P4 ATPases: Flippases in Health and Disease

Abstract
:1. Phospholipid Asymmetry in Biological Membranes
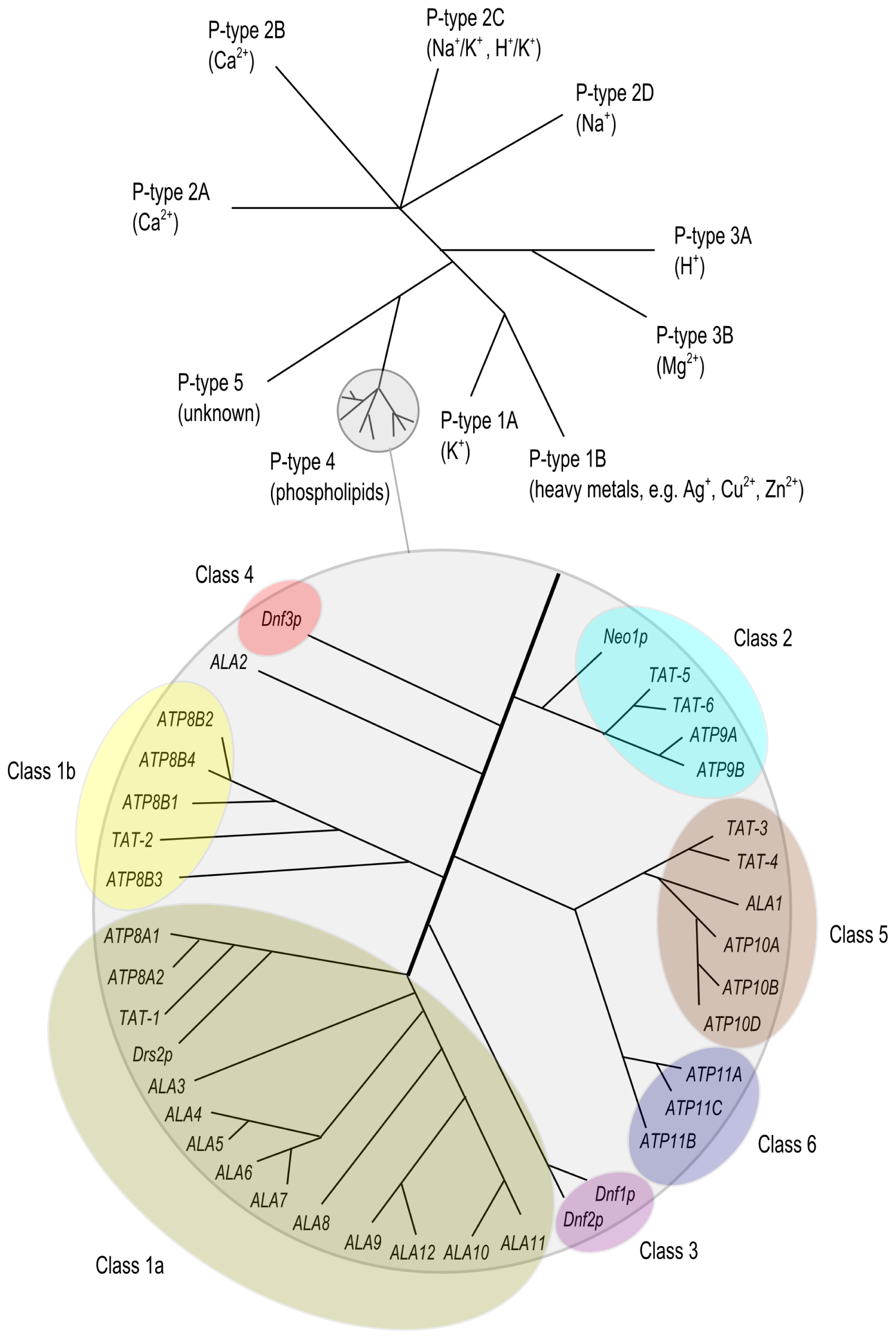
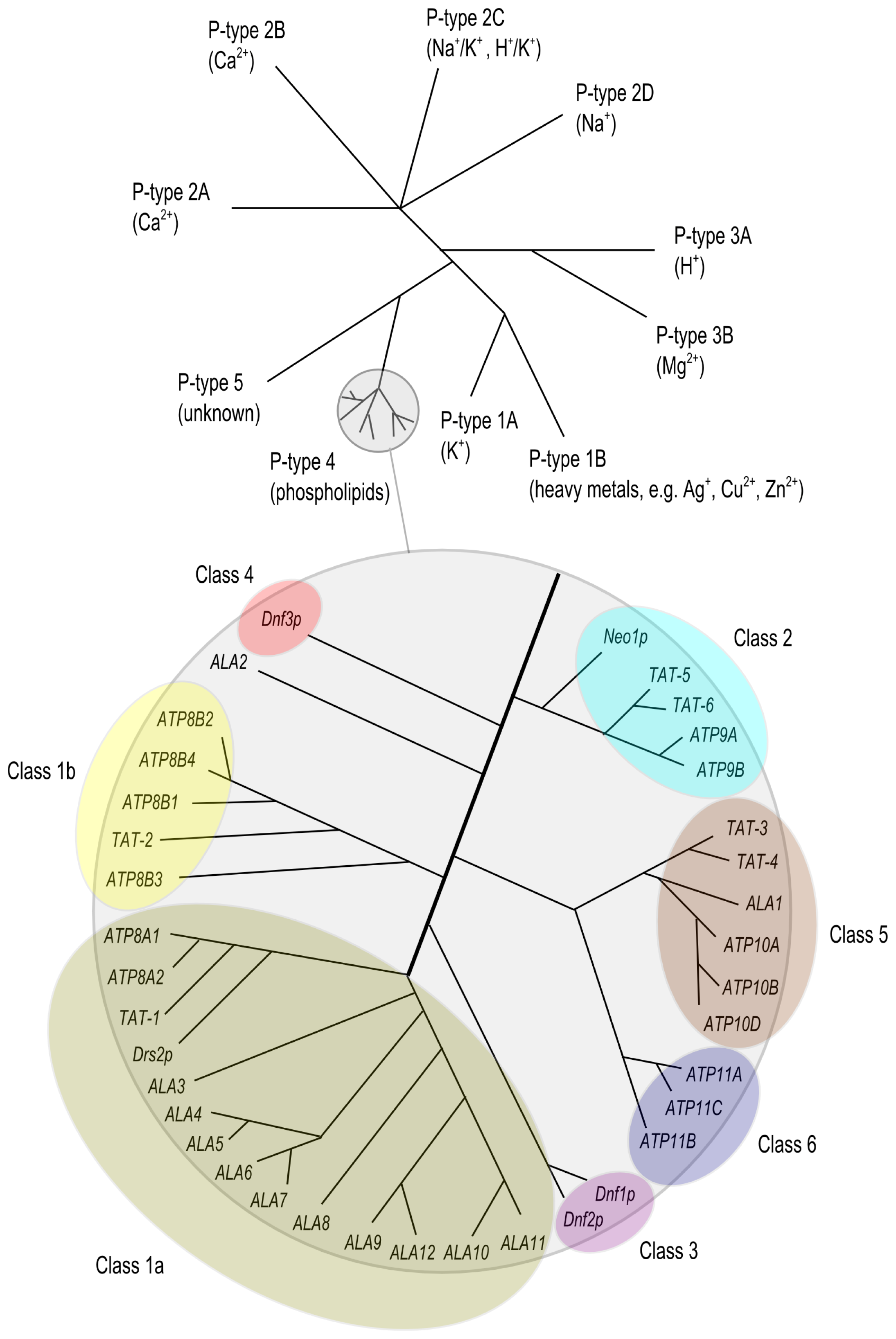
2. The P4 ATPase Family of Lipid Flippases
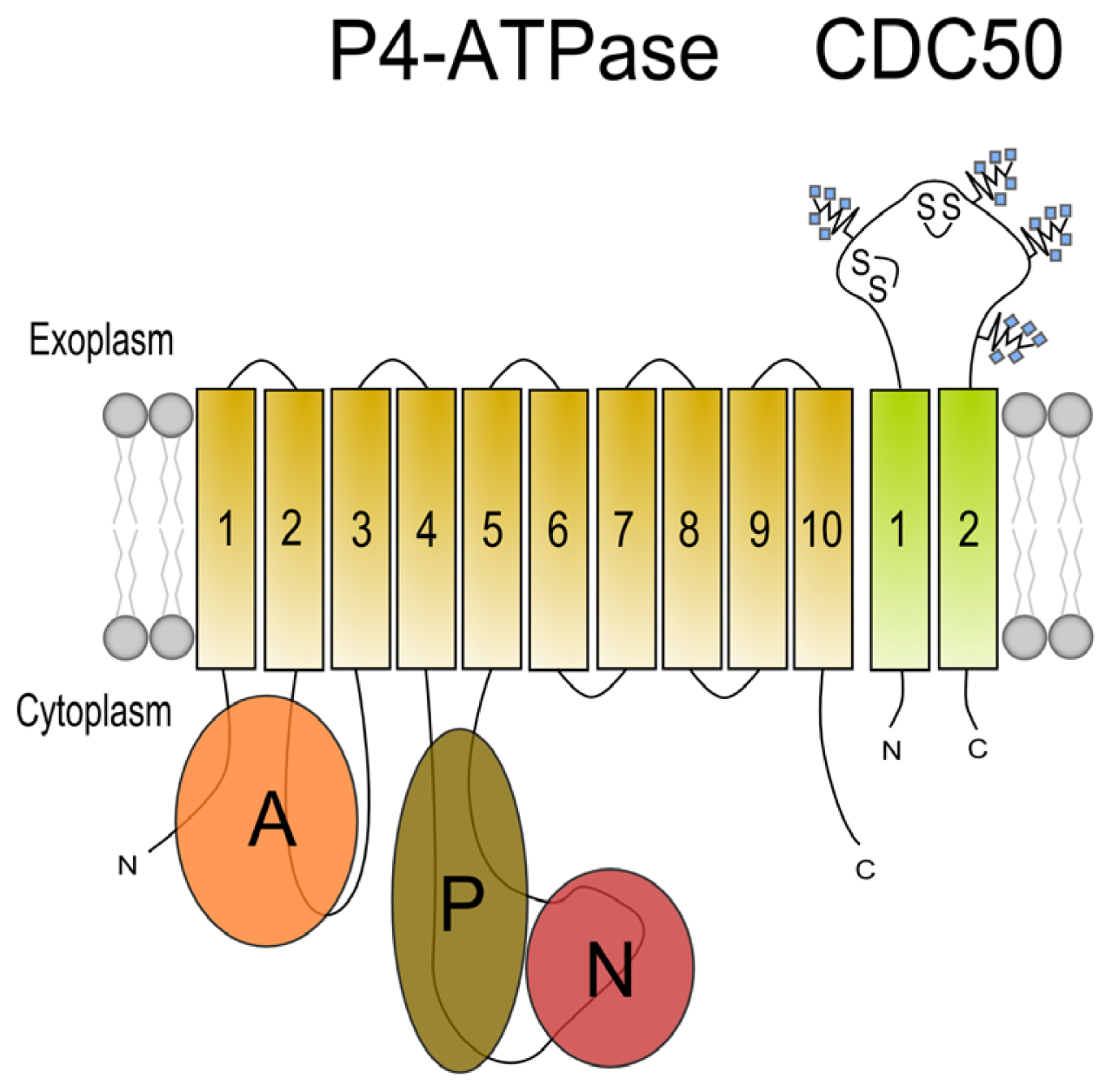
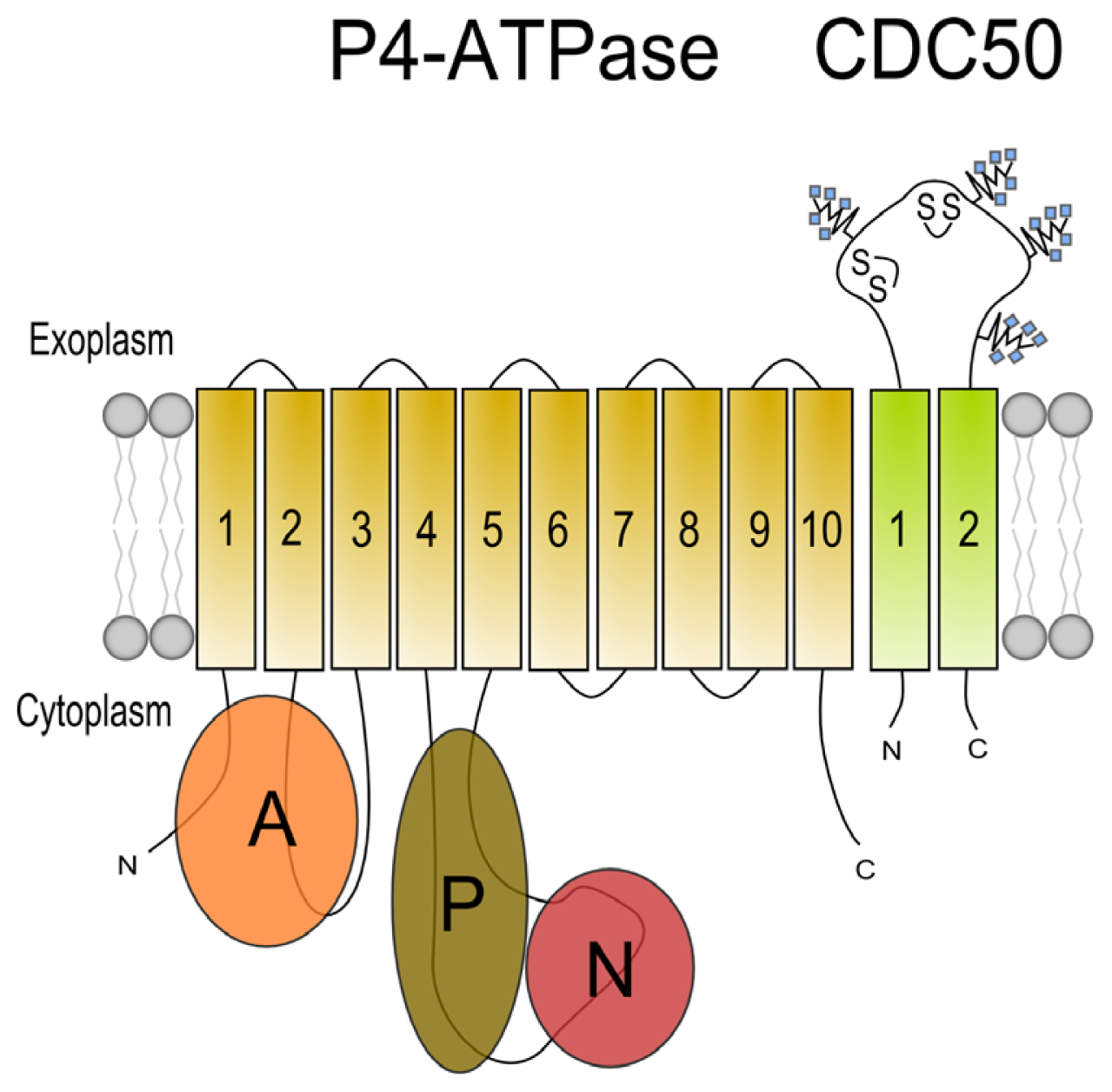
3. Beta Subunits for P4 ATPases
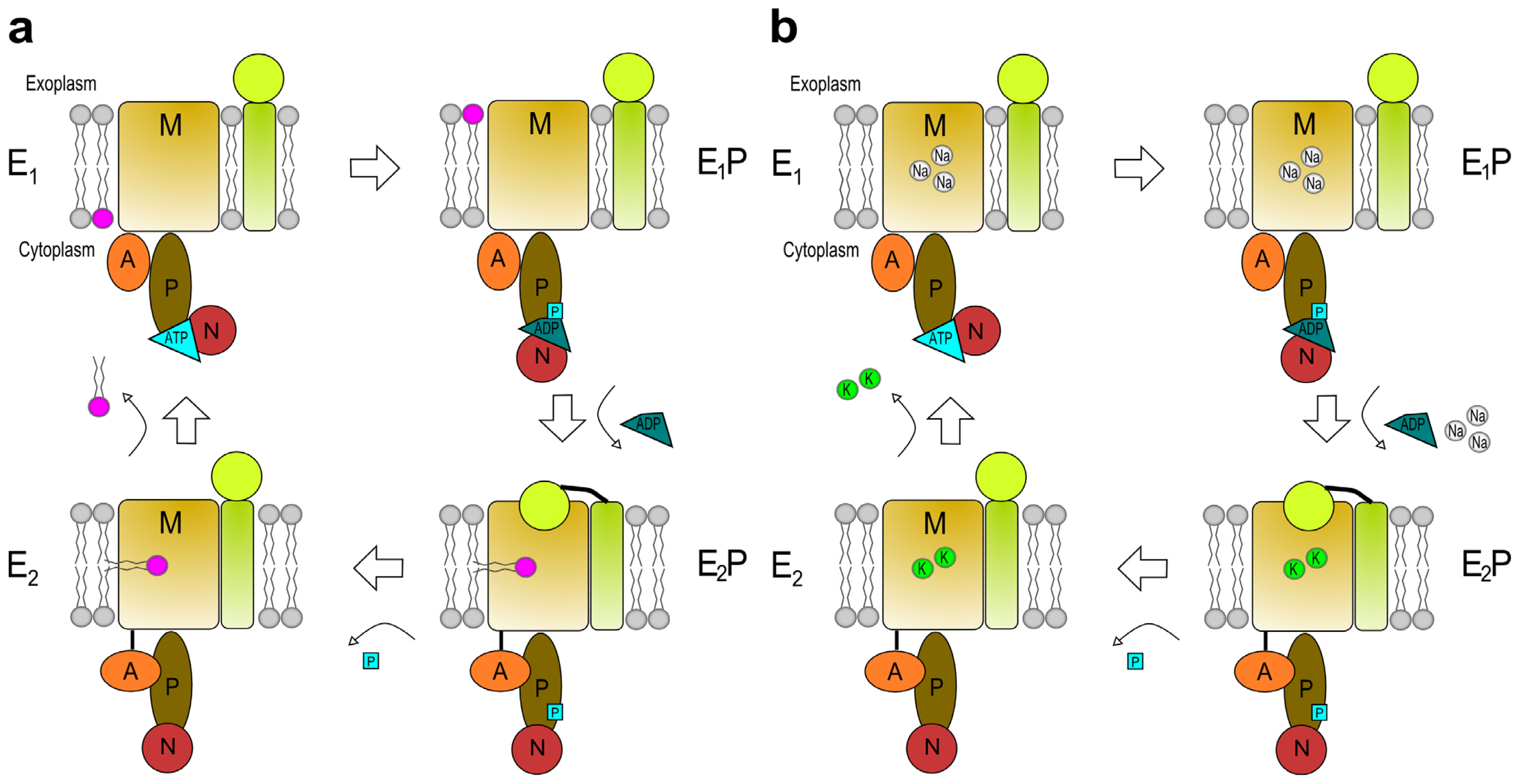
4. The Reaction Cycle
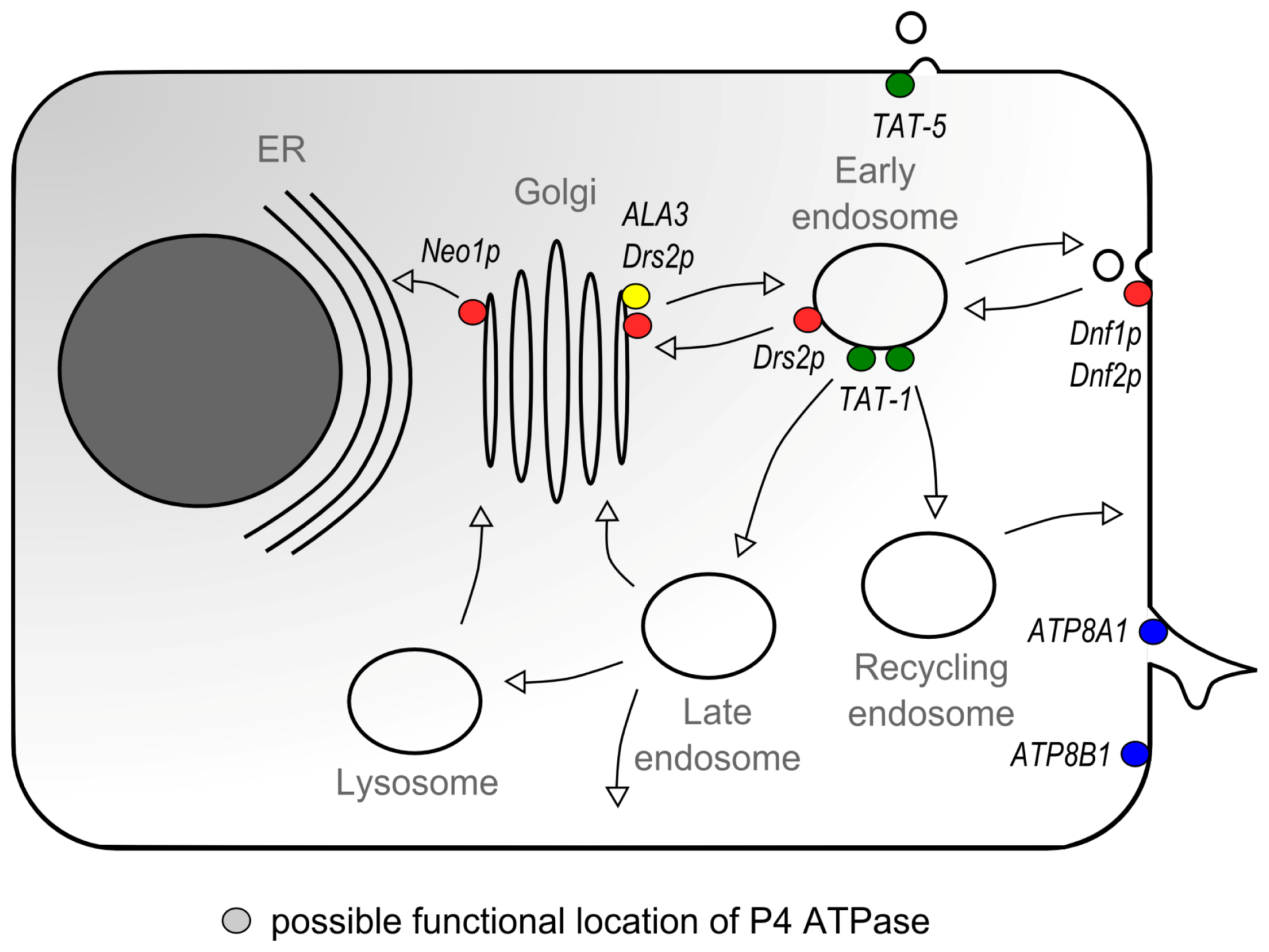
5. P4 ATPases and Vesicular Transport
6. The (Patho) Physiological Function of Mammalian P4 ATPases
7. ATP8B1 and Human Disease
8. Concluding Remarks
Conflict of Interest
References
- Gorter, E.; Grendel, F. On bimolecular layers of lipoids on the chromocytes of the blood. J. Exp. Med 1925, 41, 439–443. [Google Scholar]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol 2008, 9, 112–124. [Google Scholar]
- Van Meer, G.; de Kroon, A.I. Lipid map of the mammalian cell. J. Cell Sci 2011, 124, 5–8. [Google Scholar]
- Bevers, E.M.; Williamson, P.L. Phospholipid scramblase: An update. FEBS Lett 2010, 584, 2724–2730. [Google Scholar]
- Leonard, T.A.; Hurley, J.H. Regulation of protein kinases by lipids. Curr. Opin. Struct. Biol 2011, 21, 785–791. [Google Scholar]
- Leventis, P.A.; Grinstein, S. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys 2010, 39, 407–427. [Google Scholar]
- Bretscher, M.S. Asymmetrical lipid bilayer structure for biological membranes. Nat. New Biol 1972, 236, 11–12. [Google Scholar]
- Gordesky, S.E.; Marinetti, G.V. The asymetric arrangement of phospholipids in the human erythrocyte membrane. Biochem. Biophys. Res. Commun 1973, 50, 1027–1031. [Google Scholar]
- Verkleij, A.J.; Zwaal, R.F.; Roelofsen, B.; Comfurius, P.; Kastelijn, D.; van Deenen, L.L. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim. Biophys. Acta 1973, 323, 178–193. [Google Scholar]
- Butikofer, P.; Lin, Z.W.; Chiu, D.T.; Lubin, B.; Kuypers, F.A. Transbilayer distribution and mobility of phosphatidylinositol in human red blood cells. J. Biol. Chem 1990, 265, 16035–16038. [Google Scholar]
