The Effect of the Aerial Part of Lindera akoensis on Lipopolysaccharides (LPS)-Induced Nitric Oxide Production in RAW264.7 Cells

 , ,
, ,
Abstract
:1. Introduction
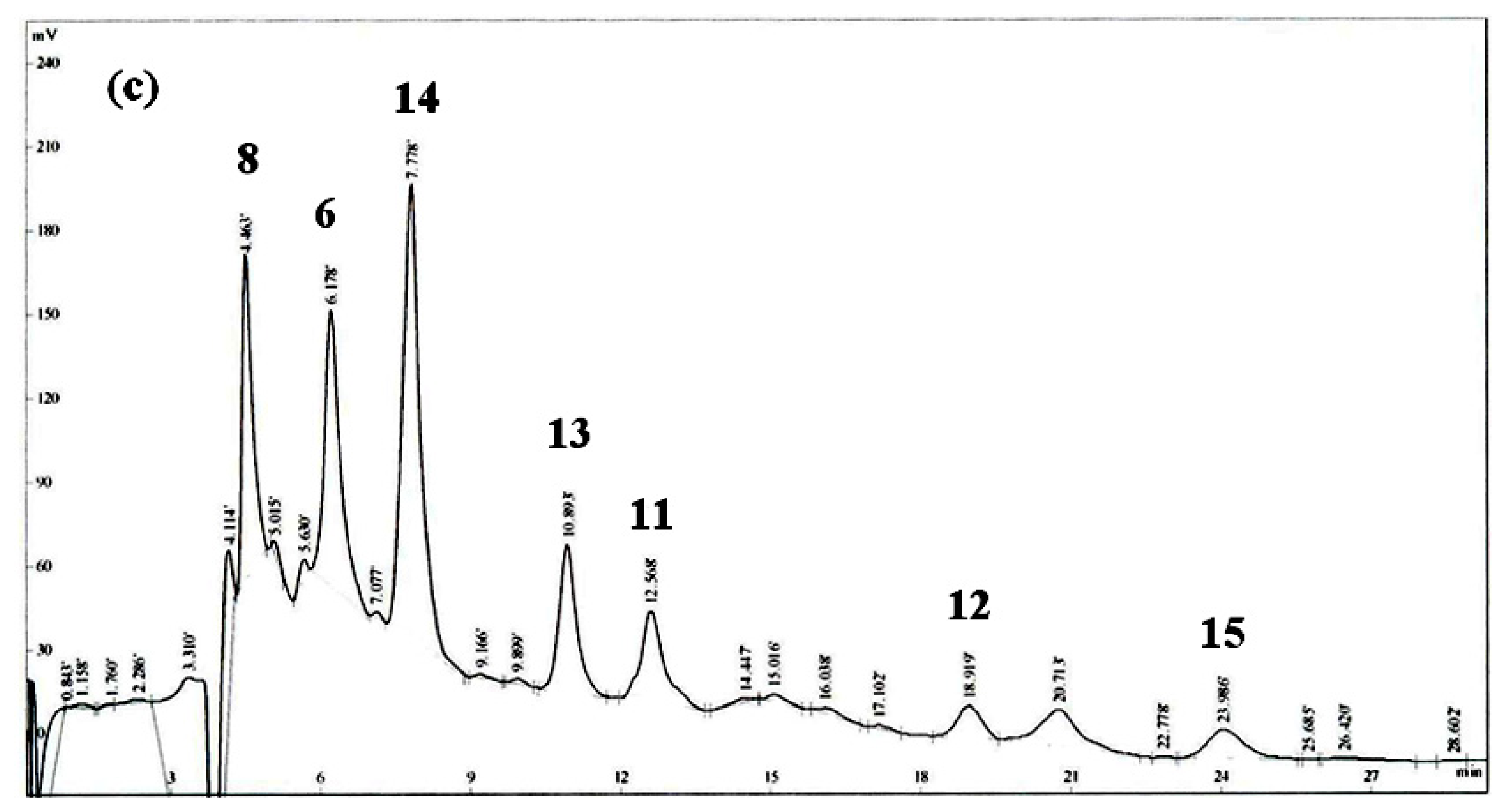
2. Results and Discussion
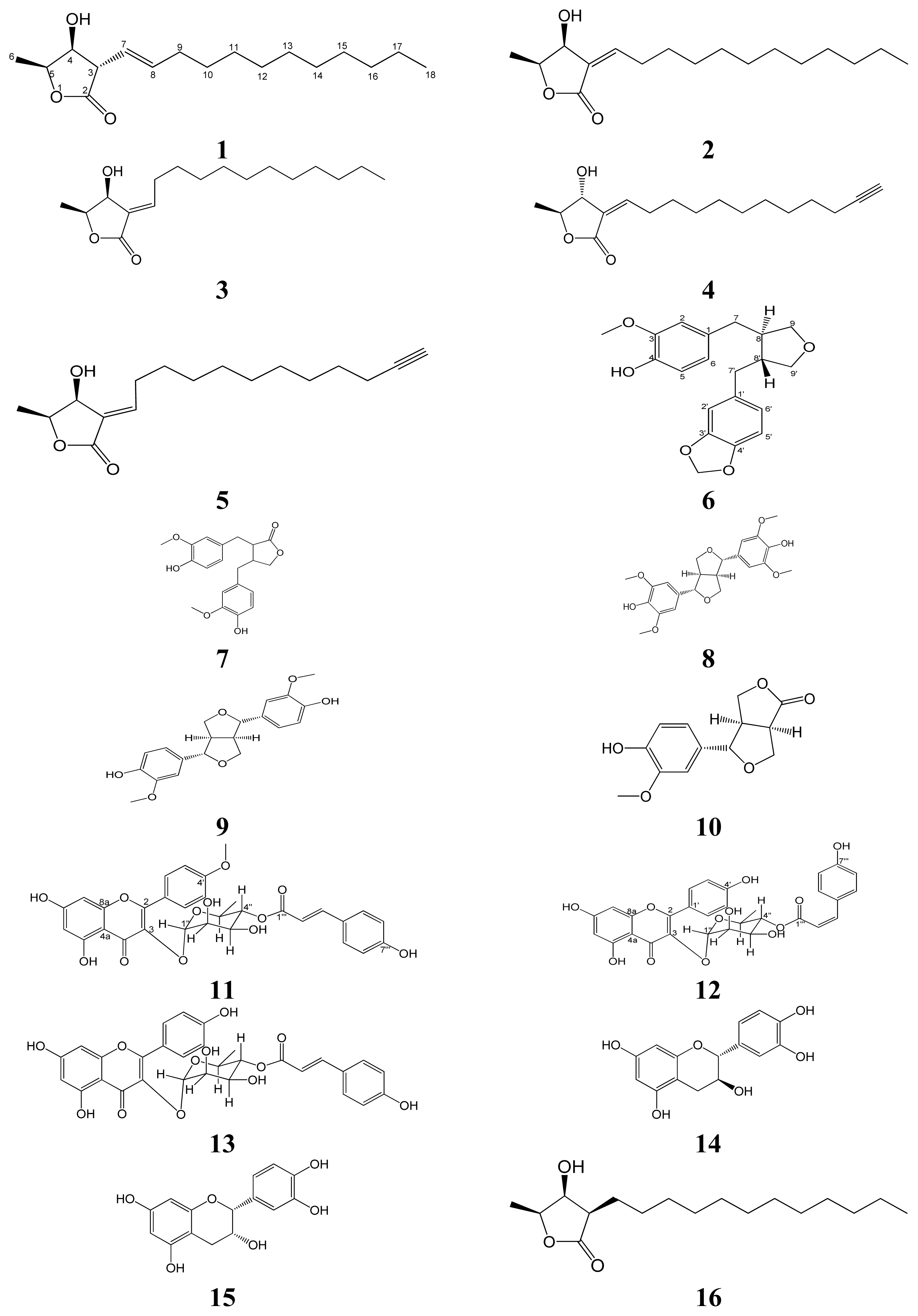
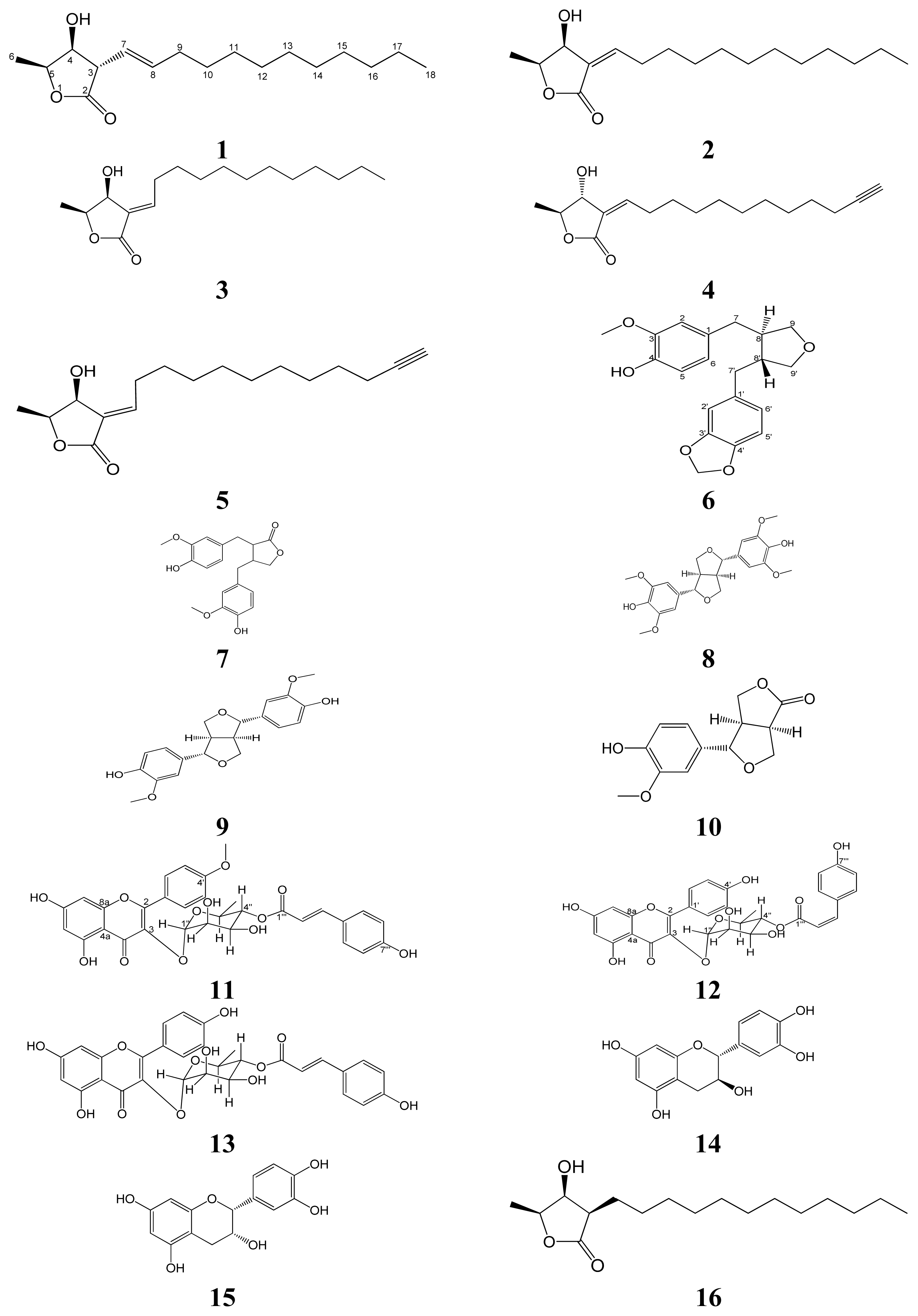
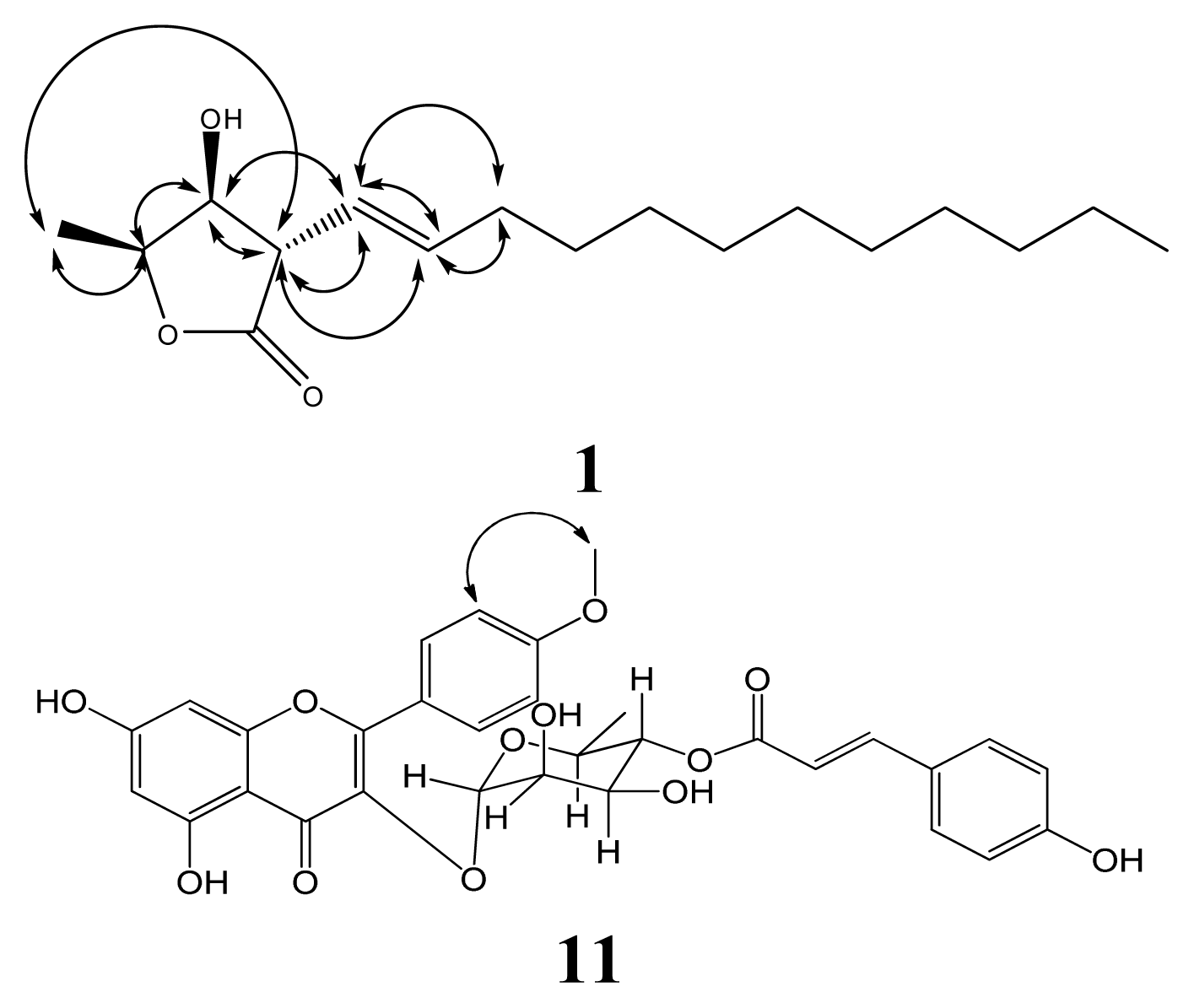
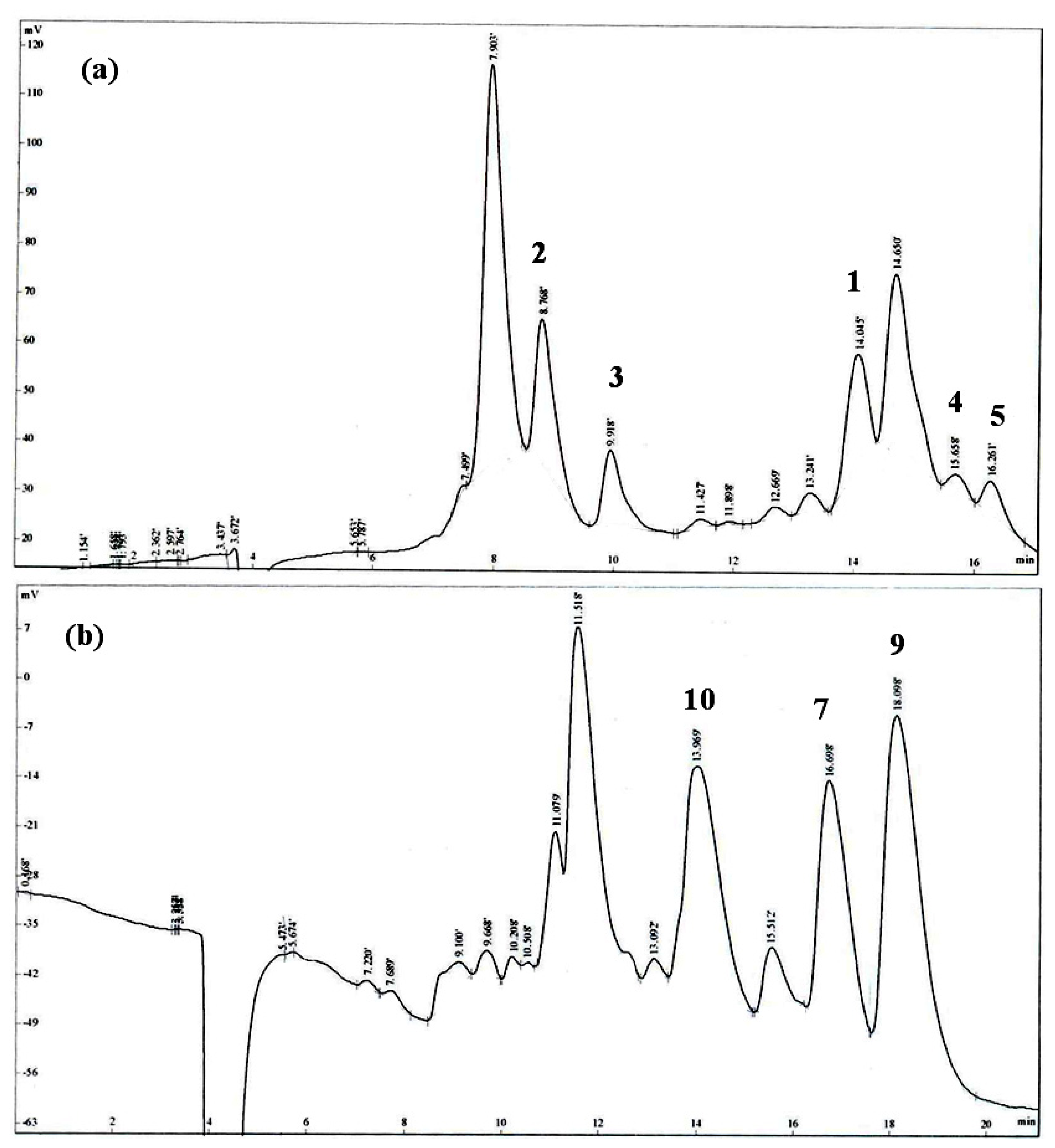
Isolation and Structural Elucidation
2.2 Anti-Inflammatory Activity
3. Experimental Section
3.1. Chemicals
3.2. General
3.3. Plant Material
3.4. Extraction and Isolation
3.5. Cell Culture
3.6. Measurement of Nitric Oxide/Nitrite
3.7. Cell Viability
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Chen, C.C.; Lin, C.F.; Huang, Y.L. Bioactive constituents from the flower buds and peduncles of Lindera megaphylla. J. Nat. Prod 1995, 9, 1423–1425. [Google Scholar]
- Chang, Y.C.; Chen, C.Y.; Chang, F.R.; Wu, Y.C. Alkaloids from Lindera glauca. J. Chin. Chem. Soc 2001, 48, 811–815. [Google Scholar]
- Cheng, X.L.; Ma, S.C.; Wei, F.; Wang, G.L.; Xiao, X.Y.; Lin, R.C. A new sesquiterpene isolated from Lindera aggregata (SIMS) KOSTERM. Chem. Pharm. Bull 2007, 55, 1390–1392. [Google Scholar]
