Biophysical Techniques for Detection of cAMP and cGMP in Living Cells

Abstract
:1. Introduction
2. Techniques to Measure Cyclic Nucleotides
2.1. Biochemical Methods
2.2. Cyclic Nucleotide Gated Channels (CNGCs)
2.3. Förster Resonance Energy Transfer (FRET) Based Sensors
2.3.1. FRET Sensors to Detect cAMP
2.3.1.1. Protein Kinase A (PKA) Based cAMP Sensors
2.3.1.2. Epac-Based cAMP Sensors
2.3.1.3. Cyclic Nucleotide Gated Channels (CNGC) Based cAMP Sensors
2.3.2. FRET Sensors to Detect cGMP
2.4. Single GFP-Linked Biosensors
2.5. Bioluminescence Resonance Energy Transfer (BRET) Based Sensors
3. Analysis of Compartmentalized Cyclic Nucleotide Signaling
4. Conclusions and Outlook
Acknowledgments
Conflict of Interest
References
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research -- still expanding after half a century. Nat. Rev. Mol. Cell Biol 2002, 3, 710–718. [Google Scholar]
- Kandel, E.R. The molecular biology of memory storage: A dialogue between genes and synapses. Science 2001, 294, 1030–1038. [Google Scholar]
- Morozov, A.; Muzzio, I.A.; Bourtchouladze, R.; Van-Strien, N.; Lapidus, K.; Yin, D.; Winder, D.G.; Adams, J.P.; Sweatt, J.D.; Kandel, E.R. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 2003, 39, 309–325. [Google Scholar]
- Holz, G.G. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes 2004, 53, 5–13. [Google Scholar]
- Tengholm, A.; Gylfe, E. Oscillatory control of insulin secretion. Mol. Cell. Endocrinol 2009, 297, 58–72. [Google Scholar]
- Leech, C.A.; Chepurny, O.G.; Holz, G.G. Epac2-dependent rap1 activation and the control of islet insulin secretion by glucagon-like peptide-1. Vitam. Horm 2010, 84, 279–302. [Google Scholar]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol 2011, 12, 141–151. [Google Scholar]
- Zagotta, W.N.; Olivier, N.B.; Black, K.D.; Young, E.C.; Olson, R.; Gouaux, E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 2003, 425, 200–205. [Google Scholar]
- Brudvik, K.W.; Tasken, K. Modulation of T cell immune functions by the prostaglandin E(2) -cAMP pathway in chronic inflammatory states. Br. J. Pharmacol 2010, 166, 411–419. [Google Scholar]
- Bodor, J.; Bopp, T.; Vaeth, M.; Klein, M.; Serfling, E.; Hunig, T.; Becker, C.; Schild, H.; Schmitt, E. Cyclic AMP underpins suppression by regulatory T cells. Eur. J. Immunol 2012, 42, 1375–1384. [Google Scholar]
- Torgersen, K.M.; Vang, T.; Abrahamsen, H.; Yaqub, S.; Tasken, K. Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell. Signal 2002, 14, 1–9. [Google Scholar]
- Biel, M.; Wahl-Schott, C.; Michalakis, S.; Zong, X. Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev 2009, 89, 847–885. [Google Scholar]
- Craven, K.B.; Zagotta, W.N. CNG and HCN channels: Two peas, one pod. Annu. Rev. Physiol 2006, 68, 375–401. [Google Scholar]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar]
- Gloerich, M.; Bos, J.L. Epac: defining a new mechanism for cAMP action. Annu. Rev. Pharmacol. Toxicol 2010, 50, 355–375. [Google Scholar]
- Tasken, K.; Aandahl, E.M. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev 2004, 84, 137–167. [Google Scholar]
- Taylor, S.S.; Buechler, J.A.; Yonemoto, W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu. Rev. Biochem 1990, 59, 971–1005. [Google Scholar]
- Krebs, E.G.; Beavo, J.A. Phosphorylation-dephosphorylation of enzymes. Annu. Rev. Biochem 1979, 48, 923–959. [Google Scholar]
- Taylor, S.S.; Kim, C.; Cheng, C.Y.; Brown, S.H.; Wu, J.; Kannan, N. Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design. Biochim. Biophys. Acta 2008, 1784, 16–26. [Google Scholar]