- Gascard, P.; Tran, D.; Sauvage, M.; Sulpice, J.C.; Fukami, K.; Takenawa, T.; Claret, M.; Giraud, F. Asymmetric distribution of phosphoinositides and phosphatidic acid in the human erythrocyte membrane. Biochim. Biophys. Acta 1991, 1069, 27–36. [Google Scholar]
- Op den Kamp, J.A. Lipid asymmetry in membranes. Annu. Rev. Biochem 1979, 48, 47–71. [Google Scholar]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol 2010, 22, 422–429. [Google Scholar]
- De Almeida, R.F.; Fedorov, A.; Prieto, M. Sphingomyelin/phosphatidylcholine/cholesterol phase diagram: Boundaries and composition of lipid rafts. Biophys. J 2003, 85, 2406–2416. [Google Scholar]
- Mondal, M.; Mesmin, B.; Mukherjee, S.; Maxfield, F.R. Sterols are mainly in the cytoplasmic leaflet of the plasma membrane and the endocytic recycling compartment in CHO cells. Mol. Biol. Cell 2009, 20, 581–588. [Google Scholar]
- Mesmin, B.; Maxfield, F.R. Intracellular sterol dynamics. Biochim. Biophys. Acta 2009, 1791, 636–645. [Google Scholar]
- Sprong, H.; van der Sluijs, P.; van Meer, G. How proteins move lipids and lipids move proteins. Nat. Rev. Mol. Cell Biol 2001, 2, 504–513. [Google Scholar]
- Ganong, B.R.; Bell, R.M. Transmembrane movement of phosphatidylglycerol and diacylglycerol sulfhydryl analogues. Biochemistry 1984, 23, 4977–4983. [Google Scholar]
- Holthuis, J.C.; Levine, T.P. Lipid traffic: Floppy drives and a superhighway. Nat. Rev. Mol. Cell Biol 2005, 6, 209–220. [Google Scholar]
- Seigneuret, M.; Devaux, P.F. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: Relation to shape changes. Proc. Natl. Acad. Sci. USA 1984, 81, 3751–3755. [Google Scholar]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010, 468, 834–838. [Google Scholar] [Green Version]
- Yang, H.; Kim, A.; David, T.; Palmer, D.; Jin, T.; Tien, J.; Huang, F.; Cheng, T.; Coughlin, S.R.; Jan, Y.N.; et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012, 151, 111–122. [Google Scholar]
