Impact of High Salt Independent of Blood Pressure on PRMT/ADMA/DDAH Pathway in the Aorta of Dahl Salt-Sensitive Rats


Abstract
:1. Introduction
2. Results
2.1. Effects of High Salt Diet on Blood Pressure and Weight of Rats
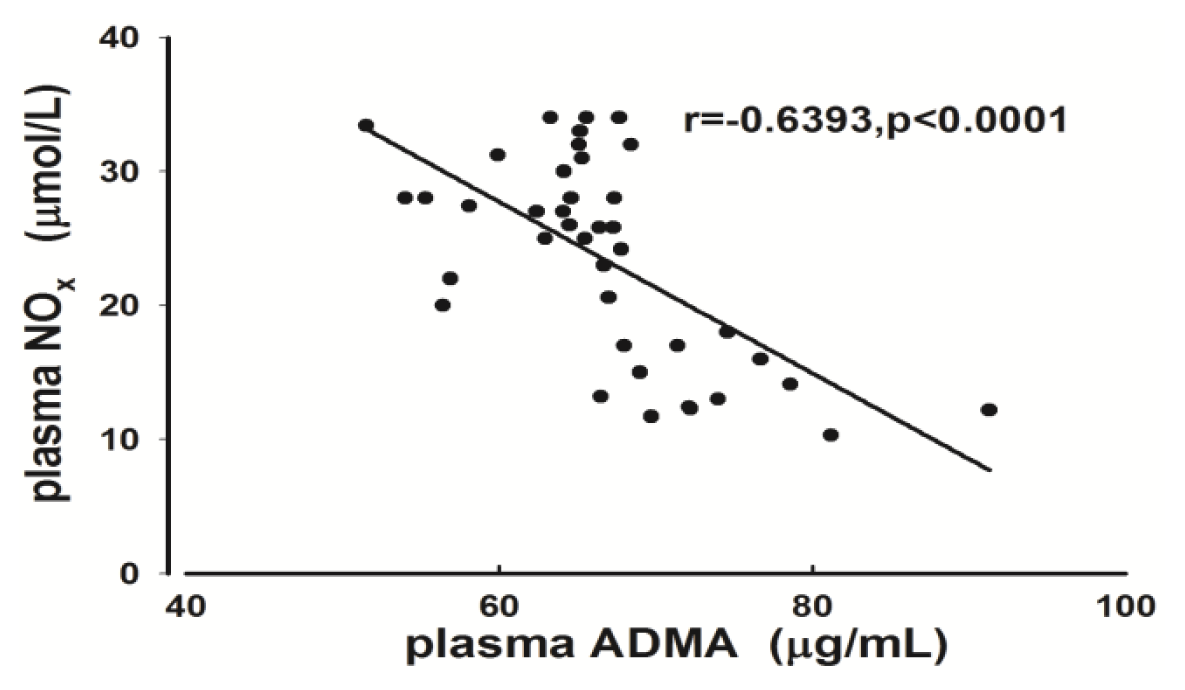
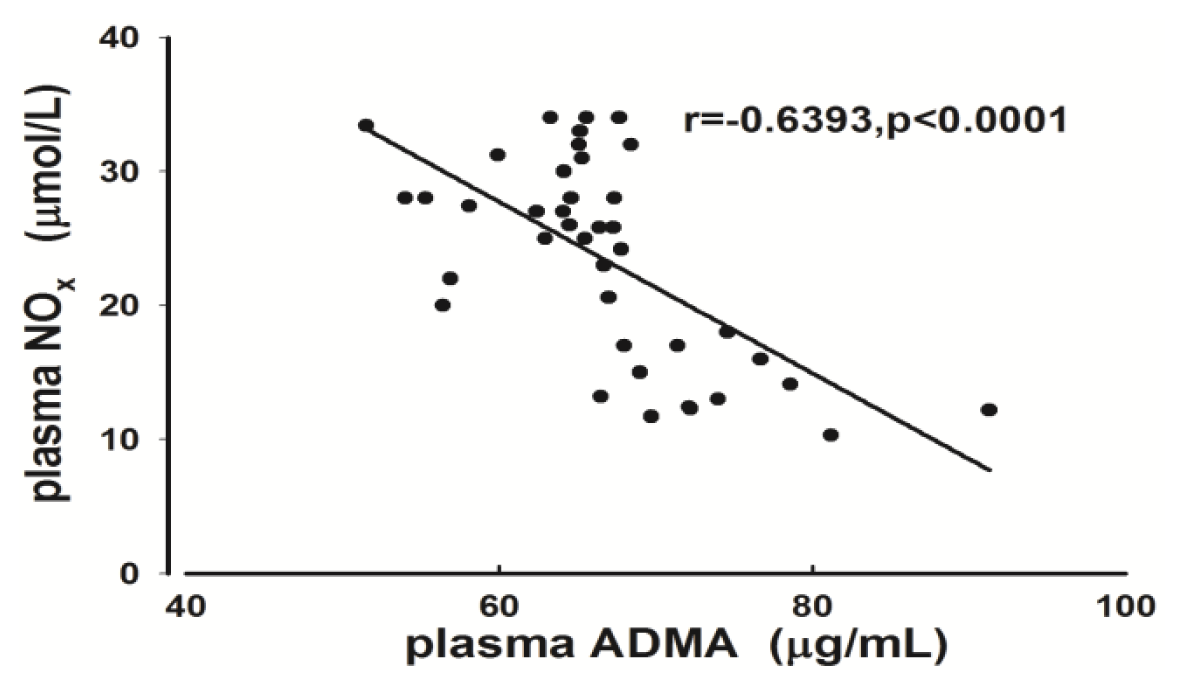
2.2. Measurement of Plasma ADMA and NOx
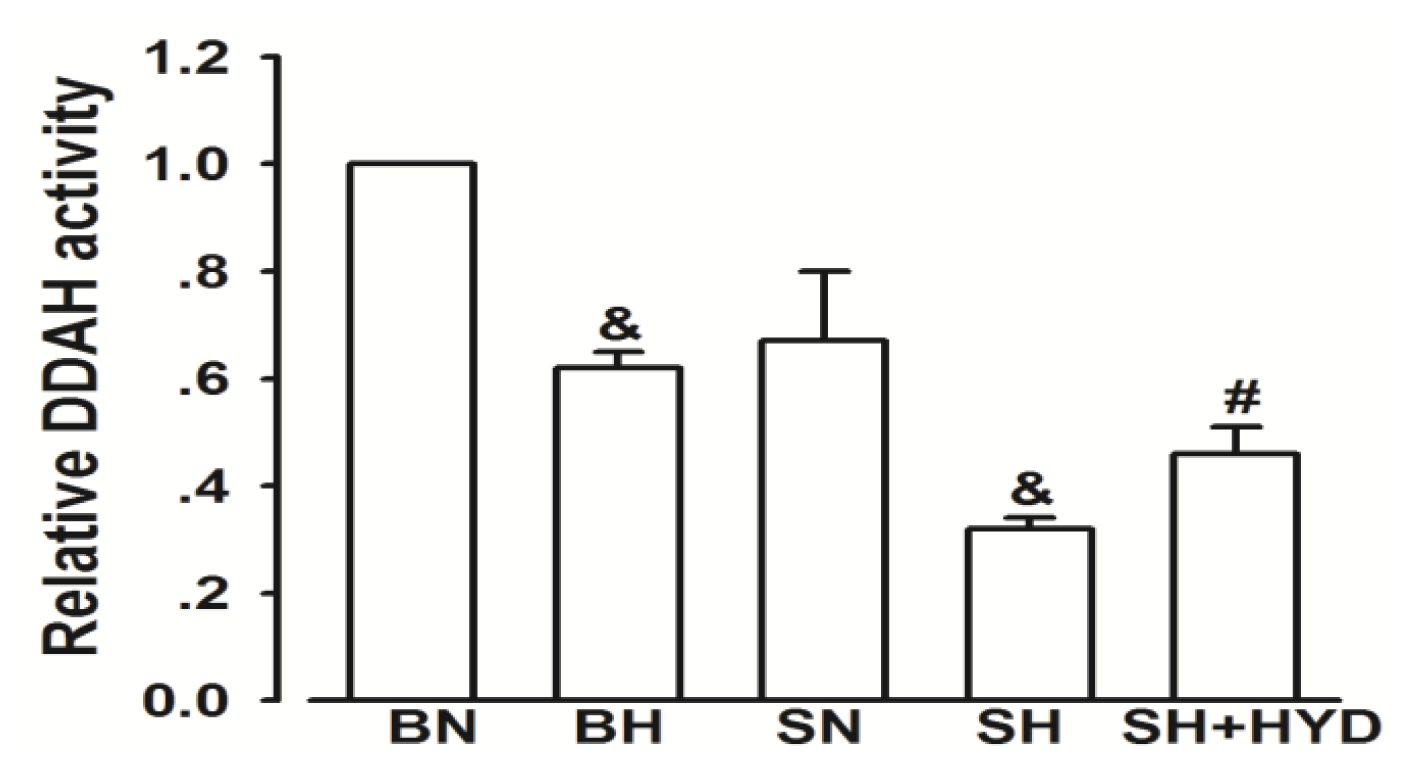
2.3. Activity of DDAH
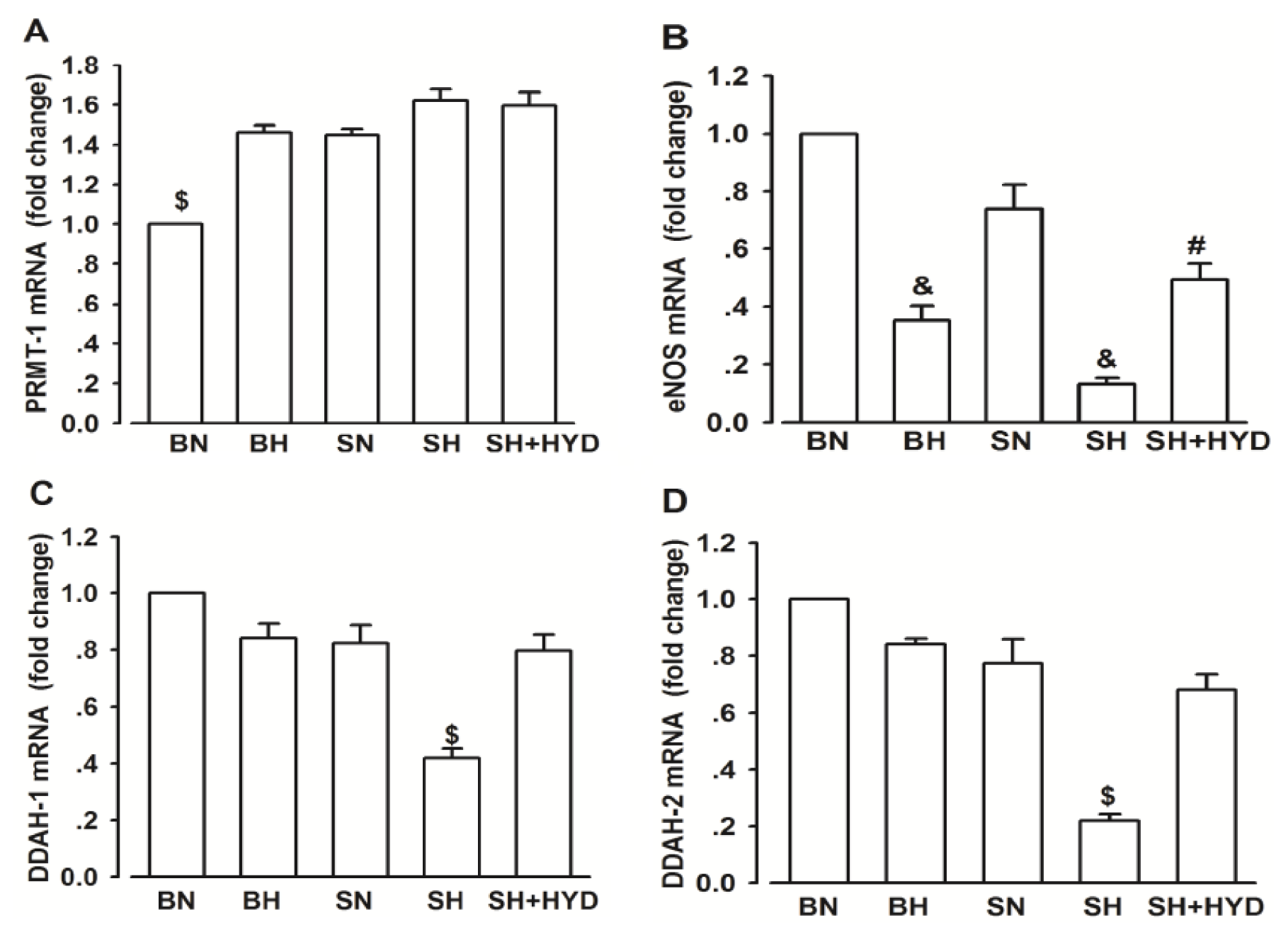
2.4. mRNA Expressions of PRMT-1, DDAH-1, DDAH-2 and eNOS
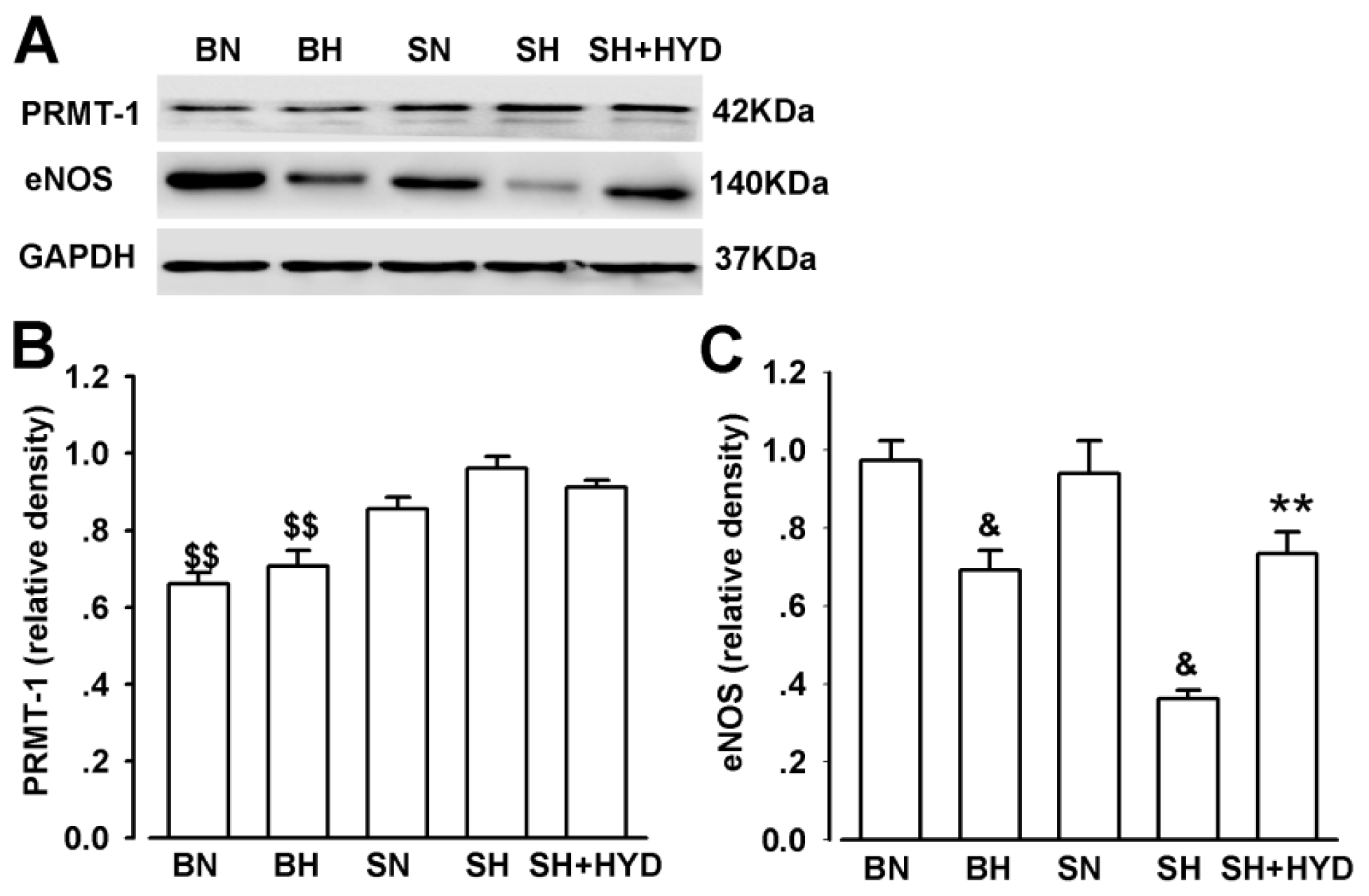
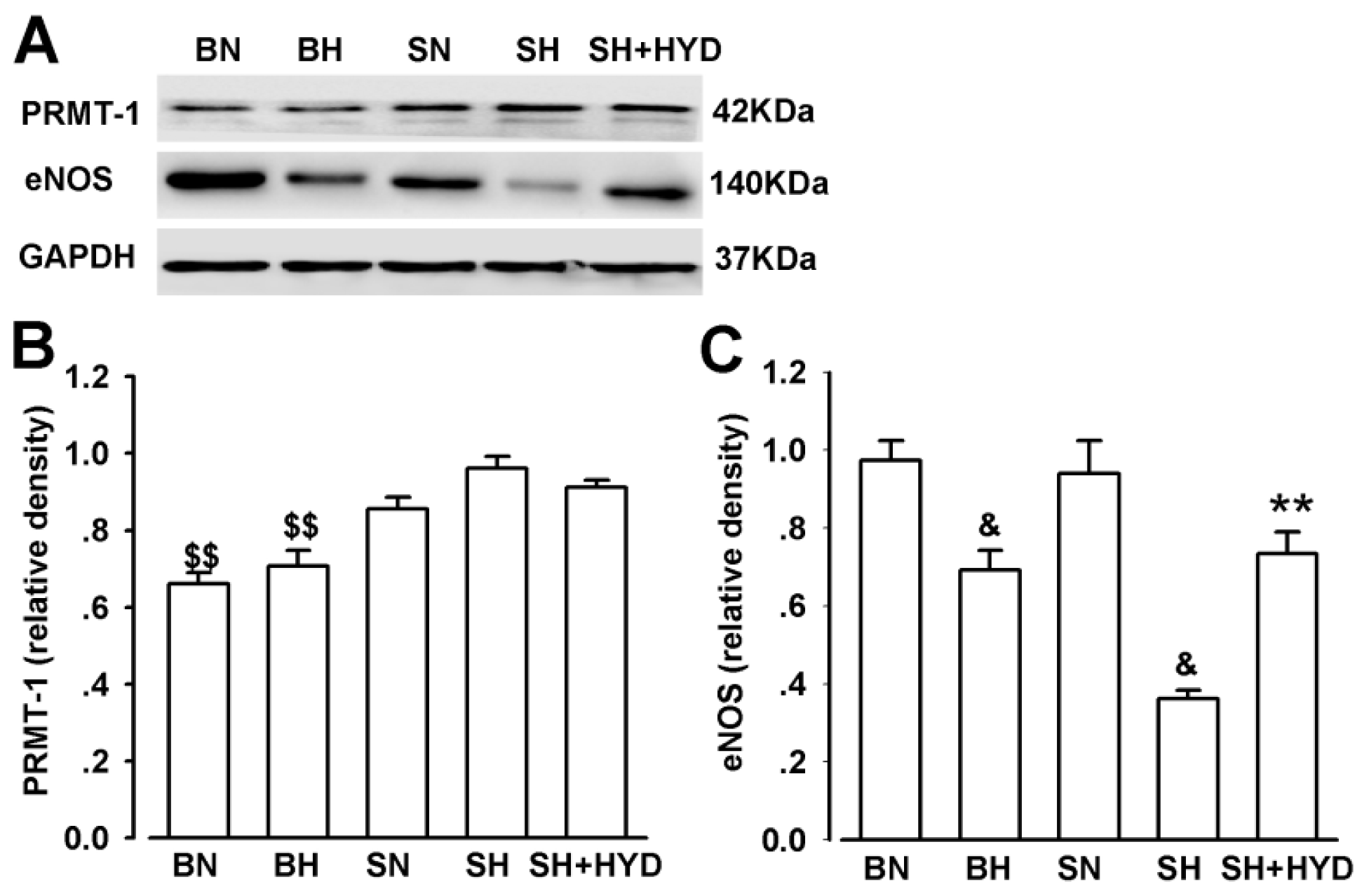
2.5. Protein Expressions of PRMT-1 and eNOS
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Procedures
4.2. Measurement of Blood Pressure and Body Weight
4.3. Measurement of Plasma ADMA
4.4. Measurement of Plasma Nitrite/Nitrate (NOx)
4.5. Measurement of DDAH Activity
4.6. RNA Quantitation
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusion
Acknowledgments
Reference
- Miyoshi, A.; Suzuki, H.; Fujiwara, M.; Masai, M.; Iwasaki, T. Impairment of endothelial function in salt-sensitive hypertension in humans. Am. J. Hypertens 1997, 10, 1083–1090. [Google Scholar]
- Vanhoutte, P.M. Endothelium and control of vascular function. State of the Art lecture. Hypertension 1989, 13, 658–667. [Google Scholar]
