Targeting Signaling Pathways in Epithelial Ovarian Cancer

 ,
, 
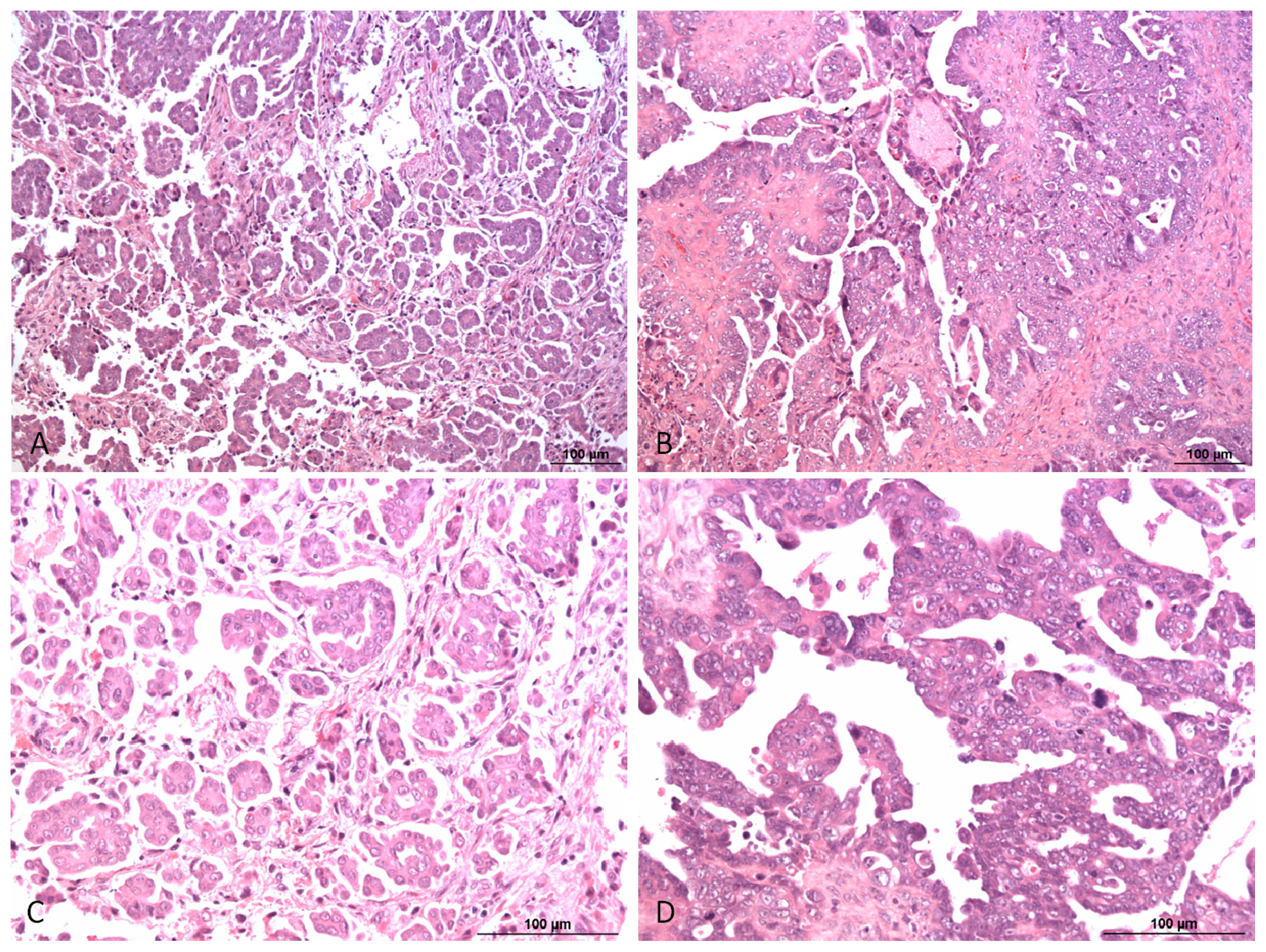
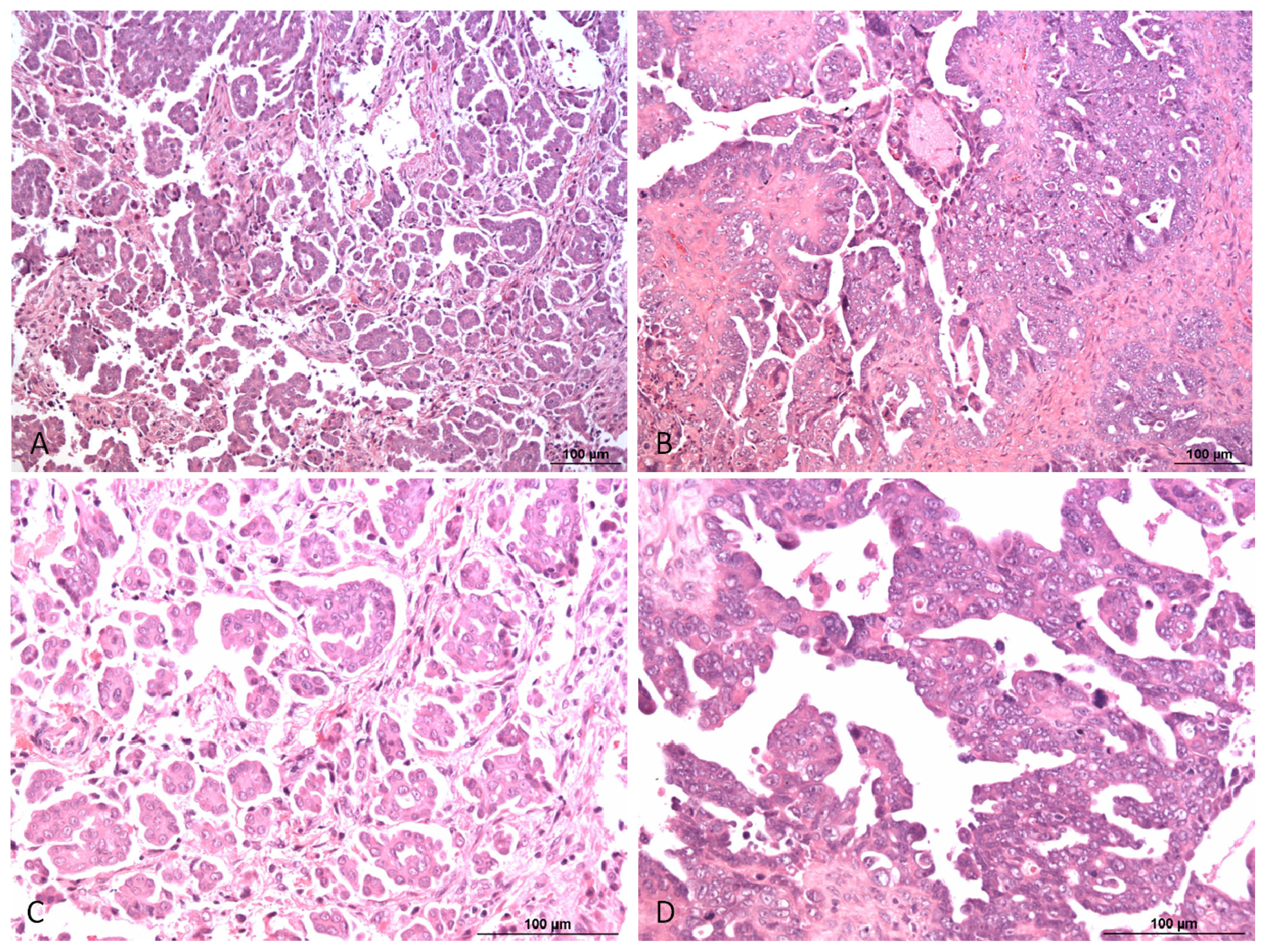{kind=link}
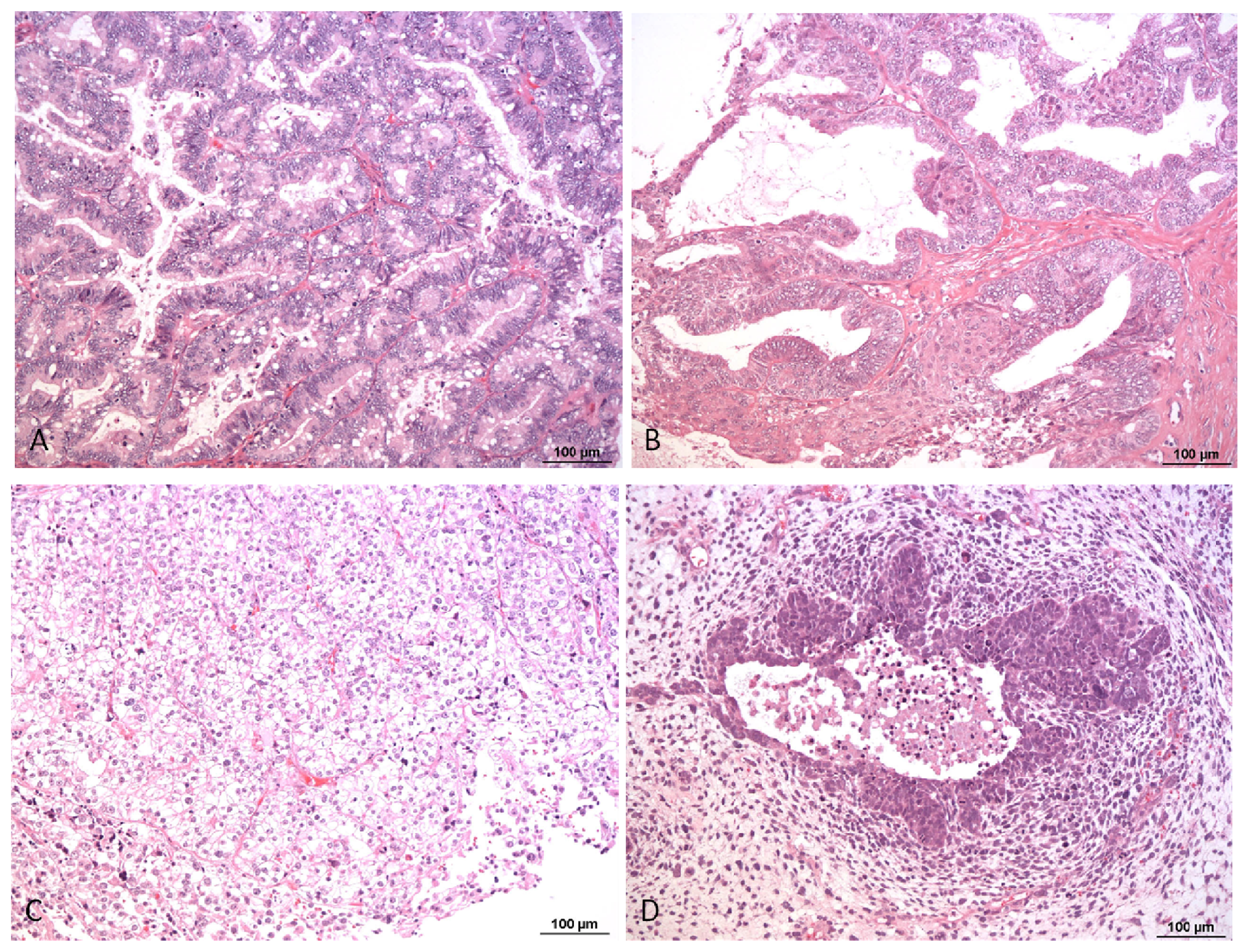{kind=link}
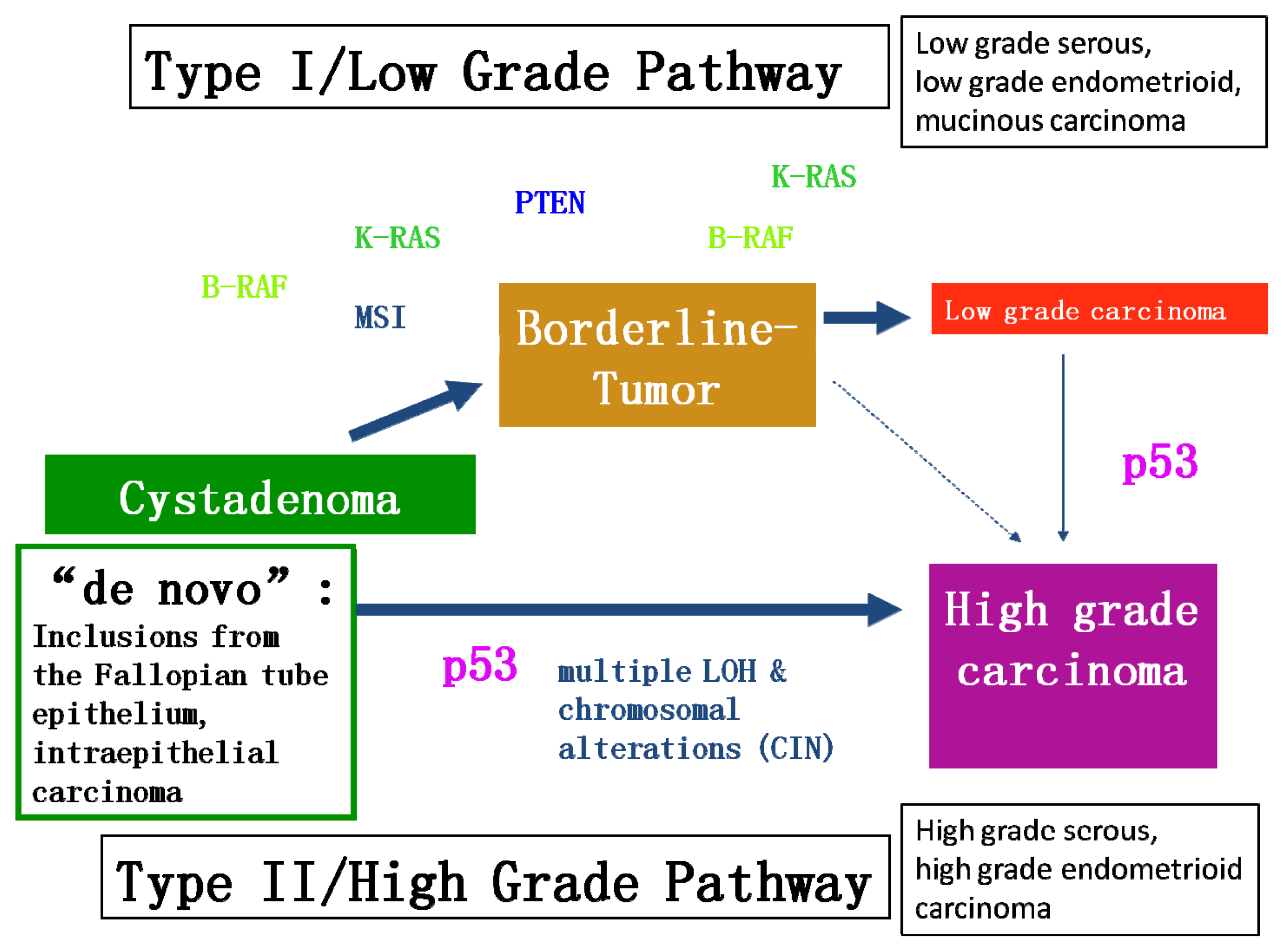{kind=link}
Abstract
:Purpose
In Depth Review of Existing Data
Conclusion
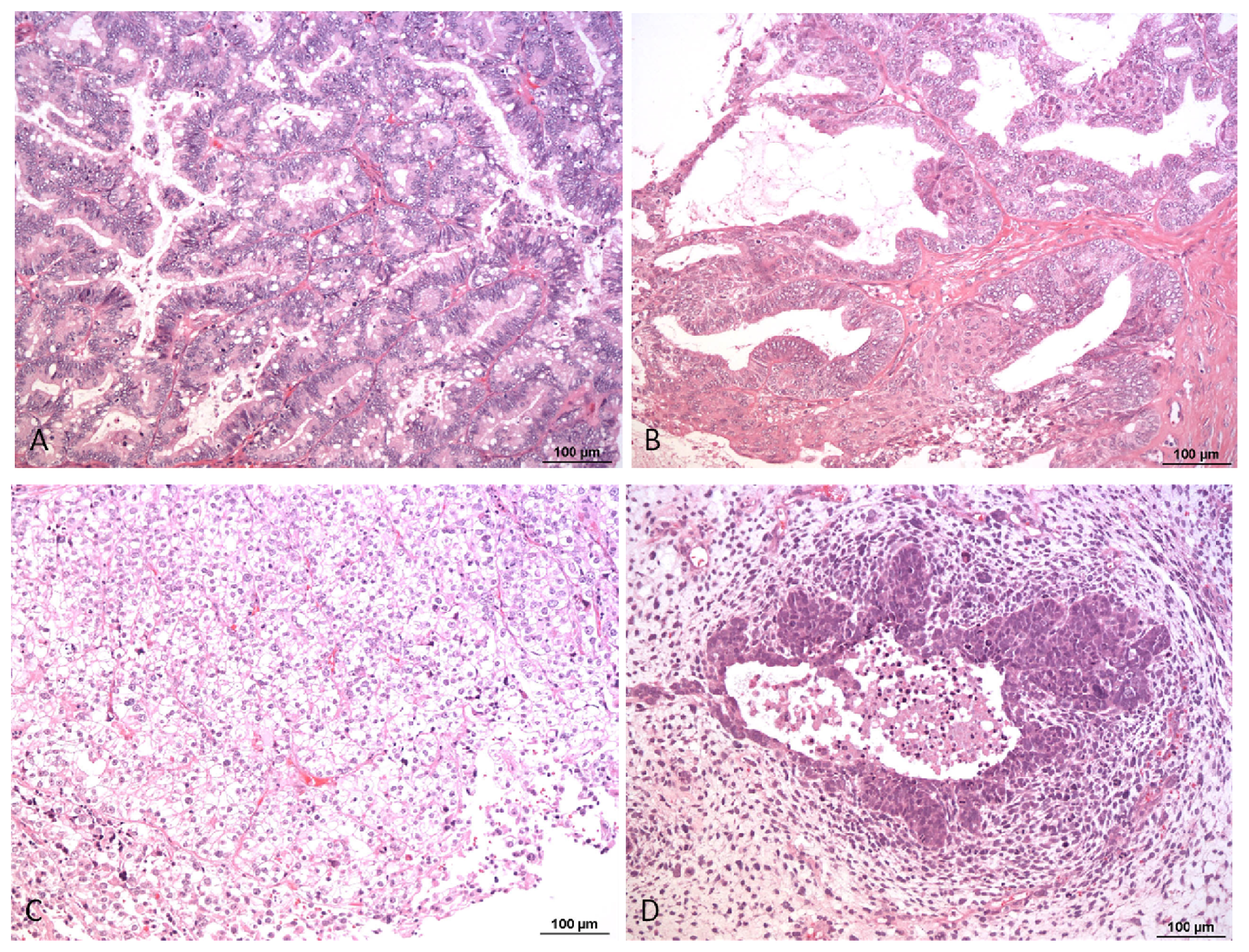
1. Pathology and Biology of Ovarian Carcinoma
2. In Depth Review of Existing Data
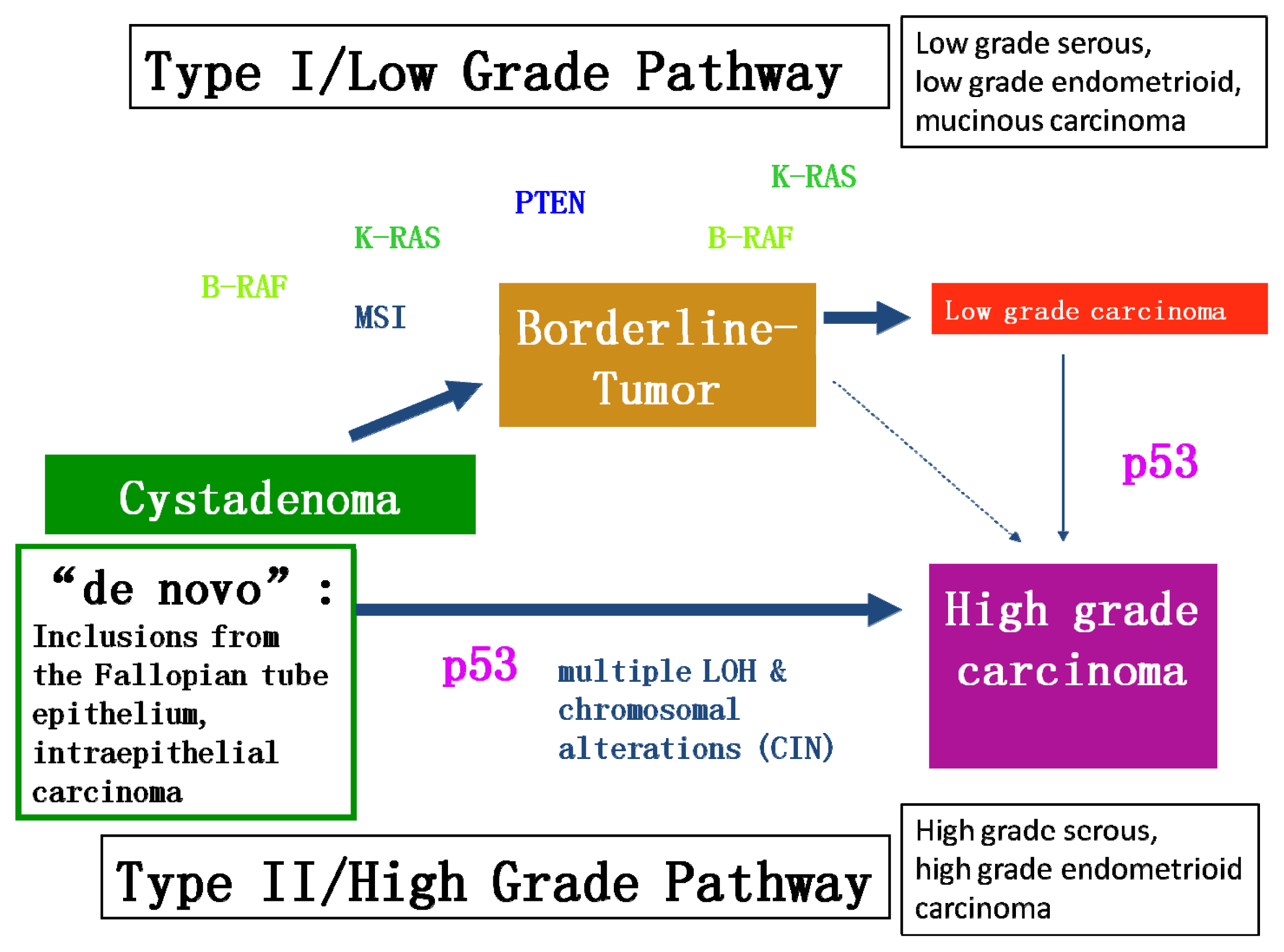
2.1. Different Molecular Genetic Pathways and Putative Molecular Targets in Ovarian Cancer
2.2. Putative Molecular Targets
2.2.1. BRCA1 and BRCA2
2.2.2. KRAS and BRAF Mutations Lead to the Activation of the MAPK/ERK Pathway
2.2.3. EGFR and the Consecutive Activation of AKT
2.2.4. Integrin Inhibitors
2.2.5. GRP78 Expression
2.2.6. The p38alpha Pathway
3. Diagnosis of Ovarian Cancer: Biomarkers and Imaging Techniques
4. Treatment Modalities
5. Discussion and Conclusion
Acknowledgements
References
- McCluggage, W.G. Morphological subtypes of ovarian carcinoma: A review with emphasis on new developments and pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar]
- Shih, I.; Ho, C.M.; Nakayama, K.; Salani, R. Pathogenesis and new therapeutic targets of ovarian cancer. J. Oncol 2012, 2012, 867512. [Google Scholar]
- Kaku, T.; Ogawa, S.; Kawano, Y.; Ohishi, Y.; Kobayashi, H.; Hirakawa, T.; Nakano, H. Histological classification of ovarian cancer. Med. Electron. Microsc 2003, 36, 9–17. [Google Scholar]
- Lax, S. Serous genital carcinoma: Molecular pathogenesis and the role of tubal fimbria. Pathologe 2009, 30, 210–216. [Google Scholar]
- Levanon, K.; Crum, C.; Drapkin, R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J. Clin. Oncol 2008, 26, 5284–5293. [Google Scholar]
- Callahan, M.J.; Crum, C.P.; Medeiros, F.; Kindelberger, D.W.; Elvin, J.A.; Garber, J.E.; Feltmate, C.M.; Berkowitz, R.S.; Muto, M.G. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J. Clin. Oncol 2007, 25, 3985–3990. [Google Scholar]
- Crum, C.P.; McKeon, F.D.; Xian, W. BRCA, the oviduct, and the space and time continuum of pelvic serous carcinogenesis. Int. J. Gynecol. Cancer 2012, 22, S29–S34. [Google Scholar]
