Diacylglycerol Kinases: Regulated Controllers of T Cell Activation, Function, and Development

{kind=link}
{kind=link}
Abstract
:1. Introduction
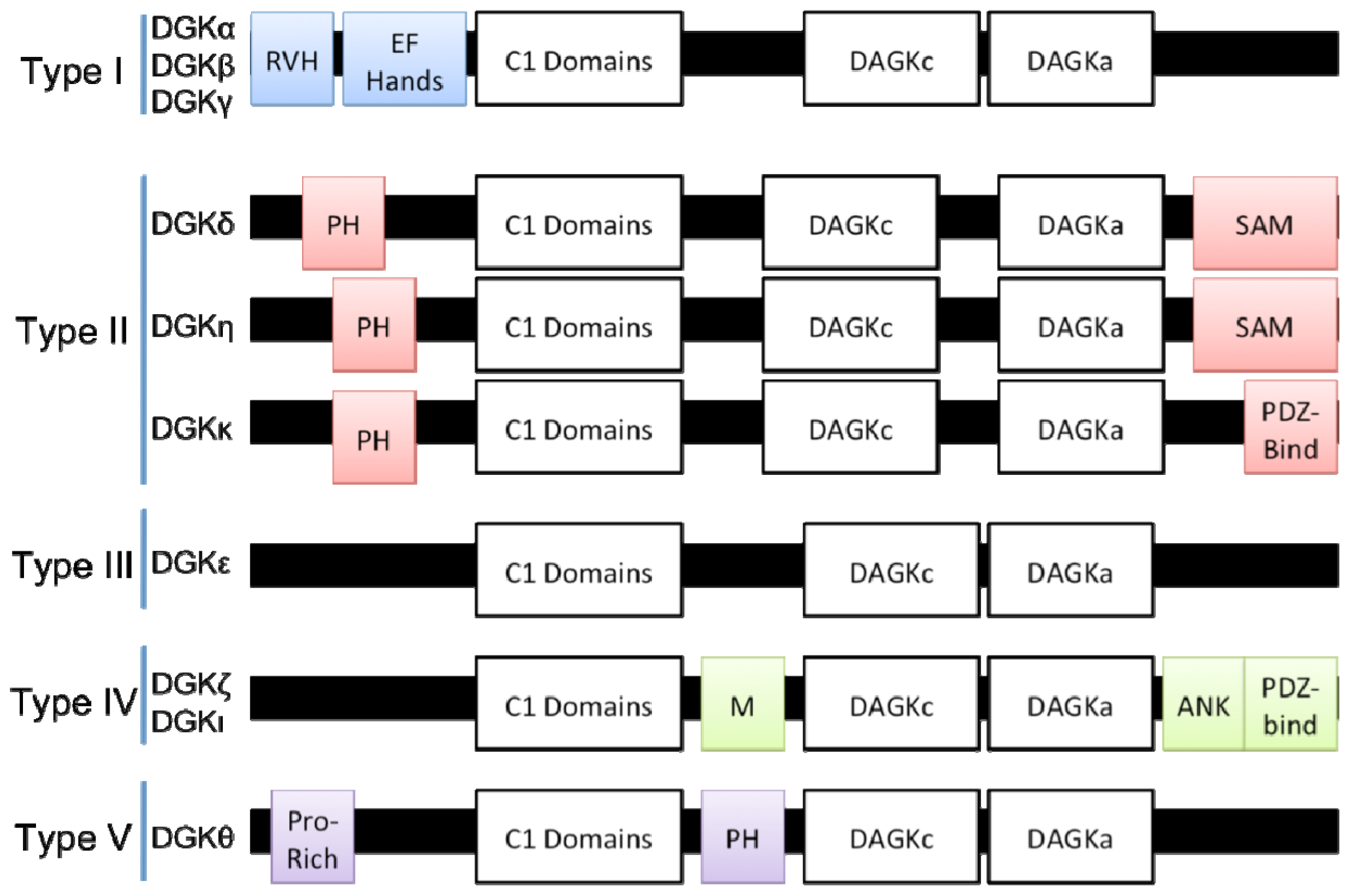
2. DGK Isotypes and Their Structures
2.1. DGK Subtypes
2.1.1. Type I DGKs (DGKα, DGKβ, and DGKγ)
2.1.2. Type II DGKs (DGKδ, DGKη, and DGKκ)
2.1.3. Type III DGKs (DGKɛ)
2.1.4. Type IV DGKs (DGKζ and DGKι)
2.1.5. Type V DGKs (DGKθ)
3. Tissue Expression of DGKs
4. Regulation of DGK Function in T Cells
4.1. Transcriptional Regulation of DGKs in T Cells
4.2. Post-Translational Regulation of DGKs in T Cells
5. DGK Regulation of Immune Cell Signaling and Activation
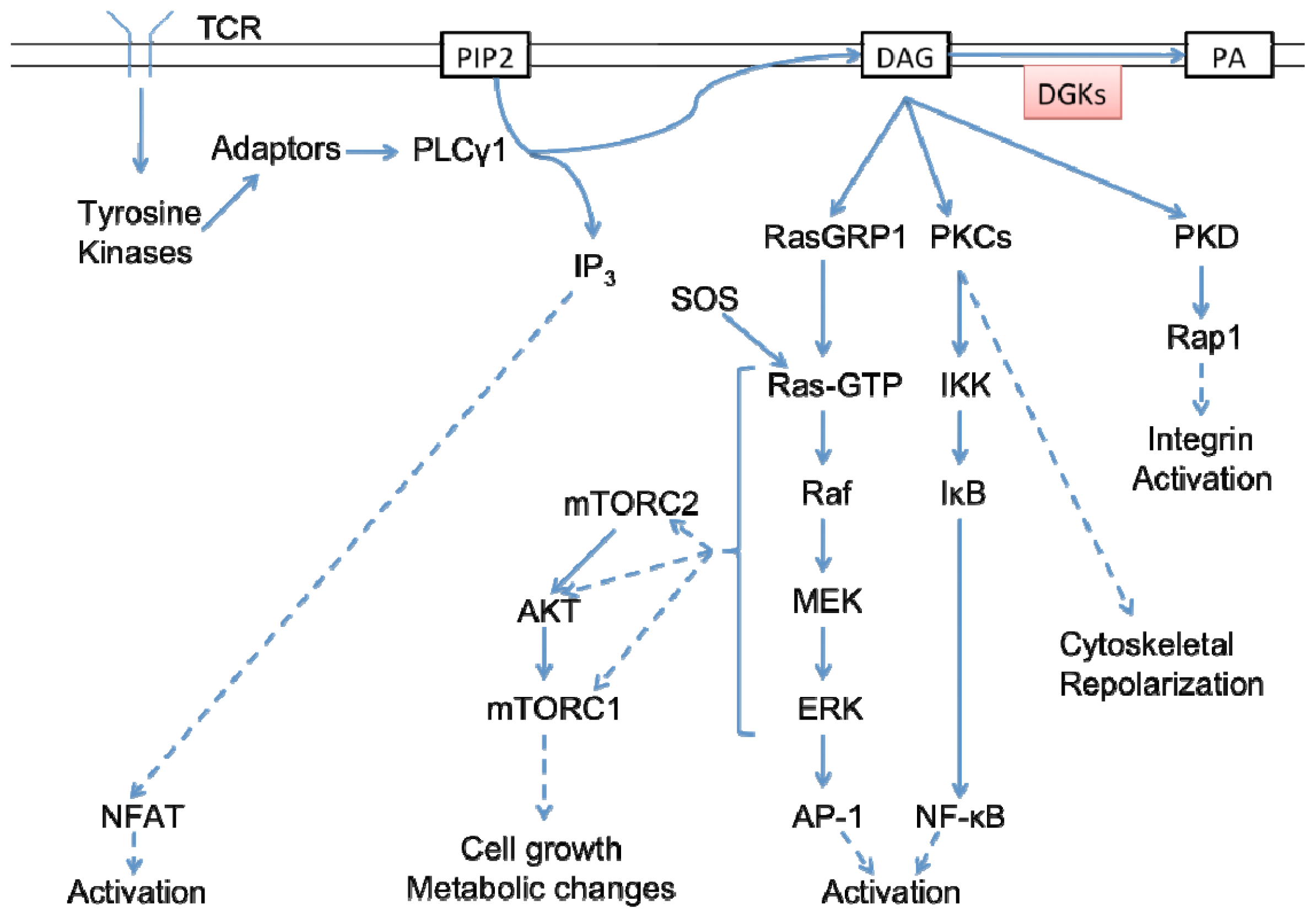
5.1. An Overview of TCR and DAG Signaling in T Cells
5.2. DGKα in T Cell Signaling and Activation
5.3. DGKζ in T Cell Signaling and Activation
5.4. DGK Regulation of AKT-mTOR Signaling in T Cells
5.5. DGKζ as a Modulator of Analog-to-Digital Signaling in T Cells
5.6. DGK at the Immunological Synapse
6. DGK Functions in T Cells
6.1. T Cell Anergy
6.2. T Cell Responses to Pathogen
6.3. T Cell Anti-Tumor Responses
6.4. T Cell Adhesion
7. DGKs and Development
7.1. T Cell Development
7.2. Invariant NKT Cell Development
8. Therapeutic Implications
9. Conclusions and Future Directions
Acknowledgments
Conflict of Interest
References
- Sanjuán, M.A.; Jones, D.R.; Izquierdo, M.; Mérida, I. Role of diacylglycerol kinase α in the attenuation of receptor signaling. J. Cell Biol 2001, 153, 207–220. [Google Scholar]
- Topham, M.K.; Bunting, M.; Zimmerman, G.A.; McIntyre, T.M.; Blackshear, P.J.; Prescott, S.M. Protein kinase C regulates the nuclear localization of diacylglycerol kinase-ζ. Nature 1998, 394, 697–700. [Google Scholar]
- Raben, D.M.; Wattenberg, B.W. Signaling at the membrane interface by the DGK/SK enzyme family. J. Lipid Res 2008, 50, S35–S39. [Google Scholar]
- Sugiura, M.; Kono, K.; Liu, H.; Shimizugawa, T.; Minekura, H.; Spiegel, S.; Kohama, T. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J. Biol. Chem 2002, 277, 23294–23300. [Google Scholar]
- Merida, I.; Avila-Flores, A.; Merino, E. Diacylglycerol kinases: At the hub of cell signalling. Biochem. J 2008, 410, 631–631. [Google Scholar]
- Hurley, J.H.; Misra, S. Signaling and subcellular targeting by membrane-binding domains. Annu. Rev. Biophys. Biomol. Struct 2000, 29, 49–79. [Google Scholar]
- Shirai, Y.; Segawa, S.; Kuriyama, M.; Goto, K.; Sakai, N.; Saito, N. Subtype-specific translocation of diacylglycerol kinase alpha and gamma and its correlation with protein kinase C. J. Biol. Chem 2000, 275, 24760–24766. [Google Scholar]
- Santos, T.; Carrasco, S.; Jones, D.R.; Mérida, I.; Eguinoa, A. Dynamics of diacylglycerol kinase ζ translocation in living T-cells. J. Biol. Chem 2002, 277, 30300–30309. [Google Scholar]
- Van Baal, J.; de Widt, J.; Divecha, N.; van Blitterswijk, W.J. Translocation of diacylglycerol kinase theta from cytosol to plasma membrane in response to activation of G protein-coupled receptors and protein kinase C. J. Biol. Chem 2005, 280, 9870–9878. [Google Scholar]
- Nelson, C.D.; Perry, S.J.; Regier, D.S.; Prescott, S.M.; Topham, M.K.; Lefkowitz, R.J. Targeting of diacylglycerol degradation to M1 muscarinic receptors by β-arrestins. Science 2007, 315, 663–666. [Google Scholar]
- Yakubchyk, Y.; Abramovici, H.; Maillet, J.-C.; Daher, E.; Obagi, C.; Parks, R.J.; Topham, M.K.; Gee, S.H. Regulation of neurite outgrowth in N1E-115 cells through PDZ-mediated recruitment of diacylglycerol kinase ζ. Mol. Cell. Biol 2005, 25, 7289–7302. [Google Scholar]