- Lüscher, T. Imbalance of endothelium-derived relaxing and contracting factors. A new concept in hypertension? Am. J. Hypertens 1990, 3, 317–330. [Google Scholar]
- Bermudez, V.; Bermudez, F.; Acosta, G.; Acosta, A.; Anez, J.; Andara, C.; Leal, E.; Cano, C.; Manuel, V.; Hernández, R. Molecular mechanisms of endothelial dysfunction: From nitric oxide synthesis to ADMA inhibition. Am. J. Ther 2008, 15, 326–333. [Google Scholar]
- Bragulat, E.; Sierra, A. Salt intake, endothelial dysfunction, and salt-sensitive hypertension. J. Clin. Hypertens 2007, 4, 41–46. [Google Scholar]
- Toda, N.; Arakawa, K. Salt-induced hemodynamic regulation mediated by nitric oxide. J. Hypertens 2011, 29, 415–424. [Google Scholar]
- Schmidlin, O.; Forman, A.; Leone, A.; Sebastian, A.; Morris, R.C., Jr. Salt sensitivity in blacks evidence that the initial pressor effect of NaCl involves inhibition of vasodilatation by asymmetrical dimethylarginine. Hypertension 2011, 58, 380–385. [Google Scholar]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine: Dimethylarginine dimethylaminohydrolase pathway. Arterioscl. Thromb. Vasc. Biol 2004, 24, 1023–1030. [Google Scholar]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar]