- Crum, C.P.; McKeon, F.D.; Xian, W. The oviduct and ovarian cancer: Causality, clinical implications, and “targeted prevention”. Clin. Obstet. Gynecol 2012, 55, 24–35. [Google Scholar]
- Harrison, M.L.; Jameson, C.; Gore, M.E. Mucinous ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 209–214. [Google Scholar]
- Pearce, C.L.; Templeman, C.; Rossing, M.A.; Lee, A.; Near, A.M.; Webb, P.M.; Nagle, C.M.; Doherty, J.A.; Cushing-Haugen, K.L.; Wicklund, K.G.; et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: A pooled analysis of case-control studies. Lancet Oncol 2012, 13, 385–394. [Google Scholar]
- McCluggage, W.G. My approach to and thoughts on the typing of ovarian carcinomas. J. Clin. Pathol 2008, 61, 152–163. [Google Scholar]
- Kurman, R.J.; Shih, I. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol 2011, 42, 918–931. [Google Scholar]
- Kshirsagar, M.; Jiang, W.; Shih, I. DNA damage response is prominent in ovarian high-grade serous carcinomas, especially those with Rsf-1 (HBXAP) overexpression. J. Oncol 2012, 2012, 621685. [Google Scholar]
- Kurman, R.J.; Shih, I. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol 2010, 34, 433–443. [Google Scholar]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med 2010, 363, 1532–1543. [Google Scholar]
- Szabo, C.I.; King, M.C. Inherited breast and ovarian cancer. Hum. Mol. Genet 1995, 4, 1811–1817. [Google Scholar]
- Collins, N.; McManus, R.; Wooster, R.; Mangion, J.; Seal, S.; Lakhani, S.R.; Ormiston, W.; Daly, P.A.; Ford, D.; Easton, D.F. Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12–13. Oncogene 1995, 10, 1673–1675. [Google Scholar]
- Smith, S.A.; Easton, D.F.; Evans, D.G.; Ponder, B.A. Allele losses in the region 17q12–21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat. Genet 1992, 2, 128–131. [Google Scholar]
- Scully, R.; Livingston, D.M. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 2000, 408, 429–432. [Google Scholar]
- Scully, R.; Puget, N.; Vlasakova, K. DNA polymerase stalling, sister chromatid recombination and the BRCA genes. Oncogene 2000, 19, 6176–6183. [Google Scholar]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol 2000, 10, 886–895. [Google Scholar]