- Paulusma, C.C.; Oude Elferink, R.P. Diseases of intramembranous lipid transport. FEBS Lett 2006, 580, 5500–5509. [Google Scholar]
- Pohl, A.; Devaux, P.F.; Herrmann, A. Function of prokaryotic and eukaryotic ABC proteins in lipid transport. Biochim. Biophys. Acta 2005, 1733, 29–52. [Google Scholar]
- Pomorski, T.; Holthuis, J.C.; Herrmann, A.; van Meer, G. Tracking down lipid flippases and their biological functions. J. Cell Sci 2004, 117, 805–813. [Google Scholar]
- Bublitz, M.; Morth, J.P.; Nissen, P. P-type ATPases at a glance. J. Cell Sci 2011, 124, 2515–2519. [Google Scholar]
- Schatzmann, H.J. ATP-dependent Ca2+-extrusion from human red cells. Experientia 1966, 22, 364–365. [Google Scholar]
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar]
- Palmgren, M.G.; Nissen, P. P-type ATPases. Annu. Rev. Biophys 2011, 40, 243–266. [Google Scholar]
- Natarajan, P.; Wang, J.; Hua, Z.; Graham, T.R. Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc. Natl. Acad. Sci. USA 2004, 101, 10614–10619. [Google Scholar]
- Tang, X.; Halleck, M.S.; Schlegel, R.A.; Williamson, P. A subfamily of P-type ATPases with aminophospholipid transporting activity. Science 1996, 272, 1495–1497. [Google Scholar]
- Chen, B.; Jiang, Y.; Zeng, S.; Yan, J.; Li, X.; Zhang, Y.; Zou, W.; Wang, X. Endocytic sorting and recycling require membrane phosphatidylserine asymmetry maintained by TAT-1/CHAT-1. PLoS. Genet 2010, 6, e1001235. [Google Scholar]
- Ray, N.B.; Durairaj, L.; Chen, B.B.; McVerry, B.J.; Ryan, A.J.; Donahoe, M.; Waltenbaugh, A.K.; O’Donnell, C.P.; Henderson, F.C.; Etscheidt, C.A.; et al. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat. Med 2010, 16, 1120–1127. [Google Scholar]
- Yabas, M.; Teh, C.E.; Frankenreiter, S.; Lal, D.; Roots, C.M.; Whittle, B.; Andrews, D.T.; Zhang, Y.; Teoh, N.C.; Sprent, J.; et al. ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat. Immunol 2011, 12, 441–449. [Google Scholar]