- Tojo, A.; Kimoto, M.; Wilcox, C.S. Renal expression of constitutive NOS and DDAH: Separate effects of salt intake and angiotensin. Kidney Int 2000, 58, 2075–2083. [Google Scholar]
- Tojo, A.; Welch, W.J.; Bremer, V.; Kimoto, M.; Kimura, K.; Omata, M.; Ogawa, T.; Vallance, P.; Wilcox, C.S. Colocalization of demethylating enzymes and NOS and functional effects of methylarginines in rat kidney. Kidney Int 1997, 52, 1593–1601. [Google Scholar]
- Arrigoni, F.I.; Vallance, P.; Haworth, S.G.; Leiper, J.M. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation 2003, 107, 1195–1201. [Google Scholar]
- Matsuoka, H.; Itoh, S.; Kimoto, M.; Kohno, K.; Tamai, O.; Wada, Y.; Yasukawa, H.; Iwami, G.; Okuda, S.; Imaizumi, T. Asymmetrical dimethylarginine, an endogenous nitric oxide synthase inhibitor, in experimental hypertension. Hypertension 1997, 29, 242–247. [Google Scholar]
- Fujiwara, N.; Osanai, T.; Kamada, T.; Katoh, T.; Takahashi, K.; Okumura, K. Study on the relationship between plasma nitrite and nitrate level and salt sensitivity in human hypertension: Modulation of nitric oxide synthesis by salt intake. Circulation 2000, 101, 856–861. [Google Scholar]
