in Malus under abiotic stress. Mol. Genet. Genomics 2012, 287, 423–436. [Google Scholar]
- Agalou, A.; Purwantomo, S.; Oevernaes, E.; Johannesson, H.; Zhu, X.; Estiati, A.; de Kam, R.J.; Engstroem, P.; Slamet-Loedin, I.H.; Zhu, Z.; et al. A genome-wide survey of HD-Zip genes in rice and analysis of drought-responsive family members. Plant Mol. Biol 2008, 66, 87–103. [Google Scholar]
- Agarwal, P.; Arora, R.; Ray, S.; Singh, A.K.; Singh, V.P.; Takatsuji, H.; Kapoor, S.; Tyagi, A.K. Genome-wide identification of C2H2 zinc-finger gene family in rice and their phylogeny and expression analysis. Plant Mol. Biol 2007, 65, 467–485. [Google Scholar]
- Amrutha, R.N.; Sekhar, P.N.; Varshney, R.K.; Kishor, P.B.K. Genome-wide analysis and identification of genes related to potassium transporter families in rice (Oryza sativa L.). Plant Sci 2007, 172, 708–721. [Google Scholar]
- Chen, R.; Jiang, Y.; Dong, J.; Zhang, X.; Xiao, H.; Xu, Z.; Gao, X. Genome-wide analysis and environmental response profiling of SOT family genes in rice (Oryza sativa). Genes Genomics 2012, 34, 549–560. [Google Scholar]
- Ding, X.; Hou, X.; Xie, K.; Xiong, L. Genome-wide identification of BURP domain-containing genes in rice reveals a gene family with diverse structures and responses to abiotic stresses. Planta 2009, 230, 149–163. [Google Scholar]
- Gollan, P.J.; Bhave, M. Genome-wide analysis of genes encoding FK506-binding proteins in rice. Plant Mol. Biol 2010, 72, 1–16. [Google Scholar]
- Huang, J.; Zhao, X.; Yu, H.; Ouyang, Y.; Wang, L.; Zhang, Q. The ankyrin repeat gene family in rice: Genome-wide identification, classification and expression profiling. Plant Mol. Biol 2009, 71, 207–226. [Google Scholar]
- Jain, M.; Tyagi, A.K.; Khurana, J.P. Genome-wide identification, classification, evolutionary expansion and expression analyses of homeobox genes in rice. FEBS J 2008, 275, 2845–2861. [Google Scholar]
- Jiang, S.-Y.; Ramamoorthy, R.; Bhalla, R.; Luan, H.-F.; Venkatesh, P.N.; Cai, M.; Ramachandran, S. Genome-wide survey of the RIP domain family in Oryza sativa and their expression profiles under various abiotic and biotic stresses. Plant Mol. Biol 2008, 67, 603–614. [Google Scholar]
- Nuruzzaman, M.; Manimekalai, R.; Sharoni, A.M.; Satoh, K.; Kondoh, H.; Ooka, H.; Kikuchi, S. Genome-wide analysis of NAC transcription factor family in rice. Gene 2010, 465, 30–44. [Google Scholar]
- Nuruzzaman, M.; Sharoni, A.M.; Satoh, K.; Al-Shammari, T.; Shimizu, T.; Sasaya, T.; Omura, T.; Kikuchi, S. The thioredoxin gene family in rice: Genome-wide identification and expression profiling under different biotic and abiotic treatments. Biochem. Biophys. Res. Commun 2012, 423, 417–423. [Google Scholar]
- Ouyang, Y.; Chen, J.; Xie, W.; Wang, L.; Zhang, Q. Comprehensive sequence and expression profile analysis of Hsp20 gene family in rice. Plant. Mol. Biol 2009, 70, 341–357. [Google Scholar]
- Vij, S.; Giri, J.; Dansana, P.K.; Kapoor, S.; Tyagi, A.K. The receptor-like cytoplasmic kinase (OsRLCK) gene family in rice: Organization, phylogenetic relationship, and expression during development and stress. Mol. Plant 2008, 1, 732–750. [Google Scholar]
- Wang, D.; Guo, Y.; Wu, C.; Yang, G.; Li, Y.; Zheng, C. Genome-wide analysis of CCCH zinc finger family in Arabidopsis and rice. BMC Genomics 2008, 9, 44. [Google Scholar]