- Muller, F.U.; Boknik, P.; Knapp, J.; Linck, B.; Luss, H.; Neumann, J.; Schmitz, W. Activation and inactivation of cAMP-response element-mediated gene transcription in cardiac myocytes. Cardiovasc. Res 2001, 52, 95–102. [Google Scholar]
- Keef, K.D.; Hume, J.R.; Zhong, J. Regulation of cardiac and smooth muscle Ca(2+) channels (Ca(V)1.2a,b) by protein kinases. Am. J. Physiol. Cell. Physiol 2001, 281, C1743–1756. [Google Scholar]
- Takasago, T.; Imagawa, T.; Shigekawa, M. Phosphorylation of the cardiac ryanodine receptor by cAMP-dependent protein kinase. J. Biochem 1989, 106, 872–877. [Google Scholar]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar]
- Kirchberger, M.A.; Tada, M.; Repke, D.I.; Katz, A.M. Cyclic adenosine 3′,5′-monophosphate-dependent protein kinase stimulation of calcium uptake by canine cardiac microsomes. J. Mol. Cell. Cardiol 1972, 4, 673–680. [Google Scholar]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell. Biol 2003, 4, 566–577. [Google Scholar]
- Lompre, A.M.; Hajjar, R.J.; Harding, S.E.; Kranias, E.G.; Lohse, M.J.; Marks, A.R. Ca2+ cycling and new therapeutic approaches for heart failure. Circulation 2010, 121, 822–830. [Google Scholar]
- Kuhn, M. Structure, regulation, and function of mammalian membrane guanylyl cyclase receptors, with a focus on guanylyl cyclase-A. Circ. Res 2003, 93, 700–709. [Google Scholar]
- Potter, L.R.; Abbey-Hosch, S.; Dickey, D.M. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr. Rev 2006, 27, 47–72. [Google Scholar]
- Pyriochou, A.; Papapetropoulos, A. Soluble guanylyl cyclase: more secrets revealed. Cell Signal 2005, 17, 407–413. [Google Scholar]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev 2006, 86, 1–23. [Google Scholar]
- Stangherlin, A.; Zaccolo, M. cGMP-cAMP interplay in cardiac myocytes: A local affair with far-reaching consequences for heart function. Biochem. Soc. Trans 2012, 40, 11–14. [Google Scholar]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res 2007, 100, 1569–1578. [Google Scholar]
- Brunton, L.L.; Hayes, J.S.; Mayer, S.E. Hormonally specific phosphorylation of cardiac troponin I and activation of glycogen phosphorylase. Nature 1979, 280, 78–80. [Google Scholar]
- Brunton, L.L.; Hayes, J.S.; Mayer, S.E. Functional compartmentation of cyclic AMP and protein kinase in heart. Adv. Cyclic Nucleotide Res 1981, 14, 391–397. [Google Scholar]
- Buxton, I.L.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem 1983, 258, 10233–10239. [Google Scholar]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevicius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ. Res 2006, 99, 816–828. [Google Scholar]
- Zaccolo, M. cAMP signal transduction in the heart: Understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol 2009, 158, 50–60. [Google Scholar]
- Diviani, D.; Dodge-Kafka, K.L.; Li, J.; Kapiloff, M.S. A-kinase anchoring proteins: scaffolding proteins in the heart. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H1742–H1753. [Google Scholar]
- Mauban, J.R.; O’Donnell, M.; Warrier, S.; Manni, S.; Bond, M. AKAP-scaffolding proteins and regulation of cardiac physiology. Physiology (Bethesda) 2009, 24, 78–87. [Google Scholar]
- Scott, J.D.; Dessauer, C.W.; Tasken, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol 2013, 53, 187–210. [Google Scholar]
- Troger, J.; Moutty, M.C.; Skroblin, P.; Klussmann, E. A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol 2012, 166, 420–433. [Google Scholar]