- Jiang, Y.; Qian, W.; Hawes, J.W.; Walsh, J.P. A domain with homology to neuronal calcium sensors is required for calcium-dependent activation of diacylglycerol kinase α. J. Biol. Chem 2000, 275, 34092–34099. [Google Scholar]
- Takahashi, M.; Yamamoto, T.; Sakai, H.; Sakane, F. Calcium negatively regulates an intramolecular interaction between the N-terminal recoverin homology and EF-hand motif domains and the C-terminal C1 and catalytic domains of diacylglycerol kinase α. Biochem. Biophys. Res. Commun 2012, 423, 571–576. [Google Scholar]
- Merino, E.; Sanjuán, M.A.; Moraga, I.; Ciprés, A.; Mérida, I. Role of the diacylglycerol kinase α-conserved domains in membrane targeting in intact T cells. J. Biol. Chem 2007, 282, 35396–35404. [Google Scholar]
- Yamada, K.; Sakane, F.; Matsushima, N.; Kanoh, H. EF-hand motifs of alpha, beta and gamma isoforms of diacylglycerol kinase bind calcium with different affinities and conformational changes. Biochem. J 1997, 321, 59–64. [Google Scholar]
- Tanino, F.; Maeda, Y.; Sakai, H.; Sakane, F. Induction of filopodia-like protrusions in N1E-115 neuroblastoma cells by diacylglycerol kinase γ independent of its enzymatic activity: Potential novel function of the C-terminal region containing the catalytic domain of diacylglycerol kinase γ. Mol. Cell. Biochem 2013, 373, 85–93. [Google Scholar]
- Imai, S.; Kai, M.; Yasuda, S.; Kanoh, H.; Sakane, F. Identification and characterization of a novel human type II diacylglycerol kinase, DGKκ. J. Biol. Chem 2005, 280, 39870–39881. [Google Scholar]
- Klauck, T.M.; Xu, X.; Mousseau, B.; Jaken, S. Cloning and characterization of a glucocorticoid-induced diacylglycerol kinase. J. Biol. Chem 1996, 271, 19781–19788. [Google Scholar]
- Sakane, F.; Imai, S.; Kai, M.; Wada, I.; Kanoh, H. Molecular cloning of a novel diacylglycerol kinase isozyme with a pleckstrin homology domain and a C-terminal tail similar to those of the EPH family of protein-tyrosine kinases. J. Biol. Chem 1996, 271, 8394–8401. [Google Scholar]
- Park, W.S.; Heo, W.D.; Whalen, J.H.; O’Rourke, N.A.; Bryan, H.M.; Meyer, T.; Teruel, M.N. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol. Cell 2008, 30, 381–392. [Google Scholar]
- Takeuchi, H.; Kanematsu, T.; Misumi, Y.; Sakane, F.; Konishi, H.; Kikkawa, U.; Watanabe, Y.; Katan, M.; Hirata, M. Distinct specificity in the binding of inositol phosphates by pleckstrin homology domains of pleckstrin, RAC-protein kinase, diacylglycerol kinase and a new 130 kDa protein. Biochim. Biophys. Acta 1997, 1359, 275–285. [Google Scholar]
- Imai, S.; Sakane, F.; Kanoh, H. Phorbol ester-regulated oligomerization of diacylglycerol kinase delta linked to its phosphorylation and translocation. J. Biol. Chem 2002, 277, 35323–35332. [Google Scholar]
- Imai, S.-I.; Kai, M.; Yamada, K.; Kanoh, H.; Sakane, F. The plasma membrane translocation of diacylglycerol kinase delta1 is negatively regulated by conventional protein kinase C-dependent phosphorylation at Ser-22 and Ser-26 within the pleckstrin homology domain. Biochem. J 2004, 382, 957–966. [Google Scholar]
- Knight, M.J.; Joubert, M.K.; Plotkowski, M.L.; Kropat, J.; Gingery, M.; Sakane, F.; Merchant, S.S.; Bowie, J.U. Zinc binding drives sheet formation by the SAM domain of diacylglycerol kinase δ. Biochemistry 2010, 49, 9667–9676. [Google Scholar]