- Coleman, J.A.; Kwok, M.C.; Molday, R.S. Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J. Biol. Chem 2009, 284, 32670–32679. [Google Scholar]
- Paulusma, C.C.; Folmer, D.E.; Ho-Mok, K.S.; de Waart, D.R.; Hilarius, P.M.; Verhoeven, A.J.; Oude Elferink, R.P. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology 2008, 47, 268–278. [Google Scholar]
- Weingartner, A.; Drobot, B.; Herrmann, A.; Sanchez-Canete, M.P.; Gamarro, F.; Castanys, S.; Gunther, P.T. Disruption of the lipid-transporting LdMT-LdRos3 complex in Leishmania donovani affects membrane lipid asymmetry but not host cell invasion. PLoS One 2010, 5, e12443. [Google Scholar]
- Lopez-Marques, R.L.; Poulsen, L.R.; Hanisch, S.; Meffert, K.; Buch-Pedersen, M.J.; Jakobsen, M.K.; Pomorski, T.G.; Palmgren, M.G. Intracellular targeting signals and lipid specificity determinants of the ALA/ALIS P4-ATPase complex reside in the catalytic ALA alpha-subunit. Mol. Biol. Cell 2010, 21, 791–801. [Google Scholar]
- Poulsen, L.R.; Lopez-Marques, R.L.; McDowell, S.C.; Okkeri, J.; Licht, D.; Schulz, A.; Pomorski, T.; Harper, J.F.; Palmgren, M.G. The Arabidopsis P4-ATPase ALA3 localizes to the golgi and requires a beta-subunit to function in lipid translocation and secretory vesicle formation. Plant Cell 2008, 20, 658–676. [Google Scholar]
- Chen, S.; Wang, J.; Muthusamy, B.P.; Liu, K.; Zare, S.; Andersen, R.J.; Graham, T.R. Roles for the Drs2p-Cdc50p complex in protein transport and phosphatidylserine asymmetry of the yeast plasma membrane. Traffic 2006, 7, 1503–1517. [Google Scholar]
- Coleman, J.A.; Molday, R.S. Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. J. Biol. Chem 2011, 286, 17205–17216. [Google Scholar]
- Coleman, J.A.; Vestergaard, A.L.; Molday, R.S.; Vilsen, B.; Peter, A.J. Critical role of a transmembrane lysine in aminophospholipid transport by mammalian photoreceptor P4-ATPase ATP8A2. Proc. Natl. Acad. Sci. USA 2012, 109, 1449–1454. [Google Scholar]
- Multiple Sequence Alignment by CLUSTALW. Available online: http://www.genome.jp/tools/clustalw/ (accessed on 1 March 2013).