- Rapundalo, S.T.; Solaro, R.J.; Kranias, E.G. Inotropic responses to isoproterenol and phosphodiesterase inhibitors in intact guinea pig hearts: Comparison of cyclic AMP levels and phosphorylation of sarcoplasmic reticulum and myofibrillar proteins. Circ. Res 1989, 64, 104–111. [Google Scholar]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Hamilton, G.; et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res 2011, 108, 929–939. [Google Scholar]
- Brooker, G.; Harper, J.F.; Terasaki, W.L.; Moylan, R.D. Radioimmunoassay of cyclic AMP and cyclic GMP. Adv. Cyclic Nucleotide Res 1979, 10, 1–33. [Google Scholar]
- Harper, J.F.; Brooker, G. Femtomole sensitive radioimmunoassay for cyclic AMP and cyclic GMP after 2′0 acetylation by acetic anhydride in aqueous solution. J. Cyclic Nucleotide Res 1975, 1, 207–218. [Google Scholar]
- Williams, C. cAMP detection methods in HTS: selecting the best from the rest. Nat. Rev. Drug Discov 2004, 3, 125–135. [Google Scholar]
- Finn, J.T.; Grunwald, M.E.; Yau, K.W. Cyclic nucleotide-gated ion channels: an extended family with diverse functions. Annu. Rev. Physiol 1996, 58, 395–426. [Google Scholar]
- Biel, M.; Zong, X.; Ludwig, A.; Sautter, A.; Hofmann, F. Structure and function of cyclic nucleotide-gated channels. Rev. Physiol. Biochem. Pharmacol 1999, 135, 151–171. [Google Scholar]
- Frings, S.; Seifert, R.; Godde, M.; Kaupp, U.B. Profoundly different calcium permeation and blockage determine the specific function of distinct cyclic nucleotide-gated channels. Neuron 1995, 15, 169–179. [Google Scholar]
- Abi-Gerges, A.; Richter, W.; Lefebvre, F.; Mateo, P.; Varin, A.; Heymes, C.; Samuel, J.L.; Lugnier, C.; Conti, M.; Fischmeister, R.; et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ. Res 2009, 105, 784–792. [Google Scholar]
- Ghigo, A.; Perino, A.; Mehel, H.; Zahradnikova, A., Jr; Morello, F.; Leroy, J.; Nikolaev, V.O.; Damilano, F.; Cimino, J.; De Luca, E.; et al. Phosphoinositide 3-kinase gamma protects against catecholamine-induced ventricular arrhythmia through protein kinase A-mediated regulation of distinct phosphodiesterases. Circulation 2012, 126, 2073–2083. [Google Scholar]
- Rochais, F.; Vandecasteele, G.; Lefebvre, F.; Lugnier, C.; Lum, H.; Mazet, J.L.; Cooper, D.M.; Fischmeister, R. Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: An in vivo study using adenovirus-mediated expression of CNG channels. J. Biol. Chem 2004, 279, 52095–52105. [Google Scholar]
- Rich, T.C.; Fagan, K.A.; Nakata, H.; Schaack, J.; Cooper, D.M.; Karpen, J.W. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol 2000, 116, 147–161. [Google Scholar]
- Rich, T.C.; Tse, T.E.; Rohan, J.G.; Schaack, J.; Karpen, J.W. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J. Gen. Physiol 2001, 118, 63–78. [Google Scholar]
- Fesenko, E.E.; Kolesnikov, S.S.; Lyubarsky, A.L. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature 1985, 313, 310–313. [Google Scholar]
- Koutalos, Y.; Nakatani, K.; Yau, K.W. The cGMP-phosphodiesterase and its contribution to sensitivity regulation in retinal rods. J. Gen. Physiol 1995, 106, 891–921. [Google Scholar]
- Baylor, D.A.; Lamb, T.D.; Yau, K.W. Responses of retinal rods to single photons. J. Physiol 1979, 288, 613–634. [Google Scholar]
- Nakatani, K.; Yau, K.W. Guanosine 3′,5′-cyclic monophosphate-activated conductance studied in a truncated rod outer segment of the toad. J. Physiol 1988, 395, 731–753. [Google Scholar]