- Kohyama-Koganeya, A.; Watanabe, M.; Hotta, Y. Molecular cloning of a diacylglycerol kinase isozyme predominantly expressed in rat retina. FEBS Lett 1997, 409, 258–264. [Google Scholar]
- De Turco, R.E.B.; Tang, W.; Topham, M.K.; Sakane, F.; Marcheselli, V.L.; Chen, C.; Taketomi, A.; Prescott, S.M.; Bazan, N.G. Diacylglycerol kinase epsilon regulates seizure susceptibility and long-term potentiation through arachidonoyl-inositol lipid signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 4740–4745. [Google Scholar]
- Prescott, S.M.; Majerus, P.W. The fatty acid composition of phosphatidylinositol from thrombin-stimulated human platelets. J. Biol. Chem 1981, 256, 579–582. [Google Scholar]
- Shulga, Y.V.; Topham, M.K.; Epand, R.M. Regulation and functions of diacylglycerol kinases. Chem. Rev 2011, 111, 6186–6208. [Google Scholar]
- Rincon, E.; Santos, T.; Ávila-Flores, A.; Albar, J.P.; Lalioti, V.; Lei, C.; Hong, W.; Mérida, I. Proteomics identification of sorting Nexin 27 as a diacylglycerol kinase ζ-associated protein. Mol. Cell. Proteomics 2007, 6, 1073–1087. [Google Scholar]
- Hogan, A.; Shepherd, L.; Chabot, J.; Quenneville, S.; Prescott, S.M.; Topham, M.K.; Gee, S.H. Interaction of γ1-syntrophin with diacylglycerol kinase-ζ. J. Biol. Chem 2001, 276, 26526–26533. [Google Scholar]
- Abramovici, H.; Hogan, A.B.; Obagi, C.; Topham, M.K.; Gee, S.H. Diacylglycerol kinase-ζ localization in skeletal muscle is regulated by phosphorylation and interaction with syntrophins. Mol. Biol. Cell 2003, 14, 4499–4511. [Google Scholar]
- Abramovici, H.; Gee, S.H. Morphological changes and spatial regulation of diacylglycerol kinase-ζ, syntrophins, and Rac1 during myoblast fusion. Cell Motil. Cytoskeleton 2007, 64, 549–567. [Google Scholar]
- Liu, Z.; Chang, G.-Q.; Leibowitz, S.F. Diacylglycerol kinase ζ in hypothalamus interacts with long form leptin receptor. J. Biol. Chem 2001, 276, 5900–5907. [Google Scholar]
- Kim, K.; Yang, J.; Zhong, X.-P.; Kim, M.-H.; Kim, Y.S.; Lee, H.W.; Han, S.; Choi, J.; Han, K.; Seo, J.; et al. Synaptic removal of diacylglycerol by DGKzeta and PSD-95 regulates dendritic spine maintenance. EMBO J 2009, 28, 1170–1179. [Google Scholar]
- Yang, J.; Seo, J.; Nair, R.; Han, S.; Jang, S.; Kim, K.; Han, K.; Paik, S.K.; Choi, J.; Lee, S.; et al. DGKι regulates presynaptic release during mGluR-dependent LTD. EMBO J 2011, 30, 165–180. [Google Scholar]
- Houssa, B.; Schaap, D.; van der Wal, J.; Goto, K.; Kondo, H.; Yamakawa, A.; Shibata, M.; Takenawa, T.; Van Blitterswijk, W.J. Cloning of a novel human diacylglycerol kinase (DGKtheta) containing three cysteine-rich domains, a proline-rich region, and a pleckstrin homology domain with an overlapping Ras-associating domain. J. Biol. Chem 1997, 272, 10422–10428. [Google Scholar]
- Los, A.P.; van Baal, J.; de Widt, J.; Divecha, N.; van Blitterswijk, W.J. Structure-activity relationship of diacylglycerol kinase theta. Biochim. Biophys. Acta 2004, 1636, 169–174. [Google Scholar]
- Crotty, T.; Cai, J.; Sakane, F.; Taketomi, A.; Prescott, S.M.; Topham, M.K. Diacylglycerol kinase δ regulates protein kinase C and epidermal growth factor receptor signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 15485–15490. [Google Scholar]
- Zha, Y.; Marks, R.; Ho, A.W.; Peterson, A.C.; Janardhan, S.; Brown, I.; Praveen, K.; Stang, S.; Stone, J.C.; Gajewski, T.F. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-[alpha]. Nat. Immunol 2006, 7, 1166–1173. [Google Scholar]