- Takamasa, O.; Akito, N.; Munehiro, N.; Makoto, I.; Li, Y.M.; Shinya, M.; Hajime, M.; Hisayoshi, F. New sesquiterpene lactones from water extract of the root of Lindera strychnifolia with cytotoxicity against the human small cell cancer, SBC-3. Tetrahedron. Lett 2005, 46, 8657–8660. [Google Scholar]
- Kouni, I.; Hirai, A.; Fukushige, A.; Jiang, Z.H.; Takashi, T. New eudesmane sesquiterpenes from the root of Lindera strychnifolia. J. Nat. Prod 2001, 64, 286–288. [Google Scholar]
- Chang, S.Y.; Chen, M.J.; Peng, C.F.; Chang, H.S.; Chen, I.S. Antimycobacterial butanolides from the root of Lindera akoensis. Chem. Biodivers 2008, 5, 2690–2698. [Google Scholar]
- Zhang, M.; Zhang, C.F.; Sun, Q.S.; Wang, Z.T. Two new compounds from Lindera chunii Merr. Chin. Chem. Lett 2006, 17, 1325–1327. [Google Scholar]
- Leong, Y.W.; Harrison, L.J.; Bennett, G.J.; Kadir, A.A.; Connolly, J.D. A dihydrochalcone from Lindera lucida. Phytochemistry 1998, 5, 891–894. [Google Scholar]
- Song, M.C.; Nigussie, F.; Jeong, T.S.; Lee, C.Y.; Regassa, F.; Markos, T.; Baek, N.I. Phenolic compounds from the roots of Lindera fruticosa. J. Nat. Prod 2006, 69, 853–855. [Google Scholar]
- Song, M.C.; Nigussie, F.; Yang, H.J.; Kim, H.H.; Kim, J.Y.; Chung, D.K.; Baek, N.I. Phenolic glycosides from Lindera fruticosa root and their inhibitory activity on osteoclast differentiation. Chrm. Pharm. Bull 2008, 5, 707–710. [Google Scholar]