- Castro, L.R.; Verde, I.; Cooper, D.M.; Fischmeister, R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 2006, 113, 2221–2228. [Google Scholar]
- Castro, L.R.; Schittl, J.; Fischmeister, R. Feedback control through cGMP-dependent protein kinase contributes to differential regulation and compartmentation of cGMP in rat cardiac myocytes. Circ. Res 2010, 107, 1232–1240. [Google Scholar]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Physik 1948, 437, 55–75. [Google Scholar]
- Wu, P.; Brand, L. Resonance energy transfer: methods and applications. Anal. Biochem 1994, 218, 1–13. [Google Scholar]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991, 349, 694–697. [Google Scholar]
- Gorbunova, Y.V.; Spitzer, N.C. Dynamic interactions of cyclic AMP transients and spontaneous Ca(2+) spikes. Nature 2002, 418, 93–96. [Google Scholar]
- Bacskai, B.J.; Hochner, B.; Mahaut-Smith, M.; Adams, S.R.; Kaang, B.K.; Kandel, E.R.; Tsien, R.Y. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science 1993, 260, 222–226. [Google Scholar]
- Hempel, C.M.; Vincent, P.; Adams, S.R.; Tsien, R.Y.; Selverston, A.I. Spatio-temporal dynamics of cyclic AMP signals in an intact neural circuitm. Nature 1996, 384, 166–169. [Google Scholar]
- Webb, R.J.; Marshall, F.; Swann, K.; Carroll, J. Follicle-stimulating hormone induces a gap junction-dependent dynamic change in [cAMP] and protein kinase a in mammalian oocytes. Dev. Biol 2002, 246, 441–454. [Google Scholar]
- Takeda, N.; Kyozuka, K.; Deguchi, R. Increase in intracellular cAMP is a prerequisite signal for initiation of physiological oocyte meiotic maturation in the hydrozoan Cytaeis uchidae. Dev. Biol 2006, 298, 248–258. [Google Scholar]
- Goaillard, J.M.; Vincent, P.V.; Fischmeister, R. Simultaneous measurements of intracellular cAMP and L-type Ca2+ current in single frog ventricular myocytes. J. Physiol 2001, 530, 79–91. [Google Scholar]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem 1998, 67, 509–544. [Google Scholar]
- Zaccolo, M.; De Giorgi, F.; Cho, C.Y.; Feng, L.; Knapp, T.; Negulescu, P.A.; Taylor, S.S.; Tsien, R.Y.; Pozzan, T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell. Biol. 2000, 2, 25–29. [Google Scholar]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res 2004, 95, 67–75. [Google Scholar]
- Lehnart, S.E.; Wehrens, X.H.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar]
- Warrier, S.; Belevych, A.E.; Ruse, M.; Eckert, R.L.; Zaccolo, M.; Pozzan, T.; Harvey, R.D. Beta-adrenergic- and muscarinic receptor-induced changes in cAMP activity in adult cardiac myocytes detected with FRET-based biosensor. Am. J. Physiol. Cell Physiol 2005, 289, C455–C461. [Google Scholar]
- Diller, T.C.; Madhusudan; Xuong, N.H.; Taylor, S.S. Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II beta regulatory subunit. Structure 2001, 9, 73–82. [Google Scholar]
- Nikolaev, V.O.; Bunemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem 2004, 279, 37215–37218. [Google Scholar]
- Allen, M.D.; Zhang, J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem. Biophys Res. Commun 2006, 348, 716–721. [Google Scholar]
- Zhang, J.; Hupfeld, C.J.; Taylor, S.S.; Olefsky, J.M.; Tsien, R.Y. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature 2005, 437, 569–573. [Google Scholar]
- Zhang, J.; Ma, Y.; Taylor, S.S.; Tsien, R.Y. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA 2001, 98, 14997–15002. [Google Scholar]