- Macián, F.; García-Cózar, F.; Im, S.-H.; Horton, H.F.; Byrne, M.C.; Rao, A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 2002, 109, 719–731. [Google Scholar]
- Olenchock, B.A.; Guo, R.; Carpenter, J.H.; Jordan, M.; Topham, M.K.; Koretzky, G.A.; Zhong, X.-P. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol 2006, 7, 1174–1181. [Google Scholar]
- Martínez-Moreno, M.; García-Liévana, J.; Soutar, D.; Torres-Ayuso, P.; Andrada, E.; Zhong, X.-P.; Koretzky, G.A.; Mérida, I.; Ávila-Flores, A. FoxO-dependent regulation of diacylglycerol kinase α gene expression. Mol. Cell. Biol 2012, 32, 4168–4180. [Google Scholar]
- Zheng, Y.; Zha, Y.; Driessens, G.; Locke, F.; Gajewski, T.F. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J. Exp. Med 2012, 209, 2157–2163. [Google Scholar]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol 2009, 27, 591–619. [Google Scholar]
- Baldanzi, G.; Pighini, A.; Bettio, V.; Rainero, E.; Traini, S.; Chianale, F.; Porporato, P.E.; Filigheddu, N.; Mesturini, R.; Song, S.; et al. SAP-mediated inhibition of diacylglycerol kinase α regulates TCR-induced diacylglycerol signaling. J. Immunol 2011, 187, 5941–5951. [Google Scholar]
- Sanjuan, M.A.; Pradet-Balade, B.; Jones, D.R.; Martinez-A, C.; Stone, J.C.; Garcia-Sanz, J.A.; Merida, I. T cell activation in vivo targets diacylglycerol kinase α to the membrane: A novel mechanism for Ras attenuation. J. Immunol 2003, 170, 2877–2883. [Google Scholar]
- Merino, E.; Ávila-Flores, A.; Shirai, Y.; Moraga, I.; Saito, N.; Mérida, I. Lck-dependent tyrosine phosphorylation of diacylglycerol kinase α regulates its membrane association in T cells. J. Immunol 2008, 180, 5805–5815. [Google Scholar]
- Gharbi, S.I.; Rincón, E.; Avila-Flores, A.; Torres-Ayuso, P.; Almena, M.; Cobos, M.A.; Albar, J.P.; Mérida, I. Diacylglycerol kinase ζ controls diacylglycerol metabolism at the immune synapse. Mol. Biol. Cell 2011, 22, 4406–4414. [Google Scholar]
- Zhang, W.; Sloan-Lancaster, J.; Kitchen, J.; Trible, R.P.; Samelson, L.E. LAT: The ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 1998, 92, 83–92. [Google Scholar]
- Motto, D.G.; Ross, S.E.; Wu, J.; Hendricks-Taylor, L.R.; Koretzky, G.A. Implication of the GRB2-associated phosphoprotein SLP-76 in T cell receptor-mediated interleukin 2 production. J. Exp. Med 1996, 183, 1937–1943. [Google Scholar]
- Imboden, J.B.; Stobo, J.D. Transmembrane signalling by the T cell antigen receptor. Perturbation of the T3-antigen receptor complex generates inositol phosphates and releases calcium ions from intracellular stores. J. Exp. Med 1985, 161, 446–456. [Google Scholar]
- Tognon, C.E.; Kirk, H.E.; Passmore, L.A.; Whitehead, I.P.; Der, C.J.; Kay, R.J. Regulation of RasGRP via a phorbol ester-responsive C1 domain. Mol. Cell. Biol 1998, 18, 6995–7008. [Google Scholar]
- Quann, E.J.; Liu, X.; Altan-Bonnet, G.; Huse, M. A cascade of protein kinase C isozymes promotes cytoskeletal polarization in T cells. Nat. Immunol 2011, 12, 647–654. [Google Scholar]
- Spitaler, M.; Emslie, E.; Wood, C.D.; Cantrell, D. Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity 2006, 24, 535–546. [Google Scholar]
- Dower, N.A.; Stang, S.L.; Bottorff, D.A.; Ebinu, J.O.; Dickie, P.; Ostergaard, H.L.; Stone, J.C. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat. Immunol 2000, 1, 317–321. [Google Scholar]