- Zhao, H.; Ma, H.; Yu, L.; Wang, X.; Zhao, J. Genome-wide survey and expression analysis of amino acid transporter gene family in rice (Oryza sativa L.). PLoS One 2012, 7, e49210. [Google Scholar]
- Wu, J.; Peng, Z.; Liu, S.; He, Y.; Cheng, L.; Kong, F.; Wang, J.; Lu, G. Genome-wide analysis of Aux/IAA gene family in Solanaceae species using tomato as a model. Mol. Genet. Genomics 2012, 287, 295–311. [Google Scholar]
- Bai, M.; Yang, G.-S.; Chen, W.-T.; Mao, Z.-C.; Kang, H.-X.; Chen, G.-H.; Yang, Y.-H.; Xie, B.-Y. Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene 2012, 501, 52–62. [Google Scholar]
- Huang, S.; Gao, Y.; Liu, J.; Peng, X.; Niu, X.; Fei, Z.; Cao, S.; Liu, Y. Genome-wide analysis of WRKY transcription factors in Solanum lycopersicum. Mol. Genet. Genomics 2012, 287, 495–513. [Google Scholar]
- Kong, F.; Wang, J.; Cheng, L.; Liu, S.; Wu, J.; Peng, Z.; Lu, G. Genome-wide analysis of the mitogen-activated protein kinase gene family in Solanum lycopersicum. Gene 2012, 499, 108–120. [Google Scholar]
- Gan, D.; Jiang, H.; Zhang, J.; Zhao, Y.; Zhu, S.; Cheng, B. Genome-wide analysis of BURP domain-containing genes in Maize and Sorghum. Mol. Biol. Rep 2011, 38, 4553–4563. [Google Scholar]
- Vannozzi, A.; Dry, I.B.; Fasoli, M.; Zenoni, S.; Lucchin, M. Genome-wide analysis of the grapevine stilbene synthase multigenic family: Genomic organization and expression profiles upon biotic and abiotic stresses. BMC Plant Biol 2012, 12, 130. [Google Scholar]
- Zhuang, J.; Peng, R.-H.; Cheng, Z.-M.; Zhang, J.; Cai, B.; Zhang, Z.; Gao, F.; Zhu, B.; Fu, X.-Y.; Jin, X.-F.; et al. Genome-wide analysis of the putative AP2/ERF family genes in Vitis vinifera. Sci. Hortic 2009, 123, 73–81. [Google Scholar]
- Cheng, Y.; Li, X.; Jiang, H.; Ma, W.; Miao, W.; Yamada, T.; Zhang, M. Systematic analysis and comparison of nucleotide-binding site disease resistance genes in maize. FEBS J 2012, 279, 2431–2443. [Google Scholar]
- Gomez-Anduro, G.; Adriana Ceniceros-Ojeda, E.; Edith Casados-Vazquez, L.; Bencivenni, C.; Sierra-Beltran, A.; Murillo-Amador, B.; Tiessen, A. Genome-wide analysis of the beta-glucosidase gene family in maize (Zea mays L. var B73). Plant Mol. Biol 2011, 77, 159–183. [Google Scholar]
- Lin, Y.-X.; Jiang, H.-Y.; Chu, Z.-X.; Tang, X.-L.; Zhu, S.-W.; Cheng, B.-J. Genome-wide identification, classification and analysis of heat shock transcription factor family in maize. BMC Genomics 2011, 12, 76. [Google Scholar]
- Peng, X.; Zhao, Y.; Cao, J.; Zhang, W.; Jiang, H.; Li, X.; Ma, Q.; Zhu, S.; Cheng, B. CCCH-type zinc finger family in Maize: Genome-wide identification, classification and expression profiling under abscisic acid and drought treatments. PLoS One 2012, 7, e40120. [Google Scholar]
- Wang, W.W.; Ma, Q.; Xiang, Y.; Zhu, S.W.; Cheng, B.J. Genome-wide analysis of immunophilin FKBP genes and expression patterns in Zea mays. Genet. Mol. Res 2012, 11, 1690–1700. [Google Scholar]
- Wei, K.-F.; Chen, J.; Chen, Y.-F.; Wu, L.-J.; Xie, D.-X. Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in Maize. DNA Res 2012, 19, 153–164. [Google Scholar]