- Depry, C.; Allen, M.D.; Zhang, J. Visualization of PKA activity in plasma membrane microdomains. Mol. Biosyst 2011, 7, 52–58. [Google Scholar]
- DiPilato, L.M.; Cheng, X.; Zhang, J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. USA 2004, 101, 16513–16518. [Google Scholar]
- Ponsioen, B.; Zhao, J.; Riedl, J.; Zwartkruis, F.; van der Krogt, G.; Zaccolo, M.; Moolenaar, W.H.; Bos, J.L.; Jalink, K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep 2004, 5, 1176–1180. [Google Scholar]
- Nikolaev, V.O.; Gambaryan, S.; Engelhardt, S.; Walter, U.; Lohse, M.J. Real-time monitoring of the PDE2 activity of live cells: hormone-stimulated cAMP hydrolysis is faster than hormone-stimulated cAMP synthesis. J. Biol. Chem 2005, 280, 1716–1719. [Google Scholar]
- Calebiro, D.; Nikolaev, V.O.; Gagliani, M.C.; de Filippis, T.; Dees, C.; Tacchetti, C.; Persani, L.; Lohse, M.J. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 2009, 7, e1000172. [Google Scholar]
- Violin, J.D.; DiPilato, L.M.; Yildirim, N.; Elston, T.C.; Zhang, J.; Lefkowitz, R.J. beta2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J. Biol. Chem 2008, 283, 2949–2961. [Google Scholar]
- Nikolaev, V.O.; Bunemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ. Res 2006, 99, 1084–1091. [Google Scholar]
- Nikolaev, V.O.; Lohse, M.J. Novel techniques for real-time monitoring of cGMP in living cells. Handb. Exp. Pharmacol. 2009, 229–243. [Google Scholar]
- Honda, A.; Adams, S.R.; Sawyer, C.L.; Lev-Ram, V.; Tsien, R.Y.; Dostmann, W.R. Spatiotemporal dynamics of guanosine 3′,5′-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proc. Natl. Acad. Sci. USA 2001, 98, 2437–2442. [Google Scholar]
- Russwurm, M.; Mullershausen, F.; Friebe, A.; Jager, R.; Russwurm, C.; Koesling, D. Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: A systematic approach. Biochem. J 2007, 407, 69–77. [Google Scholar]
- Sato, M.; Hida, N.; Ozawa, T.; Umezawa, Y. Fluorescent indicators for cyclic GMP based on cyclic GMP-dependent protein kinase Ialpha and green fluorescent proteins. Anal. Chem 2000, 72, 5918–5924. [Google Scholar]
- Cawley, S.M.; Sawyer, C.L.; Brunelle, K.F.; van der Vliet, A.; Dostmann, W.R. Nitric oxide-evoked transient kinetics of cyclic GMP in vascular smooth muscle cells. Cell Signal 2007, 19, 1023–1033. [Google Scholar]
- Honda, A.; Moosmeier, M.A.; Dostmann, W.R. Membrane-permeable cygnets: Rapid cellular internalization of fluorescent cGMP-indicators. Front. Biosci 2005, 10, 1290–1301. [Google Scholar]
- Honda, A.; Sawyer, C.L.; Cawley, S.M.; Dostmann, W.R. Cygnets: In vivo characterization of novel cGMP indicators and in vivo imaging of intracellular cGMP. Methods Mol. Biol 2005, 307, 27–43. [Google Scholar]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res 2006, 98, 226–234. [Google Scholar]
- Takimoto, E.; Champion, H.C.; Belardi, D.; Moslehi, J.; Mongillo, M.; Mergia, E.; Montrose, D.C.; Isoda, T.; Aufiero, K.; Zaccolo, M.; et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ. Res 2005, 96, 100–109. [Google Scholar]
- Nikolaev, V.O.; Gambaryan, S.; Lohse, M.J. Fluorescent sensors for rapid monitoring of intracellular cGMP. Nat. Methods 2006, 3, 23–25. [Google Scholar]
- Niino, Y.; Hotta, K.; Oka, K. Simultaneous live cell imaging using dual FRET sensors with a single excitation light. PLoS One 2009, 4, e6036. [Google Scholar]
- Iancu, R.V.; Ramamurthy, G.; Warrier, S.; Nikolaev, V.O.; Lohse, M.J.; Jones, S.W.; Harvey, R.D. Cytoplasmic cAMP concentrations in intact cardiac myocytes. Am. J. Physiol. Cell Physiol 2008, 295, C414–422. [Google Scholar]