- Wang, S.Y.; Lan, X.Y.; Xiao, J.H.; Yang, J.C.; Kao, Y.T.; Chang, S.T. Antiinflammatory activity of Lindera erythrocarpa fruits. Phytother. Res 2008, 22, 213–216. [Google Scholar]
- Ohno, T.; Takemura, G.; Murata, I.; Kagawa, T.; Akao, S.; Minatoguchi, S.; Fujiwara, T.; Fujiwara, H. Water extract of the root of Lindera strychnifolia slows down the progression of diabetic nephropathy in db/db mice. Life Sci 2005, 77, 1391–1403. [Google Scholar]
- Zhao, Q.; Zhao, Y.; Wang, K. Antinociceptive and free radical scavenging activities of alkaloids isolated from Lindera angustifolia Chen. J. Ethnopharmacol 2006, 106, 408–413. [Google Scholar]
- Department of Health, Committee on Chinese Medicine and Pharmacy, The Catologue of Medicinal Plant Resourses in Taiwan; Taipei, Taiwan, 2003.
- Kondo, S.; Mitsunaga, T. Anti-inflammatory Agents Containing Butanolides. JP 2008150347, 2008. [Google Scholar]
- Kim, N.Y.; Ryu, J.H. Butanolides from Machilus thunbergii and their inhibitory activity on nitric oxide synthesis in activated macrophages. Phytother. Res 2003, 17, 372–375. [Google Scholar]
- Lee, S.S.; Chang, S.M.; Chen, C.H. Chemical constituents from Alseodaphne andersonii. J. Nat. Prod 2001, 64, 1548–1551. [Google Scholar]
- Cheng, W.; Zhu, C.G.; Xu, W.D.; Fan, X.N.; Yang, Y.C.; Li, Y.; Cheng, X.G.; Wang, W.J.; Shi, J.G. Chemical constituents of the bark of Machilus wangchiana and their biological activities. J. Nat. Prod 2009, 72, 2145–2152. [Google Scholar]