- Fang, Y.; Mu, J.J.; He, L.C.; Wang, S.C.; Liu, Z.Q. Salt loading on plasma asymmetrical dimethylarginine and the protective role of potassium supplement in normotensive salt-sensitive Asians. Hypertension 2006, 48, 724–729. [Google Scholar]
- Du Cailar, G.; Ribstein, J.; Daures, J.P.; Mimran, A. Sodium and left ventricular mass in untreated hypertensive and normotensive subjects. Am. J. Physiol. Heart Circ. Physiol 1992, 263, H177–H181. [Google Scholar]
- Tuomilehto, J.; Jousilahti, P.; Rastenyte, D.; Moltchanov, V.; Tanskanen, A.; Pietinen, P.; Nissinen, A. Urinary sodium excretion and cardiovascular mortality in Finland: A prospective study. Lancet 2001, 357, 848–851. [Google Scholar]
- Safar, M.; Thuilliez, C.; Richard, V.; Benetos, A. Pressure-independent contribution of sodium to large artery structure and function in hypertension. Cardiovasc. Res 2000, 46, 269–276. [Google Scholar]
- Reckelhoff, J.F.; Romero, J.C. Role of oxidative stress in angiotensin-induced hypertension. Am. J. Physiol. Regul. Integr. Compar. Physiol 2003, 284, R893–R912. [Google Scholar]
- Zhu, J.; Mori, T.; Huang, T.; Lombard, J.H. Effect of high-salt diet on NO release and superoxide production in rat aorta. Am. J. Physiol. Heart Circ. Physiol 2004, 286, H575–H583. [Google Scholar]