- Willemse, M.; Janssen, E.; de Lange, F.; Wieringa, B.; Fransen, J. ATP and FRET--a cautionary note. Nat. Biotechnol 2007, 25, 170–172. [Google Scholar]
- Salonikidis, P.S.; Niebert, M.; Ullrich, T.; Bao, G.; Zeug, A.; Richter, D.W. An ion-insensitive cAMP biosensor for long term quantitative ratiometric fluorescence resonance energy transfer (FRET) measurements under variable physiological conditions. J. Biol. Chem 2011, 286, 23419–23431. [Google Scholar]
- Börner, S.; Schwede, F.; Schlipp, A.; Berisha, F.; Calebiro, D.; Lohse, M.J.; Nikolaev, V.O. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat. Protoc 2011, 6, 427–438. [Google Scholar]
- Norris, R.P.; Ratzan, W.J.; Freudzon, M.; Mehlmann, L.M.; Krall, J.; Movsesian, M.A.; Wang, H.; Ke, H.; Nikolaev, V.O.; Jaffe, L.A. Cyclic GMP from the surrounding somatic cells regulates cyclic AMP and meiosis in the mouse oocyte. Development 2009, 136, 1869–1878. [Google Scholar]
- Piggott, L.A.; Hassell, K.A.; Berkova, Z.; Morris, A.P.; Silberbach, M.; Rich, T.C. Natriuretic peptides and nitric oxide stimulate cGMP synthesis in different cellular compartments. J. Gen. Physiol 2006, 128, 3–14. [Google Scholar]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther 2009, 122, 216–238. [Google Scholar]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [Google Scholar]
- Nausch, L.W.; Ledoux, J.; Bonev, A.D.; Nelson, M.T.; Dostmann, W.R. Differential patterning of cGMP in vascular smooth muscle cells revealed by single GFP-linked biosensors. Proc. Natl. Acad. Sci. USA 2008, 105, 365–370. [Google Scholar]
- Isner, J.C.; Maathuis, F.J. Measurement of cellular cGMP in plant cells and tissues using the endogenous fluorescent reporter FlincG. Plant. J 2011, 65, 329–334. [Google Scholar]
- Isner, J.C.; Nuhse, T.; Maathuis, F.J. The cyclic nucleotide cGMP is involved in plant hormone signalling and alters phosphorylation of Arabidopsis thaliana root proteins. J. Exp. Bot 2012, 63, 3199–3205. [Google Scholar]
- Wachten, S.; Masada, N.; Ayling, L.J.; Ciruela, A.; Nikolaev, V.O.; Lohse, M.J.; Cooper, D.M. Distinct pools of cAMP centre on different isoforms of adenylyl cyclase in pituitary-derived GH3B6 cells. J. Cell. Sci 2010, 123, 95–106. [Google Scholar]
- Mohamed, T.M.; Oceandy, D.; Zi, M.; Prehar, S.; Alatwi, N.; Wang, Y.; Shaheen, M.A.; Abou-Leisa, R.; Schelcher, C.; Hegab, Z.; et al. Plasma membrane calcium pump (PMCA4)- neuronal nitric-oxide synthase complex regulates cardiac contractility through modulation of a compartmentalized cyclic nucleotide microdomain. J. Biol. Chem 2011, 286, 41520–41529. [Google Scholar]
- Boute, N.; Jockers, R.; Issad, T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol. Sci 2002, 23, 351–354. [Google Scholar]
- Willoughby, D.; Cooper, D.M. Live-cell imaging of cAMP dynamics. Nat. Methods 2008, 5, 29–36. [Google Scholar]
- Pfleger, K.D.; Eidne, K.A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 2006, 3, 165–174. [Google Scholar]
- Prinz, A.; Diskar, M.; Erlbruch, A.; Herberg, F.W. Novel, isotype-specific sensors for protein kinase A subunit interaction based on bioluminescence resonance energy transfer (BRET). Cell Signal 2006, 18, 1616–1625. [Google Scholar]