- Coleman, J.A.; Quazi, F.; Molday, R.S. Mammalian P(4)-ATPases and ABC transporters and their role in phospholipid transport. Biochim. Biophys. Acta 2013, 1831, 555–574. [Google Scholar]
- Puts, C.F.; Holthuis, J.C. Mechanism and significance of P4 ATPase-catalyzed lipid transport: Lessons from a Na+/K+-pump. Biochim. Biophys. Acta 2009, 1791, 603–611. [Google Scholar]
- Shin, J.M.; Munson, K.; Vagin, O.; Sachs, G. The gastric HK-ATPase: Structure, function, and inhibition. Pflugers Arch 2009, 457, 609–622. [Google Scholar]
- Paulusma, C.C.; Elferink, R.P. P4 ATPases—The physiological relevance of lipid flipping transporters. FEBS Lett 2010, 584, 2708–2716. [Google Scholar]
- Poulsen, L.R.; Lopez-Marques, R.L.; Palmgren, M.G. Flippases: Still more questions than answers. Cell Mol. Life Sci 2008, 65, 3119–3125. [Google Scholar]
- Blanco, G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol 2005, 25, 292–303. [Google Scholar]
- Geering, K. Functional roles of Na,K-ATPase subunits. Curr. Opin. Nephrol. Hypertens 2008, 17, 526–532. [Google Scholar]
- Muth, T.R.; Gottardi, C.J.; Roush, D.L.; Caplan, M.J. A basolateral sorting signal is encoded in the alpha-subunit of Na-K-ATPase. Am. J. Physiol 1998, 274, C688–C696. [Google Scholar]
- Vagin, O.; Turdikulova, S.; Sachs, G. The H,K-ATPase beta subunit as a model to study the role of N-glycosylation in membrane trafficking and apical sorting. J. Biol. Chem 2004, 279, 39026–39034. [Google Scholar]
- Bryde, S.; Hennrich, H.; Verhulst, P.M.; Devaux, P.F.; Lenoir, G.; Holthuis, J.C. CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J. Biol. Chem 2010, 285, 40562–40572. [Google Scholar]
- Furuta, N.; Fujimura-Kamada, K.; Saito, K.; Yamamoto, T.; Tanaka, K. Endocytic recycling in yeast is regulated by putative phospholipid translocases and the Ypt31p/32p-Rcy1p pathway. Mol. Biol. Cell 2007, 18, 295–312. [Google Scholar]
- Saito, K.; Fujimura-Kamada, K.; Furuta, N.; Kato, U.; Umeda, M.; Tanaka, K. Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol. Biol. Cell 2004, 15, 3418–3432. [Google Scholar]
- Van der Velden, L.M.; Wichers, C.G.; van Breevoort, A.E.; Coleman, J.A.; Molday, R.S.; Berger, R.; Klomp, L.W.; van de Graaf, S.F. Heteromeric interactions required for abundance and subcellular localization of human CDC50 proteins and class 1 P4-ATPases. J. Biol. Chem 2010, 285, 40088–40096. [Google Scholar]
- Baldridge, R.D.; Graham, T.R. Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc. Natl. Acad. Sci. USA 2012, 109, E290–E298. [Google Scholar]
- Folmer, D.E.; Mok, K.S.; de Wee, S.W.; Duijst, S.; Hiralall, J.K.; Seppen, J.; Oude Elferink, R.P.; Paulusma, C.C. Cellular localization and biochemical analysis of mammalian CDC50A, a glycosylated beta-subunit for P4 ATPases. J. Histochem. Cytochem 2012, 60, 205–218. [Google Scholar]
- Katoh, Y.; Katoh, M. Identification and characterization of CDC50A, CDC50B and CDC50C genes in silico. Oncol. Rep 2004, 12, 939–943. [Google Scholar]
- Xu, P.; Okkeri, J.; Hanisch, S.; Hu, R.Y.; Xu, Q.; Pomorski, T.G.; Ding, X.Y. Identification of a novel mouse P4-ATPase family member highly expressed during spermatogenesis. J. Cell Sci 2009, 122, 2866–2876. [Google Scholar]
- Takatsu, H.; Baba, K.; Shima, T.; Umino, H.; Kato, U.; Umeda, M.; Nakayama, K.; Shin, H.W. ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J. Biol. Chem 2011, 286, 38159–38167. [Google Scholar]
- Hua, Z.; Fatheddin, P.; Graham, T.R. An essential subfamily of Drs2p-related P-type ATPases is required for protein trafficking between Golgi complex and endosomal/vacuolar system. Mol. Biol. Cell 2002, 13, 3162–3177. [Google Scholar]
- Stone, A.; Chau, C.; Eaton, C.; Foran, E.; Kapur, M.; Prevatt, E.; Belkin, N.; Kerr, D.; Kohlin, T.K.; Williamson, P. Biochemical characterization of P4-ATPase mutations identified in patients with progressive familial intrahepatic cholestasis. J. Biol. Chem 2012, 287, 41139–41151. [Google Scholar]
- Jacquot, A.; Montigny, C.; Hennrich, H.; Barry, R.; le Maire, M.; Jaxel, C.; Holthuis, J.; Champeil, P.; Lenoir, G. Phosphatidylserine stimulation of Drs2p.Cdc50p lipid translocase dephosphorylation is controlled by phosphatidylinositol-4-phosphate. J. Biol. Chem 2012, 287, 13249–13261. [Google Scholar]
- Lenoir, G.; Williamson, P.; Puts, C.F.; Holthuis, J.C. Cdc50p plays a vital role in the ATPase reaction cycle of the putative aminophospholipid transporter Drs2p. J. Biol. Chem 2009, 284, 17956–17967. [Google Scholar]
- Stone, A.; Williamson, P. Outside of the box: Recent news about phospholipid translocation by P4 ATPases. J. Chem. Biol 2012, 5, 131–136. [Google Scholar]
- Albers, R.W. Biochemical aspects of active transport. Annu. Rev. Biochem 1967, 36, 727–756. [Google Scholar]
- Post, R.L.; Hegyvary, C.; Kume, S. Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem 1972, 247, 6530–6540. [Google Scholar]
- Durr, K.L.; Tavraz, N.N.; Dempski, R.E.; Bamberg, E.; Friedrich, T. Functional significance of E2 state stabilization by specific alpha/beta-subunit interactions of Na,K- and H,K-ATPase. J. Biol. Chem 2009, 284, 3842–3854. [Google Scholar]
- Natarajan, P.; Liu, K.; Patil, D.V.; Sciorra, V.A.; Jackson, C.L.; Graham, T.R. Regulation of a Golgi flippase by phosphoinositides and an ArfGEF. Nat. Cell Biol 2009, 11, 1421–1426. [Google Scholar]
- Nakano, K.; Yamamoto, T.; Kishimoto, T.; Noji, T.; Tanaka, K. Protein kinases Fpk1p and Fpk2p are novel regulators of phospholipid asymmetry. Mol. Biol. Cell 2008, 19, 1783–1797. [Google Scholar]
- Pedersen, B.P.; Buch-Pedersen, M.J.; Morth, J.P.; Palmgren, M.G.; Nissen, P. Crystal structure of the plasma membrane proton pump. Nature 2007, 450, 1111–1114. [Google Scholar]
- Baldridge, R.D.; Graham, T.R. Two-gate mechanism for phospholipid selection and transport by type IV P-type ATPases. Proc. Natl. Acad. Sci. USA 2013, 110, E358–E367. [Google Scholar]
- Sebastian, T.T.; Baldridge, R.D.; Xu, P.; Graham, T.R. Phospholipid flippases: Building asymmetric membranes and transport vesicles. Biochim. Biophys. Acta 2012, 1821, 1068–1077. [Google Scholar]
- Steinman, R.M.; Mellman, I.S.; Muller, W.A.; Cohn, Z.A. Endocytosis and the recycling of plasma membrane. J. Cell Biol 1983, 96, 1–27. [Google Scholar]
- Sheetz, M.P.; Singer, S.J. Biological membranes as bilayer couples. A molecular mechanism of drug-erythrocyte interactions. Proc. Natl. Acad. Sci. USA 1974, 71, 4457–4461. [Google Scholar]