- Henry, C.; Burrell, L.M.; Black, M.J.; Wu, L.L.; Dilley, R.J.; Cooper, M.E.; Johnston, C.I. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation 1998, 98, 2621–2628. [Google Scholar]
- Fujita, T. Mineralocorticoid receptors, salt-sensitive hypertension, and metabolic syndrome. Hypertension 2010, 55, 813–818. [Google Scholar]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease. Circulation 2006, 113, 1708–1714. [Google Scholar]
- Li, H.; Förstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, in press. [Google Scholar]
- Barton, M.; Vos, I.; Shaw, S.; Boer, P.; D’uscio, L.V.; Gröne, H.-J.; Rabelink, T.J.; Lattmann, T.; Moreau, P.; Lüscher, T.F. Dysfunctional renal nitric oxide synthase as a determinant of salt-sensitive hypertension mechanisms of renal artery endothelial dysfunction and role of endothelin for vascular hypertrophy and glomerulosclerosis. J. Am. Soc. Nephrol 2000, 11, 835–845. [Google Scholar]
- Zhou, M.S.; Adam, A.G.; Jaimes, E.A.; Raij, L. In salt-sensitive hypertension, increased superoxide production is linked to functional upregulation of angiotensin II. Hypertension 2003, 42, 945–951. [Google Scholar]
- Taylor, N.E.; Maier, K.G.; Roman, R.J.; Cowley, A.W., Jr. NO synthase uncoupling in the kidney of dahl s rats role of dihydrobiopterin. Hypertension 2006, 48, 1066–1071. [Google Scholar]