- Barak, L.S.; Salahpour, A.; Zhang, X.; Masri, B.; Sotnikova, T.D.; Ramsey, A.J.; Violin, J.D.; Lefkowitz, R.J.; Caron, M.G.; Gainetdinov, R.R. Pharmacological characterization of membrane-expressed human trace amine-associated receptor 1 (TAAR1) by a bioluminescence resonance energy transfer cAMP biosensor. Mol. Pharmacol 2008, 74, 585–594. [Google Scholar]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.M.; DeCamp, D.; Roach, T.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem 2007, 282, 10576–10584. [Google Scholar]
- Matthiesen, K.; Nielsen, J. Cyclic AMP control measured in two compartments in HEK293 cells: phosphodiesterase K(M) is more important than phosphodiesterase localization. PLoS One 2011, 6, e24392. [Google Scholar]
- Biswas, K.H.; Sopory, S.; Visweswariah, S.S. The GAF domain of the cGMP-binding, cGMP-specific phosphodiesterase (PDE5) is a sensor and a sink for cGMP. Biochemistry 2008, 47, 3534–3543. [Google Scholar]
- Jager, R.; Schwede, F.; Genieser, H.G.; Koesling, D.; Russwurm, M. Activation of PDE2 and PDE5 by specific GAF ligands: delayed activation of PDE5. Br. J. Pharmacol 2010, 161, 1645–1660. [Google Scholar]
- Hayes, J.S.; Brunton, L.L.; Brown, J.H.; Reese, J.B.; Mayer, S.E. Hormonally specific expression of cardiac protein kinase activity. Proc. Natl. Acad. Sci. USA 1979, 76, 1570–1574. [Google Scholar]
- Jurevicius, J.; Fischmeister, R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc. Natl. Acad. Sci. USA 1996, 93, 295–299. [Google Scholar]
- Rochais, F.; Abi-Gerges, A.; Horner, K.; Lefebvre, F.; Cooper, D.M.; Conti, M.; Fischmeister, R.; Vandecasteele, G. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ. Res 2006, 98, 1081–1088. [Google Scholar]
- Terrin, A.; Di Benedetto, G.; Pertegato, V.; Cheung, Y.F.; Baillie, G.; Lynch, M.J.; Elvassore, N.; Prinz, A.; Herberg, F.W.; Houslay, M.D.; et al. PGE(1) stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J. Cell. Biol 2006, 175, 441–451. [Google Scholar]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res 2008, 103, 836–844. [Google Scholar]
- Herget, S.; Lohse, M.J.; Nikolaev, V.O. Real-time monitoring of phosphodiesterase inhibition in intact cells. Cell Signal 2008, 20, 1423–1431. [Google Scholar]
- Sin, Y.Y.; Edwards, H.V.; Li, X.; Day, J.P.; Christian, F.; Dunlop, A.J.; Adams, D.R.; Zaccolo, M.; Houslay, M.D.; Baillie, G.S. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the beta-agonist induced hypertrophic response in cardiac myocytes. J. Mol. Cell Cardiol 2011, 50, 872–883. [Google Scholar]
- Liu, S.; Zhang, J.; Xiang, Y.K. FRET-based direct detection of dynamic protein kinase A activity on the sarcoplasmic reticulum in cardiomyocytes. Biochem. Biophys. Res. Commun 2011, 404, 581–586. [Google Scholar]
- Dyachok, O.; Isakov, Y.; Sagetorp, J.; Tengholm, A. Oscillations of cyclic AMP in hormone-stimulated insulin-secreting beta-cells. Nature 2006, 439, 349–352. [Google Scholar]
- Sample, V.; DiPilato, L.M.; Yang, J.H.; Ni, Q.; Saucerman, J.J.; Zhang, J. Regulation of nuclear PKA revealed by spatiotemporal manipulation of cyclic AMP. Nat. Chem. Biol 2012, 8, 375–382. [Google Scholar]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar]
- Steyer, J.A.; Almers, W. A real-time view of life within 100 nm of the plasma membrane. Nat. Rev. Mol. Cell Biol 2001, 2, 268–275. [Google Scholar]
- Dyachok, O.; Idevall-Hagren, O.; Sagetorp, J.; Tian, G.; Wuttke, A.; Arrieumerlou, C.; Akusjarvi, G.; Gylfe, E.; Tengholm, A. Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab 2008, 8, 26–37. [Google Scholar]


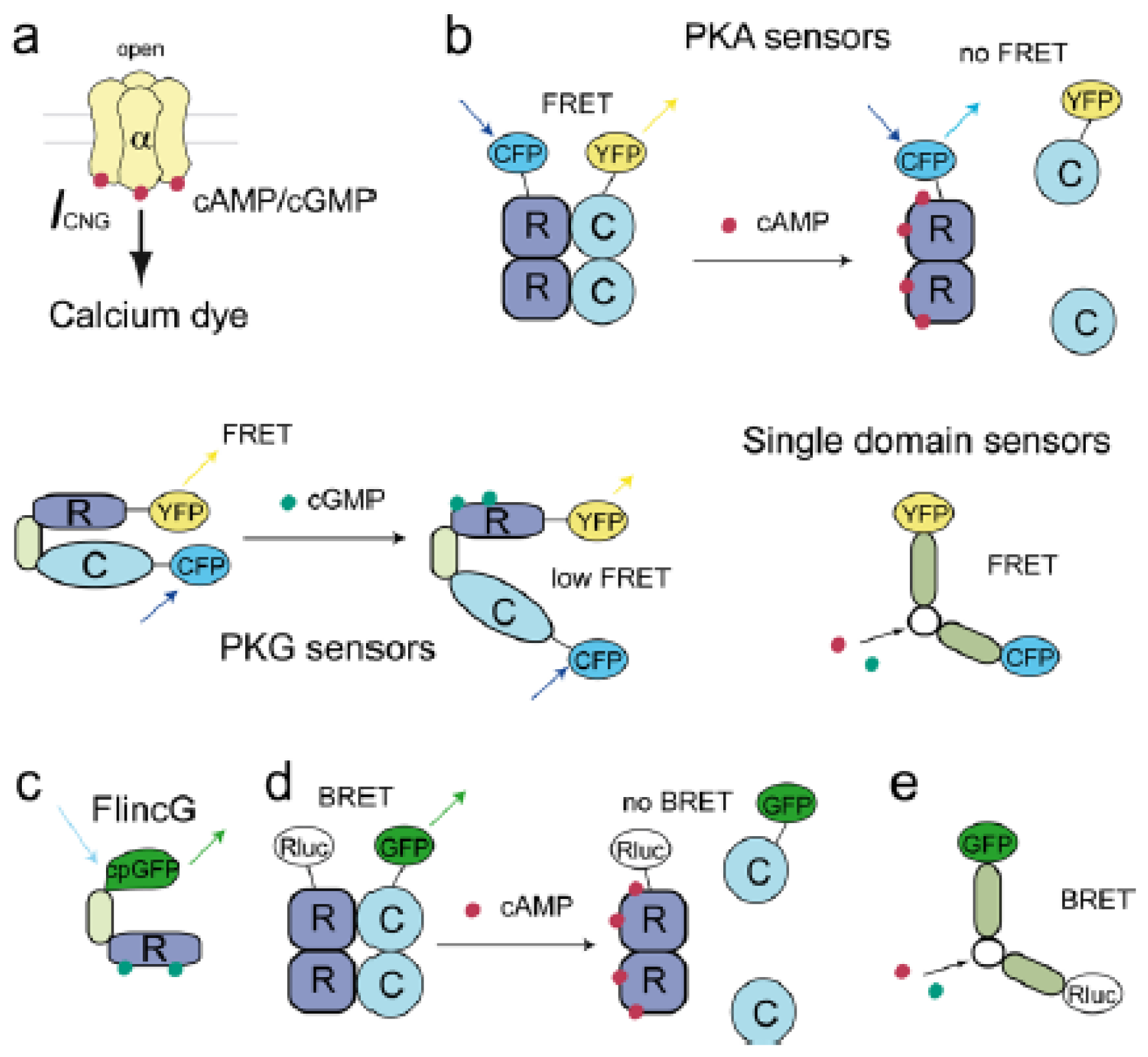{kind=link}
| Biosensor | Sensitivity | Advantages/Disadvantages | References |
|---|---|---|---|