- Tsai, I.L.; Hung, C.H.; Duh, C.Y.; Chen, J.H.; Lin, W.Y.; Chen, I.S. Cytotoxic butanolides from the stem bark of formosan Lindera communis. Planta Med 2001, 67, 865–866. [Google Scholar]
- Kaoru, U.; Ariko, S.; Masanori, K.; Akiru, U.; Takao, T. Studies on differentiation-inducers from arctium fructus. Chem. Pharm. Bull 1993, 41, 1774–1779. [Google Scholar]
- Chen, C.Y.; Wu, T.Y.; Chang, F.R.; Wu, Y.C. Lignans and kauranes from the stems of Annona Cherimola. J. Chin. Chem. Soc 1998, 45, 629–634. [Google Scholar]
- Cowan, S.; Stewart, M.; Abbiw, D.K.; Latif, Z.; Sarker, S.D.; Nash, R.J. Lignans from Strophanthus gratus. Fitoterapia 2001, 72, 80–82. [Google Scholar]
- Chang, H.S.; Lee, S.J.; Yang, C.W.; Chen, I.S. Cytotoxic Sesquiterpenes from Magnolia kachirachirai. Chem. Biodivers 2010, 7, 2737–2747. [Google Scholar]
- Walmir, S.G.; Massayoshi, Y.; Otto, R.G. Benzylisoquinoline alkaloids and flavonols from Ocotea vellosiana. Phytochemistry 1995, 39, 815–816. [Google Scholar]
- Zhao, J.; Zhou, X.W.; Chen, X.B.; Wang, Q.X. α-Glucosidase inhibitory constituents from Toona sinensis. Chem. Nat. Compd 2009, 45, 244–246. [Google Scholar]