- Swei, A.; Lacy, F.; DeLano, F.A.; Schmid-Schönbein, G.W. Oxidative stress in the Dahl hypertensive rat. Hypertension 1997, 30, 1628–1633. [Google Scholar]
- Wilcox, C.S. Asymmetric dimethylarginine and reactive oxygen species unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension 2012, 59, 375–381. [Google Scholar]
- Tian, N.; Thrasher, K.D.; Gundy, P.D.; Hughson, M.D.; Manning, R.D. Antioxidant treatment prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension. Hypertension 2005, 45, 934–939. [Google Scholar]
- Sydow, K.; Münzel, T. ADMA and oxidative stress. Atheroscl. Suppl 2003, 4, 41–51. [Google Scholar]
- Achan, V.; Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.; Vallance, P. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arteriosc. Thromb. Vasc. Biol 2003, 23, 1455–1459. [Google Scholar]
- Pope, A.J.; Karrupiah, K.; Kearns, P.N.; Xia, Y.; Cardounel, A.J. Role of dimethylarginine dimethylaminohydrolases in the regulation of endothelial nitric oxide production. J. Biol. Chem 2009, 284, 35338–35347. [Google Scholar]
- Liu, F.; Mu, J.; Liu, Z.; Shi, D.; Huang, Q.; Yuan, Z.; Lian, Q.; Zheng, S. Endothelial dysfunction in normotensive salt-sensitive subjects. J. Hum. Hypertens 2011, 26, 247–252. [Google Scholar]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Purification and properties of a new enzyme, NG, NG-dimethylarginine dimethylaminohydrolase, from rat kidney. J. Biol. Chem 1989, 264, 10205–10209. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | SBP (mmHg) | Weight (g) |
|---|---|---|
| 13-BN normal diet (n = 8) | 138.6 ± 4.8 | 345.3 ± 19.3 |
| 13-BN high salt diet (n = 8) | 147.6 ± 5.1 * | 342.6 ± 5.1 |
| SS normal diet (n = 8) | 147.3 ± 12.6 | 321.8 ± 17.1 |
| SS high salt diet (n = 8) | 160.2 ± 10.3 ** | 365.5 ± 19.4 ** |
| SS high salt diet + hydralazine (n = 8) | 144.7 ± 5.9 | 316.7 ± 17.7 |
| Group | Plasma ADMA (μg/mL)/(μmol/L) | Plasma NOx (μmol/L) |
|---|---|---|
| 13-BN normal diet (n = 8) | 64.7 ± 4.3 (320.3 ± 21.3) | 25.6 ± 2.3 |
| 13-BN high salt diet (n = 8) | 67.5 ± 8.8 (334.2 ± 43.6) | 20.2 ± 3.2 |
| SS normal diet (n = 8) | 61.4 ± 5.3 (304.0 ± 26.2) | 30.1 ± 2.4 |
| SS high salt diet (n = 8) | 75.7 ± 12.1 (374.8 ± 74.6) ** | 12.4 ± 1.7 $ |
| SS high salt diet + hydralazine (n = 8) | 65.2 ± 2.4 (322.8 ± 11.9) | 20.3 ± 4.6 ** |
| Gene | Nucleotide sequence | |
|---|---|---|
| GAPDH | Forward primer | ATGGTGAAGGTCGGTGTGAACG |
| Reverse primer | CGCTCCTGGAAGATGGTGATGG | |