- Zhang, Z.; Zhang, J.; Chen, Y.; Li, R.; Wang, H.; Wei, J. Genome-wide analysis and identification of HAK potassium transporter gene family in maize (Zea mays L.). Mol. Biol. Rep 2012, 39, 8465–8473. [Google Scholar]
- Zhou, M.-L.; Zhang, Q.; Zhou, M.; Sun, Z.-M.; Zhu, X.-M.; Shao, J.-R.; Tang, Y.-X.; Wu, Y.-M. Genome-wide identification of genes involved in raffinose metabolism in Maize. Glycobiology 2012, 22, 1775–1785. [Google Scholar]
- Wendelboe-Nelson, C.; Morris, P.C. Proteins linked to drought tolerance revealed by DIGE analysis of drought resistant and susceptible barley varieties. Proteomics 2012, 12, 3374–3385. [Google Scholar]
- Fatehi, F.; Hosseinzadeh, A.; Alizadeh, H.; Brimavandi, T.; Struik, P.C. The proteome response of salt-resistant and salt-sensitive barley genotypes to long-term salinity stress. Mol. Biol. Rep 2012, 39, 6387–6397. [Google Scholar]
- Cheng, Z.Y.; Woody, O.Z.; McConkey, B.J.; Glick, B.R. Combined effects of the plant growth-promoting bacterium Pseudomonas putida UW4 and salinity stress on the Brassica napus proteome. Appl. Soil Ecol 2012, 61, 255–263. [Google Scholar]
- Louarn, S.; Nawrocki, A.; Edelenbos, M.; Jensen, D.F.; Jensen, O.N.; Collinge, D.B.; Jensen, B. The influence of the fungal pathogen Mycocentrospora acerina on the proteome and polyacetylenes and 6-methoxymellein in organic and conventionally cultivated carrots (Daucus carota) during post harvest storage. J. Proteomics 2012, 75, 962–977. [Google Scholar]
- Deeba, F.; Pandey, A.K.; Ranjan, S.; Mishra, A.; Singh, R.; Sharma, Y.K.; Shirke, P.A.; Pandey, V. Physiological and proteomic responses of cotton (Gossypium herbaceum L.) to drought stress. Plant Physiol. Biochnol 2012, 53, 6–18. [Google Scholar]
- Zheng, M.; Wang, Y.H.; Liu, K.; Shu, H.M.; Zhou, Z.G. Protein expression changes during cotton fiber elongation in response to low temperature stress. J. Plant Physiol 2012, 169, 399–409. [Google Scholar]
- Wang, Y.H.; Zheng, M.; Gao, X.B.; Zhou, Z.G. Protein differential expression in the elongating cotton (Gossypium hirsutum L.) fiber under nitrogen stress. Sci. China Life Sci 2012, 55, 984–992. [Google Scholar]
- Li, J.; Sun, J.; Yang, Y.J.; Guo, S.R.; Glick, B.R. Identification of hypoxic-responsive proteins in cucumber roots using a proteomic approach. Plant Physiol. Biochnol 2012, 51, 74–80. [Google Scholar]
- Palmieri, M.C.; Perazzolli, M.; Matafora, V.; Moretto, M.; Bachi, A.; Pertot, I. Proteomic analysis of grapevine resistance induced by Trichoderma harzianum T39 reveals specific defence pathways activated against downy mildew. J. Exp. Bot 2012, 63, 6237–6251. [Google Scholar]
- Wang, Z.; Zhao, F.X.; Zhao, X.; Ge, H.; Chai, L.J.; Chen, S.W.; Perl, A.; Ma, H.Q. Proteomic analysis of berry-sizing effect of GA3 on seedless Vitis vinifera L. Proteomics 2012, 12, 86–94. [Google Scholar]
- Minas, I.S.; Tanou, G.; Belghazi, M.; Job, D.; Manganaris, G.A.; Molassiotis, A.; Vasilakakis, M. Physiological and proteomic approaches to address the active role of ozone in kiwifruit post-harvest ripening. J. Exp. Bot 2012, 63, 2449–2464. [Google Scholar]
- Huang, H.; Moller, I.M.; Song, S.Q. Proteomics of desiccation tolerance during development and germination of maize embryos. J. Proteomics 2012, 75, 1247–1262. [Google Scholar]