- Bau, D.T.; Fu, Y.P.; Chen, S.T.; Cheng, T.C.; Yu, J.C.; Wu, P.E.; Shen, C.Y. Breast cancer risk and the DNA double-strand break end-joining capacity of nonhomologous end-joining genes are affected by BRCA1. Cancer Res 2004, 64, 5013–5019. [Google Scholar]
- Espejel, S.; Franco, S.; Rodriguez-Perales, S.; Bouffler, S.D.; Cigudosa, J.C.; Blasco, M.A. Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. EMBO J 2002, 21, 2207–2219. [Google Scholar]
- Deng, C.X.; Scott, F. Role of the tumor suppressor gene brca1 in genetic stability and mammary gland tumor formation. Oncogene 2000, 19, 1059–1064. [Google Scholar]
- Futreal, P.A.; Liu, Q.; Shattuck-Eidens, D.; Cochran, C.; Harshman, K.; Tavtigian, S.; Bennett, L.M.; Haugen-Strano, A.; Swensen, J.; Miki, Y. BRCA1 mutations in primary breast and ovarian carcinomas. Science 1994, 266, 120–122. [Google Scholar]
- Cleton-Jansen, A.M.; Collins, N.; Lakhani, S.R.; Weissenbach, J.; Devilee, P.; Cornelisse, C.J.; Stratton, M.R. Loss of heterozygosity in sporadic breast tumours at the BRCA2 locus on chromosome 13q12–q13. Br. J. Cancer 1995, 72, 1241–1244. [Google Scholar]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet 2001, 10, 705–713. [Google Scholar]
- Singer, G.; Oldt, R., III; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih, I. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J. Natl. Cancer Inst 2003, 95, 484–486. [Google Scholar]
- Nakayama, K.; Nakayama, N.; Kurman, R.J.; Cope, L.; Pohl, G.; Samuels, Y.; Velculescu, V.E.; Wang, T.L.; Shih, I. Sequence mutations and amplification of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer. Biol. Ther 2006, 5, 779–785. [Google Scholar]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar]
- Olson, J.M.; Hallahan, A.R. P38 MAP kinase: A convergence point in cancer therapy. Trends Mol. Med 2004, 10, 125–129. [Google Scholar]
- Peyssonnaux, C.; Eychene, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar]
- Allen, L.F.; Sebolt-Leopold, J.; Meyer, M.B. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol 2003, 30, 105–116. [Google Scholar]
- Zheng, W.; Lu, J.J.; Luo, F.; Zheng, Y.; Feng, Y.; Felix, J.C.; Lauchlan, S.C.; Pike, M.C. Ovarian epithelial tumor growth promotion by follicle-stimulating hormone and inhibition of the effect by luteinizing hormone. Gynecol. Oncol 2000, 76, 80–88. [Google Scholar]