- Coudronniere, N.; Villalba, M.; Englund, N.; Altman, A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc. Natl. Acad. Sci. USA 2000, 97, 3394–3399. [Google Scholar]
- Sun, Z.; Arendt, C.W.; Ellmeier, W.; Schaeffer, E.M.; Sunshine, M.J.; Gandhi, L.; Annes, J.; Petrzilka, D.; Kupfer, A.; Schwartzberg, P.L.; et al. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 2000, 404, 402–407. [Google Scholar]
- Narayan, P.; Holt, B.; Tosti, R.; Kane, L.P. CARMA1 is required for Akt-mediated NF-kappaB activation in T cells. Mol. Cell. Biol 2006, 26, 2327–2336. [Google Scholar]
- Wang, D.; Matsumoto, R.; You, Y.; Che, T.; Lin, X.-Y.; Gaffen, S.L.; Lin, X. CD3/CD28 costimulation-induced NF-kappaB activation is mediated by recruitment of protein kinase C-theta, Bcl10, and IkappaB kinase beta to the immunological synapse through CARMA1. Mol. Cell. Biol 2004, 24, 164–171. [Google Scholar]
- Hara, H.; Bakal, C.; Wada, T.; Bouchard, D.; Rottapel, R.; Saito, T.; Penninger, J.M. The molecular adapter Carma1 controls entry of IkappaB kinase into the central immune synapse. J. Exp. Med 2004, 200, 1167–1177. [Google Scholar]
- Jun, J.E.; Wilson, L.E.; Vinuesa, C.G.; Lesage, S.; Blery, M.; Miosge, L.A.; Cook, M.C.; Kucharska, E.M.; Hara, H.; Penninger, J.M.; et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity 2003, 18, 751–762. [Google Scholar]
- Medeiros, R.B.; Dickey, D.M.; Chung, H.; Quale, A.C.; Nagarajan, L.R.; Billadeau, D.D.; Shimizu, Y. Protein kinase D1 and the beta 1 integrin cytoplasmic domain control beta 1 integrin function via regulation of Rap1 activation. Immunity 2005, 23, 213–226. [Google Scholar]
- Mor, A.; Campi, G.; Du, G.; Zheng, Y.; Foster, D.A.; Dustin, M.L.; Philips, M.R. The lymphocyte function-associated antigen-1 receptor costimulates plasma membrane Ras via phospholipase D2. Nat. Cell Biol 2007, 9, 713–719. [Google Scholar]
- Foster, D.A. Regulation of mTOR by phosphatidic acid? Cancer Res 2007, 67, 1–4. [Google Scholar]
- Foster, D.A. Phosphatidic acid signaling to mTOR: Signals for the survival of human cancer cells. Biochim. Biophys. Acta 2009, 1791, 949–955. [Google Scholar]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar]
- Avila-Flores, A. Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid. J. Biol. Chem 2005, 280, 10091–10099. [Google Scholar]
- Zhong, X.-P.; Guo, R.; Zhou, H.; Liu, C.; Wan, C.-K. Diacylglycerol kinases in immune cell function and self-tolerance. Immunol. Rev 2008, 224, 249–264. [Google Scholar]
- Zhong, X.-P.; Hainey, E.A.; Olenchock, B.A.; Zhao, H.; Topham, M.K.; Koretzky, G.A. Regulation of T cell receptor-induced activation of the Ras-ERK pathway by dacylglycerol kinase ζ. J. Biol. Chem 2002, 277, 31089–31098. [Google Scholar]
- Zhong, X.-P.; Hainey, E.A.; Olenchock, B.A.; Jordan, M.S.; Maltzman, J.S.; Nichols, K.E.; Shen, H.; Koretzky, G.A. Enhanced T cell responses due to diacylglycerol kinase [zeta] deficiency. Nat. Immunol 2003, 4, 882–890. [Google Scholar]
- Gorentla, B.K.; Wan, C.-K.; Zhong, X.-P. Negative regulation of mTOR activation by diacylglycerol kinases. Blood 2011, 117, 4022–4031. [Google Scholar]
- Chakraborty, A.K.; Das, J.; Zikherman, J.; Yang, M.; Govern, C.C.; Ho, M.; Weiss, A.; Roose, J. Molecular origin and functional consequences of digital signaling and hysteresis during Ras activation in lymphocytes. Sci. Signal. 2009, 2, pt2. [Google Scholar]