- Benesova, M.; Hola, D.; Fischer, L.; Jedelsky, P.L.; Hnilicka, F.; Wilhelmova, N.; Rothova, O.; Kocova, M.; Prochazkova, D.; Honnerova, J.; et al. The physiology and proteomics of drought tolerance in Maize: Early stomatal closure as a cause of lower tolerance to short-term dehydration? PLoS One 2012, 7, e38017. [Google Scholar]
- Fristedt, R.; Wasilewska, W.; Romanowska, E.; Vener, A.V. Differential phosphorylation of thylakoid proteins in mesophyll and bundle sheath chloroplasts from maize plants grown under low or high light. Proteomics 2012, 12, 2852–2861. [Google Scholar]
- Muneer, S.; Kim, T.H.; Qureshi, M.I. Fe modulates Cd-induced oxidative stress and the expression of stress responsive proteins in the nodules of Vigna radiata. Plant Growth Regul 2012, 68, 421–433. [Google Scholar]
- Rodrigues, S.P.; Ventura, J.A.; Aguilar, C.; Nakayasu, E.S.; Choi, H.; Sobreira, T.J.P.; Nohara, L.L.; Wermelinger, L.S.; Almeida, I.C.; Zingali, R.B.; et al. Label-free quantitative proteomics reveals differentially regulated proteins in the latex of sticky diseased Carica papaya L. plants. J. Proteomics 2012, 75, 3191–3198. [Google Scholar]
- Mohammadi, P.P.; Moieni, A.; Komatsu, S. Comparative proteome analysis of drought-sensitive and drought-tolerant rapeseed roots and their hybrid F1 line under drought stress. Amino Acids 2012, 43, 2137–2152. [Google Scholar]
- Zhu, M.M.; Dai, S.J.; Zhu, N.; Booy, A.; Simons, B.; Yi, S.; Chen, S.X. Methyl jasmonate responsive proteins in Brassica napus guard cells revealed by iTRAQ-based quantitative proteomics. J. Proteome Res 2012, 11, 3728–3742. [Google Scholar]
- Chen, J.H.; Tian, L.; Xu, H.F.; Tian, D.G.; Luo, Y.M.; Ren, C.M.; Yang, L.M.; Shi, J.S. Cold-induced changes of protein and phosphoprotein expression patterns from rice roots as revealed by multiplex proteomic analysis. Plant Omics 2012, 5, 194–199. [Google Scholar]
- Ji, K.X.; Wang, Y.Y.; Sun, W.N.; Lou, Q.J.; Mei, H.W.; Shen, S.H.; Chen, H. Drought-responsive mechanisms in rice genotypes with contrasting drought tolerance during reproductive stage. J. Plant Physiol 2012, 169, 336–344. [Google Scholar]
- Mirzaei, M.; Soltani, N.; Sarhadi, E.; Pascovici, D.; Keighley, T.; Salekdeh, G.H.; Haynes, P.A.; Atwell, B.J. Shotgun proteomic analysis of long-distance drought signaling in rice roots. J. Proteome Res 2012, 11, 348–358. [Google Scholar]
- Koga, H.; Dohi, K.; Nishiuchi, T.; Kato, T.; Takahara, H.; Mori, M.; Komatsu, S. Proteomic analysis of susceptible rice plants expressing the whole plant-specific resistance against Magnaporthe oryzae: Involvement of a thaumatin-like protein. Physiol. Mol. Plant P 2012, 77, 60–66. [Google Scholar]
- Li, Y.F.; Zhang, Z.H.; Nie, Y.F.; Zhang, L.H.; Wang, Z.Z. Proteomic analysis of salicylic acid-induced resistance to Magnaporthe oryzae in susceptible and resistant rice. Proteomics 2012, 12, 2340–2354. [Google Scholar]
- Hakeem, K.R.; Chandna, R.; Ahmad, A.; Qureshi, M.I.; Iqbal, M. Proteomic analysis for low and high nitrogen-responsive proteins in the leaves of rice genotypes grown at three nitrogen levels. Appl. Biochem. Biotechnol 2012, 168, 834–850. [Google Scholar]
- Sawada, H.; Komatsu, S.; Nanjo, Y.; Khan, N.A.; Kohno, Y. Proteomic analysis of rice response involved in reduction of grain yield under elevated ozone stress. Environ. Exp. Bot 2012, 77, 108–116. [Google Scholar]