- Choi, K.C.; Kang, S.K.; Tai, C.J.; Auersperg, N.; Leung, P.C. Follicle-stimulating hormone activates mitogen-activated protein kinase in preneoplastic and neoplastic ovarian surface epithelial cells. J. Clin. Endocrinol. Metab 2002, 87, 2245–2253. [Google Scholar]
- Zeineldin, R.; Muller, C.Y.; Stack, M.S.; Hudson, L.G. Targeting the EGF receptor for ovarian cancer therapy. J. Oncol 2010, 2010, 414676. [Google Scholar]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, S9–S15. [Google Scholar]
- Fontanini, G.; Vignati, S.; Bigini, D.; Mussi, A.; Lucchi, H.; Angeletti, C.A.; Pingitore, R.; Pepe, S.; Basolo, F.; Bevilacqua, G. Epidermal growth factor receptor (EGFr) expression in non-small cell lung carcinomas correlates with metastatic involvement of hilar and mediastinal lymph nodes in the squamous subtype. Eur. J. Cancer 1995, 31A, 178–183. [Google Scholar]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res 2003, 284, 31–53. [Google Scholar]
- Turner, T.; Chen, P.; Goodly, L.J.; Wells, A. EGF receptor signaling enhances in vivo invasiveness of du-145 human prostate carcinoma cells. Clin. Exp. Metastasis 1996, 14, 409–418. [Google Scholar]
- Casanova, M.L.; Larcher, F.; Casanova, B.; Murillas, R.; Fernandez-Acenero, M.J.; Villanueva, C.; Martinez-Palacio, J.; Ullrich, A.; Conti, C.J.; Jorcano, J.L. A critical role for ras-mediated, epidermal growth factor receptor-dependent angiogenesis in mouse skin carcinogenesis. Cancer Res 2002, 62, 3402–3407. [Google Scholar]
- Lu, Z.; Jiang, G.; Blume-Jensen, P.; Hunter, T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell. Biol 2001, 21, 4016–4031. [Google Scholar]
- Perrimon, N. Signalling pathways initiated by receptor protein tyrosine kinases in drosophila. Curr. Opin. Cell Biol 1994, 6, 260–266. [Google Scholar]
- Altomare, D.A.; Wang, H.Q.; Skele, K.L.; de Rienzo, A.; Klein-Szanto, A.J.; Godwin, A.K.; Testa, J.R. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 2004, 23, 5853–5857. [Google Scholar]
- Gao, N.; Flynn, D.C.; Zhang, Z.; Zhong, X.S.; Walker, V.; Liu, K.J.; Shi, X.; Jiang, B.H. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am. J. Physiol. Cell. Physiol 2004, 287, C281–C291. [Google Scholar]