- Yang, C.P.; Huang, G.J.; Huang, H.C.; Chen, Y.C.; Chang, C.I.; Wang, S.Y.; Chen, I.S.; Tseng, Y.H.; Chien, S.C.; Kuo, Y.H. A new anti-inflammatory butanolide from the aerial part of Lindera akoensis. Molecules 2012, 17, 6585–6592. [Google Scholar]
- Ha, T.J.; Lee, J.H.; Lee, M.H.; Lee, B.W.; Kwon, H.S.; Park, C.H.; Shim, K.B.; Kim, H.T.; Baek, I.Y.; Jang, D.S. Isolation and identification of phenolic compounds from the seeds of Perilla frutescens (L.) and their inhibitory activities against α-glucosidase and aldose reductase. Food Chem 2012, 135, 1397–1403. [Google Scholar]
- Geller, D.A.; Billiar, T.R. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev 1998, 17, 7–20. [Google Scholar]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmcol. Rev 1991, 43, 109–142. [Google Scholar]
- Luo, Y.; Liu, M.; Dai, Y.; Yao, X.; Xia, Y.; Chou, G.; Wang, Z. Norisoboldine inhibits the production of pro-inflammatory cytokines in lipopolysaccharide-stimulated RAW 264.7 cells by down-regulating the activation of MAPKs but not NF-κB. Inflammation 2010, 33, 389–397. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 6 | |||
|---|---|---|---|---|
| No. | δHa | δCb | δHc | δCd |
| 1 | 132.4 | |||
| 2 | 175.6 | 6.61 (s) | 109.3 | |
| 3 | 3.19 (dd, J = 6.5, 4.7) | 52.7 | 146.5 | |
| 4 | 4.23 (dd, J = 4.7, 4.5) | 74.6 | 143.8 | |
| 5 | 4.64 (qd, J = 6.5, 4.5) | 78.1 | 6.79 (d, J = 8.0) | 114.2 |
| 6 | 1.39 (d, J = 6.5) | 13.8 | 6.68 (d, J = 8.0) | 121.7 |