- Roose, J.P.; Mollenauer, M.; Gupta, V.A.; Stone, J.; Weiss, A. A diacylglycerol-protein kinase C-RasGRP1 pathway directs Ras activation upon antigen receptor stimulation of T cells. Mol. Cell. Biol 2005, 25, 4426–4441. [Google Scholar]
- Roose, J.P.; Mollenauer, M.; Ho, M.; Kurosaki, T.; Weiss, A. Unusual interplay of two types of Ras activators, RasGRP and SOS, establishes sensitive and robust Ras activation in lymphocytes. Mol. Cell. Biol 2007, 27, 2732–2745. [Google Scholar]
- Das, J.; Ho, M.; Zikherman, J.; Govern, C.; Yang, M.; Weiss, A.; Chakraborty, A.K.; Roose, J.P. Digital signaling and hysteresis characterize Ras activation in lymphoid cells. Cell 2009, 136, 337–351. [Google Scholar]
- Riese, M.J.; Grewal, J.; Das, J.; Zou, T.; Patil, V.; Chakraborty, A.K.; Koretzky, G.A. Decreased diacylglycerol metabolism enhances ERK activation and augments CD8+ T cell functional responses. J. Biol. Chem 2011, 286, 5254–5265. [Google Scholar]
- Quann, E.J.; Merino, E.; Furuta, T.; Huse, M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat. Immunol 2009, 10, 627–635. [Google Scholar]
- Monleón, I.; Martínez-Lorenzo, M.J.; Monteagudo, L.; Lasierra, P.; Taulés, M.; Iturralde, M.; Piñeiro, A.; Larrad, L.; Alava, M.A.; Naval, J.; et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J. Immunol 2001, 167, 6736–6744. [Google Scholar]
- Zuccato, E.; Blott, E.J.; Holt, O.; Sigismund, S.; Shaw, M.; Bossi, G.; Griffiths, G.M. Sorting of Fas ligand to secretory lysosomes is regulated by mono-ubiquitylation and phosphorylation. J. Cell Sci 2007, 120, 191–199. [Google Scholar]
- Alonso, R.; Mazzeo, C.; Rodriguez, M.C.; Marsh, M.; Fraile-Ramos, A.; Calvo, V.; Avila-Flores, A.; Merida, I.; Izquierdo, M. Diacylglycerol kinase α regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of Fas ligand-containing exosomes in T lymphocytes. Cell Death Differ 2011, 18, 1161–1173. [Google Scholar] [Green Version]
- Jenkins, M.K.; Pardoll, D.M.; Mizuguchi, J.; Chused, T.M.; Schwartz, R.H. Molecular events in the induction of a nonresponsive state in interleukin 2-producing helper T-lymphocyte clones. Proc. Natl. Acad. Sci. USA 1987, 84, 5409–5413. [Google Scholar]
- Choi, S.; Schwartz, R.H. Molecular mechanisms for adaptive tolerance and other T cell anergy models. Semin. Immunol 2007, 19, 140–152. [Google Scholar]
- Fields, P.E.; Gajewski, T.F.; Fitch, F.W. Blocked Ras activation in anergic CD4+ T cells. Science 1996, 271, 1276–1278. [Google Scholar]
- Li, W.; Whaley, C.D.; Mondino, A.; Mueller, D.L. Blocked signal transduction to the ERK and JNK protein kinases in anergic CD4+ T cells. Science 1996, 271, 1272–1276. [Google Scholar]
- Kang, S.M.; Beverly, B.; Tran, A.C.; Brorson, K.; Schwartz, R.H.; Lenardo, M.J. Transactivation by AP-1 is a molecular target of T cell clonal anergy. Science 1992, 257, 1134–1138. [Google Scholar]
- Zehn, D.; Lee, S.Y.; Bevan, M.J. Complete but curtailed T-cell response to very low-affinity antigen. Nature 2009, 458, 211–214. [Google Scholar]
- Smith-Garvin, J.E.; Burns, J.C.; Gohil, M.; Zou, T.; Kim, J.S.; Maltzman, J.S.; Wherry, E.J.; Koretzky, G.A.; Jordan, M.S. T-cell receptor signals direct the composition and function of the memory CD8+ T-cell pool. Blood 2010, 116, 5548–5559. [Google Scholar]
- Wiehagen, K.R.; Corbo, E.; Schmidt, M.; Shin, H.; Wherry, E.J.; Maltzman, J.S. Loss of tonic T-cell receptor signals alters the generation but not the persistence of CD8+ memory T cells. Blood 2010, 116, 5560–5570. [Google Scholar]