- Sawada, K.; Ohyagi-Hara, C.; Kimura, T.; Morishige, K. Integrin inhibitors as a therapeutic agent for ovarian cancer. J. Oncol 2012, 2012, 915140. [Google Scholar]
- Delie, F.; Petignat, P.; Cohen, M. GRP78 protein expression in ovarian cancer patients and perspectives for a drug-targeting approach. J. Oncol 2012, 2012, 468615. [Google Scholar]
- Grossi, V.; Simone, C. Special agents hunting down women silent killer: The emerging role of the p38alpha kinase. J. Oncol 2012, 2012, 382159. [Google Scholar]
- Badgwell, D.; Bast, R.C., Jr. Early detection of ovarian cancer. Dis. Markers 2007, 23, 397–410. [Google Scholar]
- Bast, R.C., Jr. Early detection of ovarian cancer: New technologies in pursuit of a disease that is neither common nor rare. Trans. Am. Clin. Climatol. Assoc 2004, 115, 233–247, ; discussion 247–248.. [Google Scholar]
- Marret, H. Doppler ultrasonography in the diagnosis of ovarian cysts: Indications, pertinence and diagnostic criteria. J. Gynecol. Obstet. Biol. Reprod. (Paris) 2001, 30, S20–S33. [Google Scholar]
- Vrachnis, N.; Sifakis, S.; Samoli, E.; Kappou, D.; Pavlakis, K.; Iliodromiti, Z.; Botsis, D. Three-dimensional ultrasound and three-dimensional power doppler improve the preoperative evaluation of complex benign ovarian lesions. Clin. Exp. Obstet. Gynecol 2012, 39, 474–478. [Google Scholar]
- Bruchim, I.; Ben-Harim, Z.; Piura, E.; Tepper, R.; Fishman, A. Preoperative clinical and radiological features of metastatic ovarian tumors. Arch. Gynecol. Obstet. 2013. [Google Scholar] [CrossRef]
- Kondalsamy-Chennakesavan, S.; Hackethal, A.; Bowtell, D.; Obermair, A. On behalf of the Australian Ovarian Cancer Study Group. Differentiating stage 1 epithelial ovarian cancer from benign ovarian tumours using a combination of tumour markers HE4, CA125, and CEA and patient’s age. Gynecol. Oncol 2013. [Google Scholar] [CrossRef]
- Leung, F.; Dimitromanolakis, A.; Kobayashi, H.; Diamandis, E.P.; Kulasingam, V. Folate-receptor 1 (FOLR1) protein is elevated in the serum of ovarian cancer patients. Clin. Biochem. 2013. [Google Scholar] [CrossRef]
- Huckabay, H.A.; Wildgen, S.M.; Dunn, R.C. Label-free detection of ovarian cancer biomarkers using whispering gallery mode imaging. Biosens. Bioelectron 2013, 45C, 223–229. [Google Scholar]