| 7 | 5.37 (dd, J = 15.4, 6.5) | 120.9 | 2.62 (dd, J = 12.8, 5.0) | 35.9 |
| 2.72 (dd, J = 12.8, 8.5) | ||||
| 8 | 5.72 (dt, J = 15.4, 7.4) | 136.7 | 1.85 (m) | 44.1 |
| 9 | 2.04 (q, J = 7.4) | 32.6 | 3.51 (dd, J = 11.2, 6.4) | 60.6 |
| 3.77 (dd, J = 11.2, 6.4) | ||||
| 10–15 | 1.24 (br s) | 29.0–31.9 | ||
| 16–17 | 1.24 (br s) | 22.7 | ||
| 18 | 0.86 (t, J = 6.6) | 14.1 | ||
| 1′ | 134.4 | |||
| 2′ | 6.61 (s) | 111.4 | ||
| 3′ | 147.6 | |||
| 4′ | 145.7 | |||
| 5′ | 6.62 (d, J = 8.0) | 108.1 | ||
| 6′ | 6.57 (d, J = 8.0) | 121.9 | ||
| 7′ | 2.62 (dd, J = 12.8, 5.0) | 35.9 | ||
| 2.72 (dd, J = 12.8, 8.5) | ||||
| 8′ | 1.85 (m) | 44.1 | ||
| 9′ | 3.51 (dd, J = 11.2, 6.4) | 60.5 | ||
| 3.77 (dd, J = 11.2, 6.4) | ||||
| OCH3 | 3.82 (s) | 55.9 | ||
| OCH2O | 5.90 (s) | 100.8 | ||
| Compound | Cytotoxicity IC50 (μM) | Inhibition of NO production IC50 (μM) |
|---|---|---|
| 1 | 78.0 ± 5.1 | 20.1 ± 0.3 |
| 2 | 32.6 ± 0.5 | 4.1 ± 0.1 |
| 3 | 27.7 ± 1.6 | 4.5 ± 0.1 |
| 4 | 138.8 ± 2.8 | 21.7 ± 0.4 |
| 5 | 142.8 ± 1.9 | 33.4 ± 1.0 |
| 6 | >292.4 | 196.0 ± 4.0 |
| 7 | >279.3 | 178.8 ± 12.1 |
| 8 | >239.2 | 49.7 ± 4.5 |
| 9 | >279.3 | 90.4 ± 8.6 |
| 10 | >400.0 | 311.6 ± 14.1 |
| 11 | >84.5 | 62.5 ± 2.2 |
| 12 | >86.5 | 67.9 ± 1.9 |
| 13 | >86.5 | 76.9 ± 7.3 |
| 14 | >517.2 | 413.8 ± 6.9 |
| 15 | >517.2 | 351.7 ± 37.4 |