| PRMT-1 | Forward primer | GTGACAGCCATTGAGGACCG |
| Reverse primer | TGTGGCATCGGGTGAACTCG | |
| DDAH-1 | Forward primer | ACAGTCCCCGTGGCCGATTCTT |
| Reverse primer | TGGGGTTCGGTGCAGCAAGA | |
| DDAH-2 | Forward primer | GACACGGCTCTAATCACAAG |
| Reverse primer | AGCGTAGCGTTCTCATCC | |
| eNOS | Forward primer | TTGGACAAGTCCTCACCGCC |
| Reverse primer | TGGGTGCGCAATGTGAGTCC | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cao, Y.; Mu, J.-J.; Fang, Y.; Yuan, Z.-Y.; Liu, F.-Q. Impact of High Salt Independent of Blood Pressure on PRMT/ADMA/DDAH Pathway in the Aorta of Dahl Salt-Sensitive Rats. Int. J. Mol. Sci. 2013, 14, 8062-8072. https://doi.org/10.3390/ijms14048062
Cao Y, Mu J-J, Fang Y, Yuan Z-Y, Liu F-Q. Impact of High Salt Independent of Blood Pressure on PRMT/ADMA/DDAH Pathway in the Aorta of Dahl Salt-Sensitive Rats. International Journal of Molecular Sciences. 2013; 14(4):8062-8072. https://doi.org/10.3390/ijms14048062
Chicago/Turabian StyleCao, Yu, Jian-Jun Mu, Yuan Fang, Zu-Yi Yuan, and Fu-Qiang Liu. 2013. "Impact of High Salt Independent of Blood Pressure on PRMT/ADMA/DDAH Pathway in the Aorta of Dahl Salt-Sensitive Rats" International Journal of Molecular Sciences 14, no. 4: 8062-8072. https://doi.org/10.3390/ijms14048062