- Damania, D.; Roy, H.K.; Kunte, D.; Hurteau, J.A.; Subramanian, H.; Cherkezyan, L.; Krosnjar, N.; Shah, M.; Backman, V. Insights into the field carcinogenesis of ovarian cancer based on the nanocytology of endocervical and endometrial epithelial cells. Int. J. Cancer 2013. [Google Scholar] [CrossRef]
- Han, E.S.; Lin, P.; Wakabayashi, M. Current status on biologic therapies in the treatment of epithelial ovarian cancer. Curr. Treat. Opt. Oncol 2009, 10, 54–66. [Google Scholar]
- Markman, M. Pharmaceutical management of ovarian cancer: Current status. Drugs 2008, 68, 771–789. [Google Scholar]
- Han, E.S.; Monk, B.J. Bevacizumab in the treatment of ovarian cancer. Expert Rev. Anticancer Ther 2007, 7, 1339–1345. [Google Scholar]
- Burges, A.; Schmalfeldt, B. Ovarian cancer: Diagnosis and treatment. Dtsch. Arztebl Int 2011, 108, 635–641. [Google Scholar]
- Neijt, J.P.; ten Bokkel Huinink, W.W.; van der Burg, M.E.; van Oosterom, A.T.; Willemse, P.H.; Vermorken, J.B.; van Lindert, A.C.; Heintz, A.P.; Aartsen, E.; van Lent, M. Long-term survival in ovarian cancer. Mature data from the netherlands joint study group for ovarian cancer. Eur. J. Cancer 1991, 27, 1367–1372. [Google Scholar]
- Copeland, L.J.; Vaccarello, L.; Lewandowski, G.S. Second-look laparotomy in epithelial ovarian cancer. Obstet. Gynecol. Clin. North Am 1994, 21, 155–166. [Google Scholar]
- Rubin, S.C.; Hoskins, W.J.; Hakes, T.B.; Markman, M.; Cain, J.M.; Lewis, J.L., Jr. Recurrence after negative second-look laparotomy for ovarian cancer: Analysis of risk factors. Am. J. Obstet. Gynecol 1988, 159, 1094–1098. [Google Scholar]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med 2003, 348, 203–213. [Google Scholar]
- Armstrong, D.K. Relapsed ovarian cancer: Challenges and management strategies for a chronic disease. Oncologist 2002, 7, 20–28. [Google Scholar]
- Morgan, R.J., Jr; Alvarez, R.D.; Armstrong, D.K.; Burger, R.A.; Castells, M.; Chen, L.M.; Copeland, L.; Crispens, M.A.; Gershenson, D.; Gray, H.; et al. Ovarian cancer, version 3.2012. J. Natl. Compr. Cancer Netw 2012, 10, 1339–1349. [Google Scholar]
- National Comprehensive Cancer Network (NCCN), NCCN Clinical Practice Guidelines in Oncology; National Comprehensive Cancer Network, Inc: Washington, DC, USA, 2006.