- Farge, E.; Ojcius, D.M.; Subtil, A.; Dautry-Varsat, A. Enhancement of endocytosis due to aminophospholipid transport across the plasma membrane of living cells. Am. J. Physiol 1999, 276, C725–C733. [Google Scholar]
- Graham, T.R. Flippases and vesicle-mediated protein transport. Trends Cell Biol 2004, 14, 670–677. [Google Scholar]
- Kozlov, M.M.; McMahon, H.T.; Chernomordik, L.V. Protein-driven membrane stresses in fusion and fission. Trends Biochem. Sci 2010, 35, 699–706. [Google Scholar]
- Gillon, A.D.; Latham, C.F.; Miller, E.A. Vesicle-mediated ER export of proteins and lipids. Biochim. Biophys. Acta 2012, 1821, 1040–1049. [Google Scholar]
- Horvath, C.A.; Vanden Broeck, D.; Boulet, G.A.; Bogers, J.; de Wolf, M.J. Epsin: Inducing membrane curvature. Int. J. Biochem. Cell Biol 2007, 39, 1765–1770. [Google Scholar]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol 2011, 12, 517–533. [Google Scholar]
- Pinot, M.; Goud, B.; Manneville, J.B. Physical aspects of COPI vesicle formation. Mol. Membr. Biol 2010, 27, 428–442. [Google Scholar]
- Robinson, M.S. Adaptable adaptors for coated vesicles. Trends Cell Biol 2004, 14, 167–174. [Google Scholar]
- Muthusamy, B.P.; Natarajan, P.; Zhou, X.; Graham, T.R. Linking phospholipid flippases to vesicle-mediated protein transport. Biochim. Biophys. Acta 2009, 1791, 612–619. [Google Scholar]
- Gall, W.E.; Geething, N.C.; Hua, Z.; Ingram, M.F.; Liu, K.; Chen, S.I.; Graham, T.R. Drs2p-dependent formation of exocytic clathrin-coated vesicles in vivo. Curr. Biol 2002, 12, 1623–1627. [Google Scholar]
- Liu, K.; Surendhran, K.; Nothwehr, S.F.; Graham, T.R. P4-ATPase requirement for AP-1/clathrin function in protein transport from the trans-Golgi network and early endosomes. Mol. Biol. Cell 2008, 19, 3526–3535. [Google Scholar]
- Chen, C.Y.; Ingram, M.F.; Rosal, P.H.; Graham, T.R. Role for Drs2p, a P-type ATPase and potential aminophospholipid translocase, in yeast late Golgi function. J. Cell Biol 1999, 147, 1223–1236. [Google Scholar]
- Chantalat, S.; Park, S.K.; Hua, Z.; Liu, K.; Gobin, R.; Peyroche, A.; Rambourg, A.; Graham, T.R.; Jackson, C.L. The Arf activator Gea2p and the P-type ATPase Drs2p interact at the Golgi in Saccharomyces cerevisiae. J. Cell Sci 2004, 117, 711–722. [Google Scholar]
- Tsai, P.C.; Hsu, J.W.; Liu, Y.W.; Chen, K.Y.; Lee, F.J. Arl1p regulates spatial membrane organization at the trans-Golgi network through interaction with Arf-GEF Gea2p and flippase Drs2p. Proc. Natl. Acad. Sci. USA 2013, 110, E668–E677. [Google Scholar]
- Puts, C.F.; Lenoir, G.; Krijgsveld, J.; Williamson, P.; Holthuis, J.C. A P4-ATPase protein interaction network reveals a link between aminophospholipid transport and phosphoinositide metabolism. J. Proteome. Res 2010, 9, 833–842. [Google Scholar]
- Sakane, H.; Yamamoto, T.; Tanaka, K. The functional relationship between the Cdc50p-Drs2p putative aminophospholipid translocase and the Arf GAP Gcs1p in vesicle formation in the retrieval pathway from yeast early endosomes to the TGN. Cell Struct. Funct 2006, 31, 87–108. [Google Scholar]
- Mayinger, P. Phosphoinositides and vesicular membrane traffic. Biochim. Biophys. Acta 2012, 1821, 1104–1113. [Google Scholar]
- Graham, T.R.; Burd, C.G. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol 2011, 21, 113–121. [Google Scholar]
- Wang, Y.J.; Wang, J.; Sun, H.Q.; Martinez, M.; Sun, Y.X.; Macia, E.; Kirchhausen, T.; Albanesi, J.P.; Roth, M.G.; Yin, H.L. Phosphatidylinositol 4 phosphate regulates targeting of clathrin adaptor AP-1 complexes to the Golgi. Cell 2003, 114, 299–310. [Google Scholar]
- Liu, K.; Hua, Z.; Nepute, J.A.; Graham, T.R. Yeast P4-ATPases Drs2p and Dnf1p are essential cargos of the NPFXD/Sla1p endocytic pathway. Mol. Biol. Cell 2007, 18, 487–500. [Google Scholar]
- Pomorski, T.; Lombardi, R.; Riezman, H.; Devaux, P.F.; van Meer, G.; Holthuis, J.C. Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol. Biol. Cell 2003, 14, 1240–1254. [Google Scholar]
- Alder-Baerens, N.; Lisman, Q.; Luong, L.; Pomorski, T.; Holthuis, J.C. Loss of P4 ATPases Drs2p and Dnf3p disrupts aminophospholipid transport and asymmetry in yeast post-Golgi secretory vesicles. Mol. Biol. Cell 2006, 17, 1632–1642. [Google Scholar]
- Hua, Z.; Graham, T.R. Requirement for neo1p in retrograde transport from the Golgi complex to the endoplasmic reticulum. Mol. Biol. Cell 2003, 14, 4971–4983. [Google Scholar]
- Wicky, S.; Schwarz, H.; Singer-Kruger, B. Molecular interactions of yeast Neo1p, an essential member of the Drs2 family of aminophospholipid translocases, and its role in membrane trafficking within the endomembrane system. Mol. Cell Biol 2004, 24, 7402–7418. [Google Scholar]
- Das, A.; Slaughter, B.D.; Unruh, J.R.; Bradford, W.D.; Alexander, R.; Rubinstein, B.; Li, R. Flippase-mediated phospholipid asymmetry promotes fast Cdc42 recycling in dynamic maintenance of cell polarity. Nat. Cell Biol 2012, 14, 304–310. [Google Scholar]
- Ruaud, A.F.; Nilsson, L.; Richard, F.; Larsen, M.K.; Bessereau, J.L.; Tuck, S. The C. elegans P4-ATPase TAT-1 regulates lysosome biogenesis and endocytosis. Traffic 2009, 10, 88–100. [Google Scholar]
- Wehman, A.M.; Poggioli, C.; Schweinsberg, P.; Grant, B.D.; Nance, J. The P4-ATPase TAT-5 inhibits the budding of extracellular vesicles in C. elegans embryos. Curr. Biol 2011, 21, 1951–1959. [Google Scholar]
- Kato, U.; Inadome, H.; Yamamoto, M.; Emoto, K.; Kobayashi, T.; Umeda, M. Role for phospholipid flippase complex of ATP8A1 and CDC50A in cell migration. J. Biol. Chem 2012, 288, 4922–4934. [Google Scholar]
- Levano, K.; Punia, V.; Raghunath, M.; Debata, P.R.; Curcio, G.M.; Mogha, A.; Purkayastha, S.; McCloskey, D.; Fata, J.; Banerjee, P. Atp8a1 deficiency is associated with phosphatidylserine externalization in hippocampus and delayed hippocampus-dependent learning. J. Neurochem 2012, 120, 302–313. [Google Scholar]
- Emre Onat, O.; Gulsuner, S.; Bilguvar, K.; Nazli, B.A.; Topaloglu, H.; Tan, M.; Tan, U.; Gunel, M.; Ozcelik, T. Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur. J. Hum. Genet 2012, 21, 281–285. [Google Scholar] [Green Version]
- Zhu, X.; Libby, R.T.; de Vries, W.N.; Smith, R.S.; Wright, D.L.; Bronson, R.T.; Seburn, K.L.; John, S.W. Mutations in a P-type ATPase gene cause axonal degeneration. PLoS Genet 2012, 8, e1002853. [Google Scholar]
- Cai, S.Y.; Gautam, S.; Nguyen, T.; Soroka, C.J.; Rahner, C.; Boyer, J.L. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology 2009, 136, 1060–1069. [Google Scholar]