- Wang, Y.D.; Wang, X.; Wong, Y.S. Proteomics analysis reveals multiple regulatory mechanisms in response to selenium in rice. J. Proteomics 2012, 75, 1849–1866. [Google Scholar]
- Li, D.X.; Wang, L.J.; Teng, S.L.; Zhang, G.G.; Guo, L.J.; Mao, Q.; Wang, W.; Li, M.; Chen, L. Proteomics analysis of rice proteins up-regulated in response to bacterial leaf streak disease. J. Plant Biol 2012, 55, 316–324. [Google Scholar]
- Ngara, R.; Ndimba, R.; Borch-Jensen, J.; Jensen, O.N.; Ndimba, B. Identification and profiling of salinity stress-responsive proteins in Sorghum bicolor seedlings. J. Proteomics 2012, 75, 4139–4150. [Google Scholar]
- Hossain, Z.; Hajika, M.; Komatsu, S. Comparative proteome analysis of high and low cadmium accumulating soybeans under cadmium stress. Amino Acids 2012, 43, 2393–2416. [Google Scholar]
- Mohammadi, P.P.; Moieni, A.; Hiraga, S.; Komatsu, S. Organ-specific proteomic analysis of drought-stressed soybean seedlings. J. Proteomics 2012, 75, 1906–1923. [Google Scholar]
- Salavati, A.; Khatoon, A.; Nanjo, Y.; Komatsu, S. Analysis of proteomic changes in roots of soybean seedlings during recovery after flooding. J. Proteomics 2012, 75, 878–893. [Google Scholar]
- Yanagawa, Y.; Komatsu, S. Ubiquitin/proteasome-mediated proteolysis is involved in the response to flooding stress in soybean roots, independent of oxygen limitation. Plant Sci 2012, 185, 250–258. [Google Scholar]
- Khatoon, A.; Rehman, S.; Salavati, A.; Komatsu, S. A comparative proteomics analysis in roots of soybean to compatible symbiotic bacteria under flooding stress. Amino Acids 2012, 43, 2513–2525. [Google Scholar]
- Wang, L.Q.; Ma, H.; Song, L.R.; Shu, Y.J.; Gu, W.H. Comparative proteomics analysis reveals the mechanism of pre-harvest seed deterioration of soybean under high temperature and humidity stress. J. Proteomics 2012, 75, 2109–2127. [Google Scholar]
- Wang, Y.; Yuan, X.Z.; Hu, H.; Liu, Y.; Sun, W.H.; Shan, Z.H.; Zhou, X.A. Proteomic analysis of differentially expressed proteins in resistant soybean leaves after Phakopsora pachyrhizi infection. J. Phytopathol 2012, 160, 554–560. [Google Scholar]
- Ma, H.; Song, L.; Shu, Y.; Wang, S.; Niu, J.; Wang, Z.; Yu, T.; Gu, W.; Ma, H. Comparative proteomic analysis of seedling leaves of different salt tolerant soybean genotypes. J. Proteomics 2012, 75, 1529–1546. [Google Scholar]
- Khatoon, A.; Rehman, S.; Hiraga, S.; Makino, T.; Komatsu, S. Organ-specific proteomics analysis for identification of response mechanism in soybean seedlings under flooding stress. J. Proteomics 2012, 75, 5706–5723. [Google Scholar]
- Nanjo, Y.; Skultety, L.; Uvackova, L.; Kubicova, K.; Hajduch, M.; Komatsu, S. Mass spectrometry-based analysis of proteomic changes in the root tips of flooded soybean seedlings. J. Proteome Res 2012, 11, 372–385. [Google Scholar]
- Khatoon, A.; Rehman, S.; Oh, M.W.; Woo, S.H.; Komatsu, S. Analysis of response mechanism in soybean under low oxygen and flooding stresses using gel-base proteomics technique. Mol. Biol. Rep 2012, 39, 10581–10594. [Google Scholar]