- Teixeiro, E.; Daniels, M.A.; Hamilton, S.E.; Schrum, A.G.; Bragado, R.; Jameson, S.C.; Palmer, E. Different T cell receptor signals determine CD8+ memory versus effector development. Science 2009, 323, 502–505. [Google Scholar]
- Shin, J.; O’Brien, T.F.; Grayson, J.M.; Zhong, X.-P. Differential regulation of primary and memory CD8 T cell immune responses by diacylglycerol kinases. J. Immunol 2012, 188, 2111–2117. [Google Scholar]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar]
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and eomesodermin. Immunity 2010, 32, 67–78. [Google Scholar]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol 2012, 12, 325–338. [Google Scholar]
- Staveley-O’Carroll, K.; Sotomayor, E.; Montgomery, J.; Borrello, I.; Hwang, L.; Fein, S.; Pardoll, D.; Levitsky, H. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA 1998, 95, 1178–1183. [Google Scholar]
- Prinz, P.U.; Mendler, A.N.; Masouris, I.; Durner, L.; Oberneder, R.; Noessner, E. High DGK-α and disabled MAPK pathways cause dysfunction of human tumor-infiltrating CD8+ T cells that is reversible by pharmacologic intervention. J. Immunol 2012, 188, 5990–6000. [Google Scholar]
- Hogg, N.; Patzak, I.; Willenbrock, F. The insider’s guide to leukocyte integrin signalling and function. Nat. Rev. Immunol 2011, 11, 416–426. [Google Scholar]
- Sackstein, R. The lymphocyte homing receptors: Gatekeepers of the multistep paradigm. Curr. Opin. Hematol 2005, 12, 444–450. [Google Scholar]
- Lee, D.; Kim, J.; Beste, M.T.; Koretzky, G.A.; Hammer, D.A. Diacylglycerol kinase zeta negatively regulates CXCR4-stimulated T lymphocyte firm arrest to ICAM-1 under shear flow. Integr. Biol 2012, 4, 606–614. [Google Scholar]
- Priatel, J.J.; Chen, X.; Dhanji, S.; Abraham, N.; Teh, H.-S. RasGRP1 transmits prodifferentiation TCR signaling that is crucial for CD4 T cell development. J. Immunol 2006, 177, 1470–1480. [Google Scholar]
- Daniels, M.A.; Teixeiro, E.; Gill, J.; Hausmann, B.; Roubaty, D.; Holmberg, K.; Werlen, G.; Hollander, G.A.; Gascoigne, N.R.J.; Palmer, E. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature 2006, 444, 724–729. [Google Scholar]
- Mariathasan, S.; Zakarian, A.; Bouchard, D.; Michie, A.M.; Zúñiga-Pflücker, J.C.; Ohashi, P.S. Duration and strength of extracellular signal-regulated kinase signals are altered during positive versus negative thymocyte selection. J. Immunol 2001, 167, 4966–4973. [Google Scholar]
- Werlen, G.; Hausmann, B.; Palmer, E. A motif in the alphabeta T-cell receptor controls positive selection by modulating ERK activity. Nature 2000, 406, 422–426. [Google Scholar]
- Outram, S.V.; Crompton, T.; Merida, I.; Varas, A.; Martinez-A, C. Diacylglycerol kinase α activity promotes survival of CD4+ 8+ double positive cells during thymocyte development. Immunology 2002, 105, 391–398. [Google Scholar]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W., III; et al. I. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar]
- Lattin, J.E.; Schroder, K.; Su, A.I.; Walker, J.R.; Zhang, J.; Wiltshire, T.; Saijo, K.; Glass, C.K.; Hume, D.A.; Kellie, S.; et al. Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res 2008, 4, 5. [Google Scholar]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar]
- Joshi, R.P.; Koretzky, G.A. Abramson Family Cancer Research Institute, University of Pennsylvania: Philadelphia PA, USA, Unpublished work; 2013.