- Paulusma, C.C.; Groen, A.; Kunne, C.; Ho-Mok, K.S.; Spijkerboer, A.L.; de Waart, D.R.; Hoek, F.J.; Vreeling, H.; Hoeben, K.A.; van Marle, J.; et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology 2006, 44, 195–204. [Google Scholar]
- Paulusma, C.C.; Elferink, R.P.; Jansen, P.L. Progressive familial intrahepatic cholestasis type 1. Semin. Liver Dis 2010, 30, 117–124. [Google Scholar]
- Pawlikowska, L.; Groen, A.; Eppens, E.F.; Kunne, C.; Ottenhoff, R.; Looije, N.; Knisely, A.S.; Killeen, N.P.; Bull, L.N.; Elferink, R.P.; et al. A mouse genetic model for familial cholestasis caused by ATP8B1 mutations reveals perturbed bile salt homeostasis but no impairment in bile secretion. Hum. Mol. Genet 2004, 13, 881–892. [Google Scholar]
- Stapelbroek, J.M.; Peters, T.A.; van Beurden, D.H.; Curfs, J.H.; Joosten, A.; Beynon, A.J.; van Leeuwen, B.M.; van der Velden, L.M.; Bull, L.; Oude Elferink, R.P.; et al. ATP8B1 is essential for maintaining normal hearing. Proc. Natl. Acad. Sci. USA 2009, 106, 9709–9714. [Google Scholar]
- Gong, E.Y.; Park, E.; Lee, H.J.; Lee, K. Expression of Atp8b3 in murine testis and its characterization as a testis specific P-type ATPase. Reproduction 2009, 137, 345–351. [Google Scholar]
- Wang, L.; Beserra, C.; Garbers, D.L. A novel aminophospholipid transporter exclusively expressed in spermatozoa is required for membrane lipid asymmetry and normal fertilization. Dev. Biol 2004, 267, 203–215. [Google Scholar]
- Li, H.; Wetten, S.; Li, L.; St Jean, P.L.; Upmanyu, R.; Surh, L.; Hosford, D.; Barnes, M.R.; Briley, J.D.; Borrie, M.; et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol 2008, 65, 45–53. [Google Scholar]
- Dhar, M.S.; Sommardahl, C.S.; Kirkland, T.; Nelson, S.; Donnell, R.; Johnson, D.K.; Castellani, L.W. Mice heterozygous for Atp10c, a putative amphipath, represent a novel model of obesity and type 2 diabetes. J. Nutr 2004, 134, 799–805. [Google Scholar]
- Dhar, M.S.; Yuan, J.S.; Elliott, S.B.; Sommardahl, C. A type IV P-type ATPase affects insulin-mediated glucose uptake in adipose tissue and skeletal muscle in mice. J. Nutr. Biochem 2006, 17, 811–820. [Google Scholar]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet 2010, 42, 105–116. [Google Scholar] [Green Version]
- Irvin, M.R.; Wineinger, N.E.; Rice, T.K.; Pajewski, N.M.; Kabagambe, E.K.; Gu, C.C.; Pankow, J.; North, K.E.; Wilk, J.B.; Freedman, B.I.; et al. Genome-wide detection of allele specific copy number variation associated with insulin resistance in African Americans from the HyperGEN study. PLoS One 2011, 6, e24052. [Google Scholar]
- Milagro, F.I.; Campion, J.; Cordero, P.; Goyenechea, E.; Gomez-Uriz, A.M.; Abete, I.; Zulet, M.A.; Martinez, J.A. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB J 2011, 25, 1378–1389. [Google Scholar]
- Flamant, S.; Pescher, P.; Lemercier, B.; Clement-Ziza, M.; Kepes, F.; Fellous, M.; Milon, G.; Marchal, G.; Besmond, C. Characterization of a putative type IV aminophospholipid transporter P-type ATPase. Mamm. Genome 2003, 14, 21–30. [Google Scholar]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Nagai, K.; Uemura, M.; Sekimoto, M.; Doki, Y.; Mori, M. ATP11A is a novel predictive marker for metachronous metastasis of colorectal cancer. Oncol. Rep 2010, 23, 505–510. [Google Scholar]
- Siggs, O.M.; Schnabl, B.; Webb, B.; Beutler, B. X-linked cholestasis in mouse due to mutations of the P4-ATPase ATP11C. Proc. Natl. Acad. Sci. USA 2011, 108, 7890–7895. [Google Scholar]
- Siggs, O.M.; Arnold, C.N.; Huber, C.; Pirie, E.; Xia, Y.; Lin, P.; Nemazee, D.; Beutler, B. The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat. Immunol 2011, 12, 434–440. [Google Scholar]
- Soupene, E.; Kemaladewi, D.U.; Kuypers, F.A. ATP8A1 activity and phosphatidylserine transbilayer movement. J. Receptor Ligand. Channel. Res 2008, 1, 1–10. [Google Scholar]
- Paulusma, C.C.; de Waart, D.R.; Kunne, C.; Mok, K.S.; Elferink, R.P. Activity of the bile salt export pump (ABCB11) is critically dependent on canalicular membrane cholesterol content. J. Biol. Chem 2009, 284, 9947–9954. [Google Scholar]
- Xu, Q.; Yang, G.Y.; Liu, N.; Xu, P.; Chen, Y.L.; Zhou, Z.; Luo, Z.G.; Ding, X. P4-ATPase ATP8A2 acts in synergy with CDC50A to enhance neurite outgrowth. FEBS Lett 2012, 586, 1803–1812. [Google Scholar]
- Cacciagli, P.; Haddad, M.R.; Mignon-Ravix, C.; El-Waly, B.; Moncla, A.; Missirian, C.; Chabrol, B.; Villard, L. Disruption of the ATP8A2 gene in a patient with a t(10;13) de novo balanced translocation and a severe neurological phenotype. Eur. J. Hum. Genet 2010, 18, 1360–1363. [Google Scholar]
- Hurst, S.E.; Minkin, S.C.; Biggerstaff, J.; Dhar, M.S. Transient Silencing of a Type IV P-Type ATPase, Atp10c, Results in Decreased Glucose Uptake in C2C12 Myotubes. J. Nutr. Metab 2012, 2012, 152902. [Google Scholar]
- Roshwalb, S.; Gorman, S.; Hurst, S.; Bartges, J.; Agarwal, S.; Sommardahl, C.; Odoi, A.; Dhar, M.S. mRNA expression of canine ATP10C, a P4-type ATPase, is positively associated with body condition score. Vet. J 2011, 190, 173–175. [Google Scholar]
- Surwit, R.S.; Feinglos, M.N.; Rodin, J.; Sutherland, A.; Petro, A.E.; Opara, E.C.; Kuhn, C.M.; Rebuffe-Scrive, M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism 1995, 44, 645–651. [Google Scholar]
- Andrew Nesbit, M.; Bowl, M.R.; Harding, B.; Schlessinger, D.; Whyte, M.P.; Thakker, R.V. X-linked hypoparathyroidism region on Xq27 is evolutionarily conserved with regions on 3q26 and 13q34 and contains a novel P-type ATPase. Genomics 2004, 84, 1060–1070. [Google Scholar]
- Bull, L.N.; van Eijk, M.J.; Pawlikowska, L.; DeYoung, J.A.; Juijn, J.A.; Liao, M.; Klomp, L.W.; Lomri, N.; Berger, R.; Scharschmidt, B.F.; et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet 1998, 18, 219–224. [Google Scholar]
- Klomp, L.W.; Vargas, J.C.; van Mil, S.W.; Pawlikowska, L.; Strautnieks, S.S.; van Eijk, M.J.; Juijn, J.A.; Pabon-Pena, C.; Smith, L.B.; DeYoung, J.A.; et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 2004, 40, 27–38. [Google Scholar]
- Clayton, R.J.; Iber, F.L.; Ruebner, B.H.; McKusick, V.A. Byler disease. Fatal familial intrahepatic cholestasis in an Amish kindred. Am. J. Dis. Child 1969, 117, 112–124. [Google Scholar]
- Summerskill, W.H.; Walshe, J.M. Benign recurrent intrahepatic “obstructive” jaundice. Lancet 1959, 2, 686–690. [Google Scholar]
- Orphanet: Progressive familial intrahepatic cholestasis type 1. Available online: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=79306 (accessed on 1 March 2013).