- Koehler, G.; Wilson, R.C.; Goodpaster, J.V.; Sonsteby, A.; Lai, X.; Witzmann, F.A.; You, J.S.; Rohloff, J.; Randall, S.K.; Alsheikh, M. Proteomic study of low-temperature responses in strawberry cultivars (Fragaria x ananassa) that differ in cold tolerance. Plant Physiol 2012, 159, 1787–1805. [Google Scholar]
- Fang, X.P.; Chen, W.Y.; Xin, Y.; Zhang, H.M.; Yan, C.Q.; Yu, H.; Liu, H.; Xiao, W.F.; Wang, S.Z.; Zheng, G.Z.; et al. Proteomic analysis of strawberry leaves infected with Colletotrichum fragariae. J. Proteomics 2012, 75, 4074–4090. [Google Scholar]
- Zhou, G.; Yang, L.T.; Li, Y.R.; Zou, C.L.; Huang, L.P.; Qiu, L.H.; Huang, X.; Srivastava, M.K. Proteomic analysis of osmotic stress-responsive proteins in sugarcane leaves. Plant Mol. Biol. Rep 2012, 30, 349–359. [Google Scholar]
- Shah, P.; Powell, A.L.T.; Orlando, R.; Bergmann, C.; Gutierrez-Sanchez, G. Proteomic analysis of ripening tomato fruit infected by Botrytis cinerea. J. Proteome Res 2012, 11, 2178–2192. [Google Scholar]
- Ge, P.; Ma, C.; Wang, S.; Gao, L.; Li, X.; Guo, G.; Ma, W.; Yan, Y. Comparative proteomic analysis of grain development in two spring wheat varieties under drought stress. Anal. Bioanal. Chem 2012, 402, 1297–1313. [Google Scholar]
- Kang, G.Z.; Li, G.Z.; Xu, W.; Peng, X.Q.; Han, Q.X.; Zhu, Y.J.; Guo, T.C. Proteomics reveals the effects of salicylic acid on growth and tolerance to subsequent drought stress in wheat. J. Proteome Res 2012, 11, 6066–6079. [Google Scholar]
- Vitamvas, P.; Prasil, I.T.; Kosova, K.; Planchon, S.; Renaut, J. Analysis of proteome and frost tolerance in chromosome 5A and 5B reciprocal substitution lines between two winter wheats during long-term cold acclimation. Proteomics 2012, 12, 68–85. [Google Scholar]
- Gunnaiah, R.; Kushalappa, A.C.; Duggavathi, R.; Fox, S.; Somers, D.J. Integrated metabolo-proteomic approach to decipher the mechanisms by which wheat QTL (Fhb1) contributes to resistance against Fusarium graminearum. PLoS One 2012, 7, e40695. [Google Scholar]
- Ravalason, H.; Grisel, S.; Chevret, D.; Favel, A.; Berrin, J.G.; Sigoillot, J.C.; Herpoel-Gimbert, I. Fusarium verticillioides secretome as a source of auxiliary enzymes to enhance saccharification of wheat straw. Bioresour. Technol 2012, 114, 589–596. [Google Scholar]
- Kang, G.Z.; Li, G.Z.; Zheng, B.B.; Han, Q.X.; Wang, C.Y.; Zhu, Y.J.; Guo, T.C. Proteomic analysis on salicylic acid-induced salt tolerance in common wheat seedlings (Triticum aestivum L.). Biochim. Biophys. Acta 2012, 1824, 1324–1333. [Google Scholar]

{kind=link}
| Database name | URL | Reference |
|---|---|---|
| GenBank | http://www.ncbi.nlm.nih.gov/genbank/ | [4] |
| ENA | http://www.ebi.ac.uk/ena/ | [5] |
| DDBJ | http://www.ddbj.nig.ac.jp/ | [6] |
| Phytozome | http://www.phytozome.net/ | [7] |
| Gramene | http://www.gramene.org/ | [8] |
| KEGG | http://www.genome.jp/kegg/ | [9] |
| PlantGDB | http://www.plantgdb.org/ | [10] |
| EnsemblPlants | http://plants.ensembl.org/index.html | [11] |
| VISTA | http://genome.lbl.gov/vista/index.shtml | [12] |
| PLAZA | http://bioinformatics.psb.ugent.be/plaza/ | [13] |
| GigaDB | http://gigadb.org/ | [14] |
| SGN | http://solgenomics.net/ | [15] |
| GrainGenes | http://wheat.pw.usda.gov | [16] |
| LIS | http://www.comparative-legumes.org/ | [17] |
| Crop | Database name | URL of related database | Ref |