| CNGC -subunit wildtype C460W/E583M mutant | cAMP EC50 = 36 μM cGMP EC50 = 1.6 μM cAMP EC50 = 1 μM | Low cAMP/cGMP selectivity. Restriction to the subsarcolemmal compartment | [53,55] |
| FRET based biosensors: | |||
| FlCRhR (PKA based) | cAMP EC50 = 90 nM | Chemical labeling, purification and microinjection Relatively slow kinetics | [64] |
| R-CFP, C-YFP (PKA based) | cAMP EC50 = 0.5–0.9 μM | Multimeric. Here and below: genetically encoded | [72–74] |
| PKA-camps (PKA based) | cAMP EC50 = 1.9 μM | Single-chain architecture | [78] |
| AKAR1-3 | Not applicable | Measures PKA catalytic activity in real time | [79–81] |
| AKAR4 | Not applicable | Improved dynamic range | [82] |
| Epac1/2-camps (Epac based) | cAMP EC50 = 2.4/0.9 μM | Single-chain. Faster kinetics than for multimeric sensors | [78] |
| Epac2-camp300 CFP-Epac-YFP ( DEP,CD) | cAMP EC50 = 300 nM cAMP EC50 ~ 50 μM cAMP EC50 ~ 15 μM | High sensitivity Single-chain. Relatively low sensitivity | [104] [84,111,112] |
| ICUE1/2 (Epac based) | cAMP EC50 ~ 10–50 μM | As above | [83,87] |
| HCN2-camps (CNGC based) | cAMP EC50 = 6 μM | Good for cells with high basal cAMP concentrations | [88] |
| CGY-Del1 | cGMP EC50 = 20 nM | Low cGMP/cAMP selectivity | [92,98] |
| Cygnet-1/2 (PKG based) | cGMP EC50 = 1.5/1.9 μM | Single-chain. Relatively low sensitivity and temporal resolution | [90] |
| cGES-DE2/5 (PDE2/5 based) | cGMP EC50 = 0.9/1.5 μM | Small size. Relatively low sensitivity | [98] |
| redcGES-DE5 (PDE5 based) | cGMP EC50 = 40 nM | High sensitivity | [99] |
| cGi-500/3000/6000 (PKG based) | cGMP EC50 = 500/3000/6000 nM | Small size. Relatively high sensitivity and dynamic range. Fast kinetics | [91] |
| Non-FRET sensors: FlincGs cGMP | EC50 = 150nM (δ-FlincG) | Good dynamic range. Rapid kinetics | [108] |
| Targeted biosensor | Structure | Microdomain | References |
|---|---|---|---|
| RI_epac and RII_epac | N-terminal dimerization-docking domains of RI or RII fused to Epac1-camps | PKA-RI and PKA-RII | [126] |
| Epac1-camps-PDE3/4 | Fusion of Epac1-camps to N-terminus of PDEs | PDE3/4 | [127] |
| cGES-DE2-PDE5 | Fusion of cGES-DE2 to N-terminus of PDE5 | PDE5 | [127] |
| Epac1-camps-Hsp20 | Fusion of Epac1-camps to Hsp20 | Hsp20 | [128] |
| SR-AKAR3 | Fusion of AKAR3 to the N-terminal helical transmembrane domain of phospholamban | SR membrane | [129] |
| pm PKA-RII-CFP/C-YFP | 26 amino acid CAAX box sequence fused to the C-terminus of PKA-CFP | Subsarcolemmal | [130] |
| pmEpac2-camps and AC8-Epac2-camps pmEpac1-camps | 10 amino acid sequence form Lyn kinase or AC8 are fused to N-terminus of Epac1/2-camps | Subsarcolemmal caveolar or associated with AC8 | [111,112] |
| pm ICUE, NLS-ICUE, | mitoICUE Fusions of ICUE to CAAX box, nuclear localization signals or two different mitochondrial sequences | Subsarcolemmal, nuclear, mitochondrial | [83,131] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sprenger, J.U.; Nikolaev, V.O. Biophysical Techniques for Detection of cAMP and cGMP in Living Cells. Int. J. Mol. Sci. 2013, 14, 8025-8046. https://doi.org/10.3390/ijms14048025
Sprenger JU, Nikolaev VO. Biophysical Techniques for Detection of cAMP and cGMP in Living Cells. International Journal of Molecular Sciences. 2013; 14(4):8025-8046. https://doi.org/10.3390/ijms14048025
Chicago/Turabian StyleSprenger, Julia U., and Viacheslav O. Nikolaev. 2013. "Biophysical Techniques for Detection of cAMP and cGMP in Living Cells" International Journal of Molecular Sciences 14, no. 4: 8025-8046. https://doi.org/10.3390/ijms14048025