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. Progressive familial intrahepatic cholestasis. Orphanet. J. Rare. Dis 2009, 4, 1. [Google Scholar]
- Summerskill, W.H. The syndrome of benign recurrent cholestasis. Am. J. Med 1965, 38, 298–305. [Google Scholar]
- Bustorff-Silva, J.; Sbraggia, N.L.; Olimpio, H.; de Alcantara, R.V.; Matsushima, E.; de Tommaso, A.M.; Brandao, M.A.; Hessel, G. Partial internal biliary diversion through a cholecystojejunocolonic anastomosis—A novel surgical approach for patients with progressive familial intrahepatic cholestasis: A preliminary report. J. Pediatr. Surg 2007, 42, 1337–1340. [Google Scholar]
- Emond, J.C.; Whitington, P.F. Selective surgical management of progressive familial intrahepatic cholestasis (Byler’s disease). J. Pediatr. Surg 1995, 30, 1635–1641. [Google Scholar]
- Lykavieris, P.; van Mil, S.; Cresteil, D.; Fabre, M.; Hadchouel, M.; Klomp, L.; Bernard, O.; Jacquemin, E. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: No catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J. Hepatol 2003, 39, 447–452. [Google Scholar]
- Morotti, R.A.; Suchy, F.J.; Magid, M.S. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: A review of the liver pathology findings. Semin. Liver Dis 2011, 31, 3–10. [Google Scholar]
- Pawlikowska, L.; Strautnieks, S.; Jankowska, I.; Czubkowski, P.; Emerick, K.; Antoniou, A.; Wanty, C.; Fischler, B.; Jacquemin, E.; Wali, S.; et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J. Hepatol 2010, 53, 170–178. [Google Scholar]
- Folmer, D.E.; van der Mark, V.A.; Ho-Mok, K.S.; Oude Elferink, R.P.; Paulusma, C.C. Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology 2009, 50, 1597–1605. [Google Scholar]
- Van Mil, S.W.; Klomp, L.W.; Bull, L.N.; Houwen, R.H. FIC1 disease: A spectrum of intrahepatic cholestatic disorders. Semin. Liver Dis 2001, 21, 535–544. [Google Scholar]
- Eppens, E.F.; van Mil, S.W.; de Vree, J.M.; Mok, K.S.; Juijn, J.A.; Oude Elferink, R.P.; Berger, R.; Houwen, R.H.; Klomp, L.W. FIC1, the protein affected in two forms of hereditary cholestasis, is localized in the cholangiocyte and the canalicular membrane of the hepatocyte. J. Hepatol 2001, 35, 436–443. [Google Scholar]
- Van Mil, S.W.; van Oort, M.M.; van den Berg, I.E.; Berger, R.; Houwen, R.H.; Klomp, L.W. Fic1 is expressed at apical membranes of different epithelial cells in the digestive tract and is induced in the small intestine during postnatal development of mice. Pediatr. Res 2004, 56, 981–987. [Google Scholar]
- Groen, A.; Kunne, C.; Paulusma, C.C.; Kramer, W.; Agellon, L.B.; Bull, L.N.; Oude Elferink, R.P. Intestinal bile salt absorption in Atp8b1 deficient mice. J. Hepatol 2007, 47, 114–122. [Google Scholar]
- Groen, A.; Kunne, C.; Jongsma, G.; van den Oever, K.; Mok, K.S.; Petruzzelli, M.; Vrins, C.L.; Bull, L.; Paulusma, C.C.; Oude Elferink, R.P. Abcg5/8 independent biliary cholesterol excretion in Atp8b1-deficient mice. Gastroenterology 2008, 134, 2091–2100. [Google Scholar]
- Paulusma, C.C.; Houwen, R.H.; Williamson, P.L. The flip side of cardiolipin import. Nat. Med 2011, 17, 413–414. [Google Scholar]
- Frankenberg, T.; Miloh, T.; Chen, F.Y.; Ananthanarayanan, M.; Sun, A.Q.; Balasubramaniyan, N.; Arias, I.; Setchell, K.D.; Suchy, F.J.; Shneider, B.L. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology 2008, 48, 1896–1905. [Google Scholar]
- Chen, F.; Ellis, E.; Strom, S.C.; Shneider, B.L. ATPase Class I Type 8B Member 1 and protein kinase C zeta induce the expression of the canalicular bile salt export pump in human hepatocytes. Pediatr. Res 2010, 67, 183–187. [Google Scholar]
- Chen, F.; Ananthanarayanan, M.; Emre, S.; Neimark, E.; Bull, L.N.; Knisely, A.S.; Strautnieks, S.S.; Thompson, R.J.; Magid, M.S.; Gordon, R.; et al. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology 2004, 126, 756–764. [Google Scholar]
- Koh, S.; Takada, T.; Kukuu, I.; Suzuki, H. FIC1-mediated stimulation of FXR activity is decreased with PFIC1 mutations in HepG2 cells. J. Gastroenterol 2009, 44, 592–600. [Google Scholar]
- Verhulst, P.M.; van der Velden, L.M.; Oorschot, V.; van Faassen, E.E.; Klumperman, J.; Houwen, R.H.; Pomorski, T.G.; Holthuis, J.C.; Klomp, L.W. A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology 2010, 51, 2049–2060. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | P4 ATPase | Pathophysiology in mice | Pathophysiology in humans | References |
|---|---|---|---|---|
| 1A | ATP8A1 | impaired learning, increased physical activity | [105] | |
| ATP8A2 | neurodegenerative disease, axonal degeneration, growth retardation | mental retardation, hypotonia, CAMRQ | [106,107] | |
| 1B | ATP8B1 | intrahepatic cholestasis, hearing loss | PFIC1, BRIC1 | [108–112] |
| ATP8B2 | ||||
| ATP8B3 | sperm capacitation anomalies | [113,114] | ||
| ATP8B4 | Alzheimer’s disease | [115] | ||
| ATP8B5 | not present in humans | |||
| 2 | ATP9A | |||
| ATP9B | ||||
| 5 | ATP10A | insulin resistance, diet-induced obesity, hyperlipidemia, hyperinsulinemia | type 2 diabetes, insulin resistance in African Americans, diet-induced obesity | [116–120] |
| ATP10B | ||||
| ATP10D | diet-induced obesity, hyperinsulinemia, hyperglycemia | [121] | ||
| 6 | ATP11A | metastasis in colorectal cancer | [122] | |
| ATP11B | ||||
| ATP11C | arrested B cell development, dystocia, anemia, hepatocellular carcinoma, conjugated hyperbilirubinemia, unconjugated hypercholanemia | [34,123,124] | ||
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Van der Mark, V.A.; Elferink, R.P.J.O.; Paulusma, C.C. P4 ATPases: Flippases in Health and Disease. Int. J. Mol. Sci. 2013, 14, 7897-7922. https://doi.org/10.3390/ijms14047897
Van der Mark VA, Elferink RPJO, Paulusma CC. P4 ATPases: Flippases in Health and Disease. International Journal of Molecular Sciences. 2013; 14(4):7897-7922. https://doi.org/10.3390/ijms14047897
Chicago/Turabian StyleVan der Mark, Vincent A., Ronald P.J. Oude Elferink, and Coen C. Paulusma. 2013. "P4 ATPases: Flippases in Health and Disease" International Journal of Molecular Sciences 14, no. 4: 7897-7922. https://doi.org/10.3390/ijms14047897