| indomethacin | 182.9 ± 5.5 |
| 11 | 12 | |||
|---|---|---|---|---|
| No. | δHa | δCb | δHa | δCb |
| 2 | 159.1 | 159.6 | ||
| 3 | 135.7 | 135.8 | ||
| 4 | 179.6 | 179.9 | ||
| 4a | 106.1 | 106.1 | ||
| 5 | 158.7 | 158.8 | ||
| 6 | 6.22, d, J = 2.0 | 100.1 | 6.21, d, J = 2.0 | 100.1 |
| 7 | 166.2 | 166.1 | ||
| 8 | 6.38, d, J = 2.0 | 95.0 | 6.38, d, J = 2.0 | 95.0 |
| 8a | 163.3 | 163.4 | ||
| 1′ | 124.1 | 122.7 | ||
| 2′ | 7.84, d, J = 8.8 | 132.0 | 7.73, d, J = 8.5 | 132.1 |
| 3′ | 7.14, d, J = 8.8 | 115.4 | 6.94, d, J = 8.5 | 116.6 |
| 4′ | 163.6 | 161.8 | ||
| 5′ | 7.14, d, J = 8.8 | 115.4 | 6.94, d, J = 8.5 | 116.6 |
| 6′ | 7.84, d, J = 8.8 | 132.0 | 7.73, d, J = 8.5 | 132.1 |
| 1″ | 5.62, br s | 102.4 | 5.51, d, J = 1.0 | 102.9 |
| 2″ | 4.23, br s | 71.9 | 4.23, dd, J = 3.0, 1.0 | 72.0 |
| 3″ | 3.91, dd, J = 9.7, 2.9 | 70.2 | 3.89, dd, J = 9.7, 3.0 | 70.3 |
| 4″ | 4.91, t, J = 9.7 | 74.9 | 4.90, t, J = 9.7 | 74.6 |
| 5″ | 3.18, m | 69.8 | 3.28, m | 69.9 |
| 6″ | 0.78, d, J = 6.3 | 17.8 | 0.78, d, J = 6.3 | 17.8 |
| 1‴ | 168.8 | 167.8 | ||
| 2‴ | 6.25, d, J = 16.0 | 115.3 | 5.75, d, J = 12.8 | 116.0 |
| 3‴ | 7.53, d, J = 16.0 | 146.8 | 6.87, d, J = 12.8 | 145.8 |
| 4‴ | 127.3 | 127.7 | ||
| 5‴ | 7.49, d, J =8.6 | 131.4 | 7.66, d, J = 8.6 | 134.0 |
| 6‴ | 6.84, d, J = 8.6 | 117.0 | 6.74, d, J = 8.6 | 116.0 |