|---|---|---|---|
| Rice | RAP-DB | http://rapdb.dna.affrc.go.jp/ | [23] |
| Maize | MaizeGDB | http://www.maizegdb.org/ | [24] |
| Medicago | Medicago truncatula SEQUENCING RESOURCE | http://www.medicago.org/genome/index_old.php | - |
| Wheat | GrainGenes | http://wheat.pw.usda.gov/ | [16] |
| Potato | Solanaceae Genomics Resource | http://solanaceae.plantbiology.msu.edu/index.shtml | - |
| Soybean | SoyBase | http://soybase.org | [25] |
| Tomato | TOMATO FUNCTIONAL GENOMICS DATABASE | http://ted.bti.cornell.edu/ | [26] |
| Domain | Prefix | Description | Reference/website |
|---|---|---|---|
| Plant Environmental Conditions | EO | Controlled vocabulary for the representation of plant environmental conditions | http://www.gramene.org/db/ontology/search?id=EO:0007359 |
| Gene Ontology | GO | Controlled vocabulary for genes and gene products | [28] |
| Taxonomy Ontology | GR_tax | Representation of the taxonomic tree of plants in the ontology format | http://www.gramene.org/db/ontology/search?id=GR_tax:090165 |
| The Plant-Associated Microbe Gene Ontology | PAMGO | Controlled vocabulary for the interaction of microbes with their hosts | [29] |
| Plant Ontology | PO | Controlled vocabulary for anatomy, morphology and stages of development for all plants | [30] |
| Sequence Ontology | SO | Controlled vocabulary for sequence annotations, for the exchange of annotation data and for the description of sequence objects in databases | [31] |
| Plant Trait Ontology | TO | Controlled vocabulary for phenotypic traits in plants | http://www.gramene.org/db/ontology/search?id=TO:0000387 |
| Software | Description | Download URL | Reference |
|---|---|---|---|
| ABMapper | RNA-seq data alignment | http://hkbic.cuhk.edu.hk/software/abmapper | [91] |
| Bowtie | RNA-seq data alignment | http://bowtie-io.sourceforge.net/bowtie2/index.shtml | [92] |
| Cufflinks | Transcript assembly | http://cufflinks.cbcb.umd.edu/ | [93] |
| DEGseq | Differential gene expression detection | http://www.bioconductor.org/packages/2.11/bioc/html/DEGseq.html | [94] |
| Infernal | RNA-seq data alignment | http://infernal.janelia.org/ | [95] |
| Oases | De novo assembly | www.ebi.ac.uk/~zerbino/oases/ | [96] |
| Tophat | RNA-seq data alignment & Alternative splicing detection | http://tophat.cbcb.umd.edu/ | [97] |
| Trans-AByss | De novo assembly | http://www.bcgsc.ca/platform/bioinfo/software/ | [98] |
| Trinity | De novo assembly | http://trinityrnaseq.sourceforge.net/ | [99] |
| Species | Stress | Strategy | Reference |
|---|---|---|---|
| Rice | Abiotic and biotic | Using stress components as bait in Y2H | [142,148] |
| Wheat | Cold and dehydration | Using stress components as bait in Y2H | [149] |
| Soybean | SCN infection | In silico prediction | [150] |
| Tools | Descriptions | Reference |
|---|---|---|
| BiQ Analyzer HT | Quantitative study of locus-specific DNA methylation patterns from bisulfite sequencing data. | [166] |
| Bismark | Mapping of bisulfite-sequencing reads and methylation calling. | [167] |
| BRAT-BW | Genome-wide single base-resolution methylation data analysis | [168] |
| BSmooth | Providing estimate of methylation profiles with low-coverage whole-genome bisulfite-sequencing data. | [169] |