- Guo, R.; Wan, C.-K.; Carpenter, J.H.; Mousallem, T.; Boustany, R.-M.N.; Kuan, C.-T.; Burks, A.W.; Zhong, X.-P. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase α and ζ. Proc. Natl. Acad. Sci. USA 2008, 105, 11909–11914. [Google Scholar]
- Klinger, M.B.; Guilbault, B.; Goulding, R.E.; Kay, R.J. Deregulated expression of RasGRP1 initiates thymic lymphomagenesis independently of T-cell receptors. 2004, 24, 2695–2704. [Google Scholar]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol 2007, 25, 297–336. [Google Scholar]
- Bendelac, A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J. Exp. Med 1995, 182, 2091–2096. [Google Scholar]
- Wei, D.G.; Lee, H.; Park, S.-H.; Beaudoin, L.; Teyton, L.; Lehuen, A.; Bendelac, A. Expansion and long-range differentiation of the NKT cell lineage in mice expressing CD1d exclusively on cortical thymocytes. J. Exp. Med 2005, 202, 239–248. [Google Scholar]
- Nichols, K.E.; Hom, J.; Gong, S.-Y.; Ganguly, A.; Ma, C.S.; Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L.; Koretzky, G.A.; Stein, P.L. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat. Med 2005, 11, 340–345. [Google Scholar]
- Pasquier, B.; Yin, L.; Fondanèche, M.-C.; Relouzat, F.; Bloch-Queyrat, C.; Lambert, N.; Fischer, A.; de Saint-Basile, G.; Latour, S. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J. Exp. Med 2005, 201, 695–701. [Google Scholar]
- Griewank, K.; Borowski, C.; Rietdijk, S.; Wang, N.; Julien, A.; Wei, D.G.; Mamchak, A.A.; Terhorst, C.; Bendelac, A. Homotypic interactions mediated by Slamf1 and Slamf6 receptors control NKT cell lineage development. Immunity 2007, 27, 751–762. [Google Scholar]
- Eberl, G.; Lowin-Kropf, B.; MacDonald, H.R. Cutting edge: NKT cell development is selectively impaired in Fyn-deficient mice. J. Immunol 1999, 163, 4091–4094. [Google Scholar]
- Gadue, P.; Morton, N.; Stein, P.L. The Src family tyrosine kinase Fyn regulates natural killer T cell development. J. Exp. Med 1999, 190, 1189–1196. [Google Scholar]
- Shen, S.; Wu, J.; Srivatsan, S.; Gorentla, B.K.; Shin, J.; Xu, L.; Zhong, X.-P. Tight regulation of diacylglycerol-mediated signaling is critical for proper invariant NKT cell development. J. Immunol 2011, 187, 2122–2129. [Google Scholar]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Trans. Med 2011, 3, 95r, a73.. [Google Scholar]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N. Engl. J. Med 2011, 365, 725–733. [Google Scholar]
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric antigen receptors for T cell immunotherapy: Current understanding and future directions. J. Gene Med 2012, 14, 405–415. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Joshi, R.P.; Koretzky, G.A. Diacylglycerol Kinases: Regulated Controllers of T Cell Activation, Function, and Development. Int. J. Mol. Sci. 2013, 14, 6649-6673. https://doi.org/10.3390/ijms14046649
Joshi RP, Koretzky GA. Diacylglycerol Kinases: Regulated Controllers of T Cell Activation, Function, and Development. International Journal of Molecular Sciences. 2013; 14(4):6649-6673. https://doi.org/10.3390/ijms14046649
Chicago/Turabian StyleJoshi, Rohan P., and Gary A. Koretzky. 2013. "Diacylglycerol Kinases: Regulated Controllers of T Cell Activation, Function, and Development" International Journal of Molecular Sciences 14, no. 4: 6649-6673. https://doi.org/10.3390/ijms14046649