| 7‴ | 161.4 | 160.3 | ||
| 8‴ | 6.84, d, J = 8.6 | 117.0 | 6.74, d, J = 8.6 | 116.0 |
| 9‴ | 7.49, d, J = 8.6 | 131.4 | 7.66, d, J = 8.6 | 134.0 |
| OCH3 | 3.85, s | 56.3 | ||
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, C.-P.; Huang, G.-J.; Huang, H.-C.; Chen, Y.-C.; Chang, C.-I.; Wang, S.-Y.; Chang, H.-S.; Tseng, Y.-H.; Chien, S.-C.; Kuo, Y.-H. The Effect of the Aerial Part of Lindera akoensis on Lipopolysaccharides (LPS)-Induced Nitric Oxide Production in RAW264.7 Cells. Int. J. Mol. Sci. 2013, 14, 9168-9181. https://doi.org/10.3390/ijms14059168
Yang C-P, Huang G-J, Huang H-C, Chen Y-C, Chang C-I, Wang S-Y, Chang H-S, Tseng Y-H, Chien S-C, Kuo Y-H. The Effect of the Aerial Part of Lindera akoensis on Lipopolysaccharides (LPS)-Induced Nitric Oxide Production in RAW264.7 Cells. International Journal of Molecular Sciences. 2013; 14(5):9168-9181. https://doi.org/10.3390/ijms14059168
Chicago/Turabian StyleYang, Chung-Ping, Guan-Jhong Huang, Hui-Chi Huang, Yu-Chang Chen, Chi-I Chang, Sheng-Yang Wang, Hsun-Shuo Chang, Yen-Hsueh Tseng, Shih-Chang Chien, and Yueh-Hsiung Kuo. 2013. "The Effect of the Aerial Part of Lindera akoensis on Lipopolysaccharides (LPS)-Induced Nitric Oxide Production in RAW264.7 Cells" International Journal of Molecular Sciences 14, no. 5: 9168-9181. https://doi.org/10.3390/ijms14059168