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res 2012, 31, 14. [Google Scholar]
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Lisyanskaya, A.S.; Makhson, A.N.; Rolski, J.; et al. Trabectedin plus pegylated liposomal doxorubicin in recurrent ovarian cancer. J. Clin. Oncol 2010, 28, 3107–3114. [Google Scholar]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med 2011, 365, 2484–2496. [Google Scholar]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol 2012, 30, 2039–2045. [Google Scholar]
- Chen, S.S.; Michael, A.; Butler-Manuel, S.A. Advances in the treatment of ovarian cancer: A potential role of antiinflammatory phytochemicals. Discov. Med 2012, 13, 7–17. [Google Scholar]
- Tagawa, T.; Morgan, R.; Yen, Y.; Mortimer, J. Ovarian cancer: Opportunity for targeted therapy. J. Oncol 2012, 2012, 682480. [Google Scholar]
- Mabuchi, S.; Kawase, C.; Altomare, D.A.; Morishige, K.; Sawada, K.; Hayashi, M.; Tsujimoto, M.; Yamoto, M.; Klein-Szanto, A.J.; Schilder, R.J.; et al. MTOR is a promising therapeutic target both in cisplatin-sensitive and cisplatin-resistant clear cell carcinoma of the ovary. Clin. Cancer Res 2009, 15, 5404–5413. [Google Scholar]
- Moroney, J.; Fu, S.; Moulder, S.; Falchook, G.; Helgason, T.; Levenback, C.; Hong, D.; Naing, A.; Wheler, J.; Kurzrock, R. Phase I study of the antiangiogenic antibody bevacizumab and the mTOR/hypoxia-inducible factor inhibitor temsirolimus combined with liposomal doxorubicin: Tolerance and biological activity. Clin. Cancer Res 2012, 18, 5796–5805. [Google Scholar]
- Alvarez, E.A.; Brady, W.E.; Walker, J.; Rotmensch, J.; Zhou, X.C.; Kendrick, J.E.; Yamada, S.D.; Schilder, J.M.; Cohn, D.; Harrison, C.R.; et al. Phase II trial of combination bevacizumab and temsirolimus in the treatment of recurrent or persistent endometrial carcinoma: A gynecologic oncology group study. Gynecol. Oncol 2012, 129, 22–27. [Google Scholar]
- Tewari, K.S. American Society of Clinical Oncology 2012 Annual Meeting: highlights from the gynecologic oncology track. Int. J. Gynecol. Cancer 2012, 22, 1634–1639. [Google Scholar]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.B.; Rustin, G.J.S.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Phase II randomized placebo-controlled study of olaparib (AZD2281) in patients with platinum-sensitive relapsed serous ovarian cancer (PSR SOC). J. Clin. Oncol 2011, 29, 5003. [Google Scholar]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 2011, 12, 852–861. [Google Scholar]
- Sessa, C. Update on PARP1 inhibitors in ovarian cancer. Ann. Oncol. 2011, 22, viii72–viii76. [Google Scholar]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; de Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol 2010, 28, 2512–2519. [Google Scholar]
- Gui, T.; Shen, K. The epidermal growth factor receptor as a therapeutic target in epithelial ovarian cancer. Cancer Epidemiol 2012, 36, 490–496. [Google Scholar]
- Konner, J.; Schilder, R.J.; DeRosa, F.A.; Gerst, S.R.; Tew, W.P.; Sabbatini, P.J.; Hensley, M.L.; Spriggs, D.R.; Aghajanian, C.A. A phase II study of cetuximab/paclitaxel/carboplatin for the initial treatment of advanced-stage ovarian, primary peritoneal, or fallopian tube cancer. Gynecol. Oncol 2008, 110, 140–145. [Google Scholar]
- Vasey, P.A.; Gore, M.; Wilson, R.; Rustin, G.; Gabra, H.; Guastalla, J.P.; Lauraine, E.P.; Paul, J.; Carty, K.; Kaye, S.; et al. A phase Ib trial of docetaxel, carboplatin and erlotinib in ovarian, fallopian tube and primary peritoneal cancers. Br. J. Cancer 2008, 98, 1774–1780. [Google Scholar]
- Weroha, S.J.; Oberg, A.L.; Ziegler, K.L.; Dakhilm, S.R.; Rowland, K.M.; Hartmann, L.C.; Moore, D.F., Jr; Keeney, G.L.; Peethambaram, P.P.; Haluska, P. Phase II trial of lapatinib and topotecan (LapTop) in patients with platinum-refractory/resistant ovarian and primary peritoneal carcinoma. Gynecol. Oncol 2011, 122, 116–120. [Google Scholar]
- Secord, A.A.; Blessing, J.A.; Armstrong, D.K.; Rodgers, W.H.; Miner, Z.; Barnes, M.N.; Lewandowski, G.; Mannel, R.S. Gynecologic Oncology Group. Phase II trial of cetuximab and carboplatin in relapsed platinum-sensitive ovarian cancer and evaluation of epidermal growth factor receptor expression: A gynecologic oncology group study. Gynecol. Oncol 2008, 108, 493–499. [Google Scholar]
- Mano, M.S.; Awada, A.; di Leo, A.; Durbecq, V.; Paesmans, M.; Cardoso, F.; Larsimont, D.; Piccart, M. Rates of topoisomerase II-Alpha and HER-2 gene amplification and expression in epithelial ovarian carcinoma. Gynecol. Oncol 2004, 92, 887–895. [Google Scholar]
- Bookman, M.A.; Darcy, K.M.; Clarke-Pearson, D.; Boothby, R.A.; Horowitz, I.R. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: A phase II trial of the gynecologic oncology group. J. Clin. Oncol 2003, 21, 283–290. [Google Scholar]
- Gordon, M.S.; Matei, D.; Aghajanian, C.; Matulonis, U.A.; Brewer, M.; Fleming, G.F.; Hainsworth, J.D.; Garcia, A.A.; Pegram, M.D.; Schilder, R.J.; et al. Clinical activity of pertuzumab (rhuMAb 2C4), a HER dimerization inhibitor, in advanced ovarian cancer: Potential predictive relationship with tumor HER2 activation status. J. Clin. Oncol 2006, 24, 4324–4332. [Google Scholar]