| BS seeker | Mapping of bisulfite-sequencing reads. | [170] |
| BSMAP | Bisulfite reads mapping algorithm. | [171] |
| CpG_MPs | Analysis of biosulfite-sequencing read and identification of genome-wide methylation pattern | [172] |
| GBSA | Both gene-centric or gene-independent analyses of whole-genome bisulfite sequencing data | [173] |
| Kismeth | Analysis of plant bisulfite sequencing results, with a tool for designing bisulfite sequencing primers. | [174] |
| QUMA | Quantification tool for methylation analysis | [175] |
| RRBSMAP | Derivative of BSMAP—a specific tool for reduced-representation bisulfite sequencing | [176] |
| Species | Databases | URL |
|---|---|---|
| Barley | Barley DB | http://www.shigen.nig.ac.jp/barley/ |
| Barley | NordGen Plant Collection | http://www.nordgen.org |
| Maize | RescueMu Maize Mutant Phenotype Database | http://maizegdb.org/rescuemu-phenotype.php |
| Rice | Oryza Tag Line | http://oryzatagline.cirad.fr/ |
| Rice | Rice Tos17 Insertion Mutant Database | http://pfg101.nias.affrc.go.jp/ |
| Soybean | SoyBase—Fast Neutron Mutants | http://www.soybase.org/mutants/index.php |
| Tomato | Genes that Make Tomatoes | http://zamir.sgn.cornell.edu/mutants/index.html |
| Tomato | LycoTILL | http://www.agrobios.it/tilling/index.html |
| Tomato | TOMATOMA | http://tomatoma.nbrp.jp/index.jsp |
| Tomato/Pea | URGV TILLING Database | http://urgv.evry.inra.fr/UTILLdb |
| Tomato/Potato | SOL genomics network | http://solgenomics.net/ |
| Wheat | The Scottish Wheat Variety Database | http://wheat.agricrops.org/menu.php |
| Database | URL | Reference |
|---|---|---|
| AOCS Lipid Library | http://lipidlibrary.aocs.org/index.html | - |
| Golm Metabolome Database | http://gmd.mpimp-golm.mpg.de/ | [205] |
| Lipidomics Gateway | http://www.lipidmaps.org/ | [206] |
| Madison-Qingdao Metabolomics Consortium Database | http://mmcd.nmrfam.wisc.edu/ | [207] |
| Manchester Metabolomics Database | http://dbkgroup.org/MMD/ | [208] |
| MassBank | http://www.massbank.jp/index.html | [209] |
| Metabolome Express | https://www.metabolome-express.org/ | [210] |
| METLIN | http://metlin.scripps.edu/ | [211] |
| NIST Chemistry WebBook | http://webbook.nist.gov/chemistry/#Notes | - |
| Software/Tools | URL | Reference |
| AMDIS | http://www.amdis.net/ | - |
| COLMAR | http://spinportal.magnet.fsu.edu/ | [212] |
| MetDat | http://smbl.nus.edu.sg/METDAT2/ | [213] |
| MetaboSearch | http://omics.georgetown.edu/MetaboSearch.html | [214] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, M.-W.; Qi, X.; Ni, M.; Lam, H.-M. Silicon Era of Carbon-Based Life: Application of Genomics and Bioinformatics in Crop Stress Research. Int. J. Mol. Sci. 2013, 14, 11444-11483. https://doi.org/10.3390/ijms140611444
Li M-W, Qi X, Ni M, Lam H-M. Silicon Era of Carbon-Based Life: Application of Genomics and Bioinformatics in Crop Stress Research. International Journal of Molecular Sciences. 2013; 14(6):11444-11483. https://doi.org/10.3390/ijms140611444
Chicago/Turabian StyleLi, Man-Wah, Xinpeng Qi, Meng Ni, and Hon-Ming Lam. 2013. "Silicon Era of Carbon-Based Life: Application of Genomics and Bioinformatics in Crop Stress Research" International Journal of Molecular Sciences 14, no. 6: 11444-11483. https://doi.org/10.3390/ijms140611444