- Spannuth, W.A.; Sood, A.K.; Coleman, R.L. Farletuzumab in epithelial ovarian carcinoma. Expert Opin. Biol. Ther 2010, 10, 431–437. [Google Scholar]
- Jelovac, D.; Armstrong, D.K. Role of farletuzumab in epithelial ovarian carcinoma. Curr. Pharm. Des 2012, 18, 3812–3815. [Google Scholar]
- Farrell, C.; Schweizer, C.; Wustner, J.; Weil, S.; Namiki, M.; Nakano, T.; Nakai, K.; Phillips, M.D. Population pharmacokinetics of farletuzumab, a humanized monoclonal antibody against folate receptor alpha, in epithelial ovarian cancer. Cancer Chemother. Pharmacol 2012, 70, 727–734. [Google Scholar]
- Eskander, R.N.; Tewari, K.S. Emerging treatment options for management of malignant ascites in patients with ovarian cancer. Int. J. Womens Health 2012, 4, 395–404. [Google Scholar]
- Frampton, J.E. Catumaxomab: In malignant ascites. Drugs 2012, 72, 1399–1410. [Google Scholar]
- Matei, D.; Fang, F.; Shen, C.; Schilder, J.; Arnold, A.; Zeng, Y.; Berry, W.A.; Huang, T.; Nephew, K.P. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res 2012, 72, 2197–2205. [Google Scholar]
- Holmes, W.F.; Soprano, D.R.; Soprano, K.J. Early events in the induction of apoptosis in ovarian carcinoma cells by CD437: Activation of the p38 MAP kinase signal pathway. Oncogene 2003, 22, 6377–6386. [Google Scholar]
- Brozovic, A.; Osmak, M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett 2007, 251, 1–16. [Google Scholar]
- Chen, S. Natural products triggering biological targets—A review of the anti-inflammatory phytochemicals targeting the arachidonic acid pathway in allergy asthma and rheumatoid arthritis. Curr. Drug Targets 2011, 12, 288–301. [Google Scholar]
- Liu, Z.; Ouyang, L.; Peng, H.; Zhang, W.Z. Oridonin: Targeting programmed cell death pathways as an anti-tumour agent. Cell Prolif 2012, 45, 499–507. [Google Scholar]
- Habtemariam, S. Natural inhibitors of tumour necrosis factor-alpha production, secretion and function. Planta Med 2000, 66, 303–313. [Google Scholar]
- Piao, H.Z.; Jin, S.A.; Chun, H.S.; Lee, J.C.; Kim, W.K. Neuroprotective effect of wogonin: Potential roles of inflammatory cytokines. Arch. Pharm. Res 2004, 27, 930–936. [Google Scholar]
- Kim, D.S.; Park, S.S.; Nam, B.H.; Kim, I.H.; Kim, S.Y. Reversal of drug resistance in breast cancer cells by transglutaminase 2 inhibition and nuclear factor-kappaB inactivation. Cancer Res 2006, 66, 10936–10943. [Google Scholar]
- Jung, E.M.; Lim, J.H.; Lee, T.J.; Park, J.W.; Choi, K.S.; Kwon, T.K. Curcumin sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through reactive oxygen species-mediated upregulation of death receptor 5 (DR5). Carcinogenesis 2005, 26, 1905–1913. [Google Scholar]
- Fang, J.; Xia, C.; Cao, Z.; Zheng, J.Z.; Reed, E.; Jiang, B.H. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J 2005, 19, 342–353. [Google Scholar]
- Lanas, A.; Ferrandez, A. NSAIDs and the colon. Curr. Opin. Gastroenterol 2009, 25, 44–49. [Google Scholar]
- Bonovas, S.; Filioussi, K.; Sitaras, N.M. Do nonsteroidal anti-inflammatory drugs affect the risk of developing ovarian cancer? A meta-analysis. Br. J. Clin. Pharmacol 2005, 60, 194–203. [Google Scholar]
- Wernli, K.J.; Newcomb, P.A.; Hampton, J.M.; Trentham-Dietz, A.; Egan, K.M. Inverse association of NSAID use and ovarian cancer in relation to oral contraceptive use and parity. Br. J. Cancer 2008, 98, 1781–1783. [Google Scholar]
- Sempere, L.F.; Christensen, M.; Silahtaroglu, A.; Bak, M.; Heath, C.V.; Schwartz, G.; Wells, W.; Kauppinen, S.; Cole, C.N. Altered microRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res 2007, 67, 11612–11620. [Google Scholar]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. Let-7 Regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar]
- Cho, K.R. Ovarian cancer update: Lessons from morphology, molecules, and mice. Arch. Pathol. Lab. Med 2009, 133, 1775–1781. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smolle, E.; Taucher, V.; Pichler, M.; Petru, E.; Lax, S.; Haybaeck, J. Targeting Signaling Pathways in Epithelial Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 9536-9555. https://doi.org/10.3390/ijms14059536
Smolle E, Taucher V, Pichler M, Petru E, Lax S, Haybaeck J. Targeting Signaling Pathways in Epithelial Ovarian Cancer. International Journal of Molecular Sciences. 2013; 14(5):9536-9555. https://doi.org/10.3390/ijms14059536
Chicago/Turabian StyleSmolle, Elisabeth, Valentin Taucher, Martin Pichler, Edgar Petru, Sigurd Lax, and Johannes Haybaeck. 2013. "Targeting Signaling Pathways in Epithelial Ovarian Cancer" International Journal of Molecular Sciences 14, no. 5: 9536-9555. https://doi.org/10.3390/ijms14059536
APA StyleSmolle, E., Taucher, V., Pichler, M., Petru, E., Lax, S., & Haybaeck, J. (2013). Targeting Signaling Pathways in Epithelial Ovarian Cancer. International Journal of Molecular Sciences, 14(5), 9536-9555. https://doi.org/10.3390/ijms14059536