Mechanisms Involved in the Pro-Apoptotic Effect of Melatonin in Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
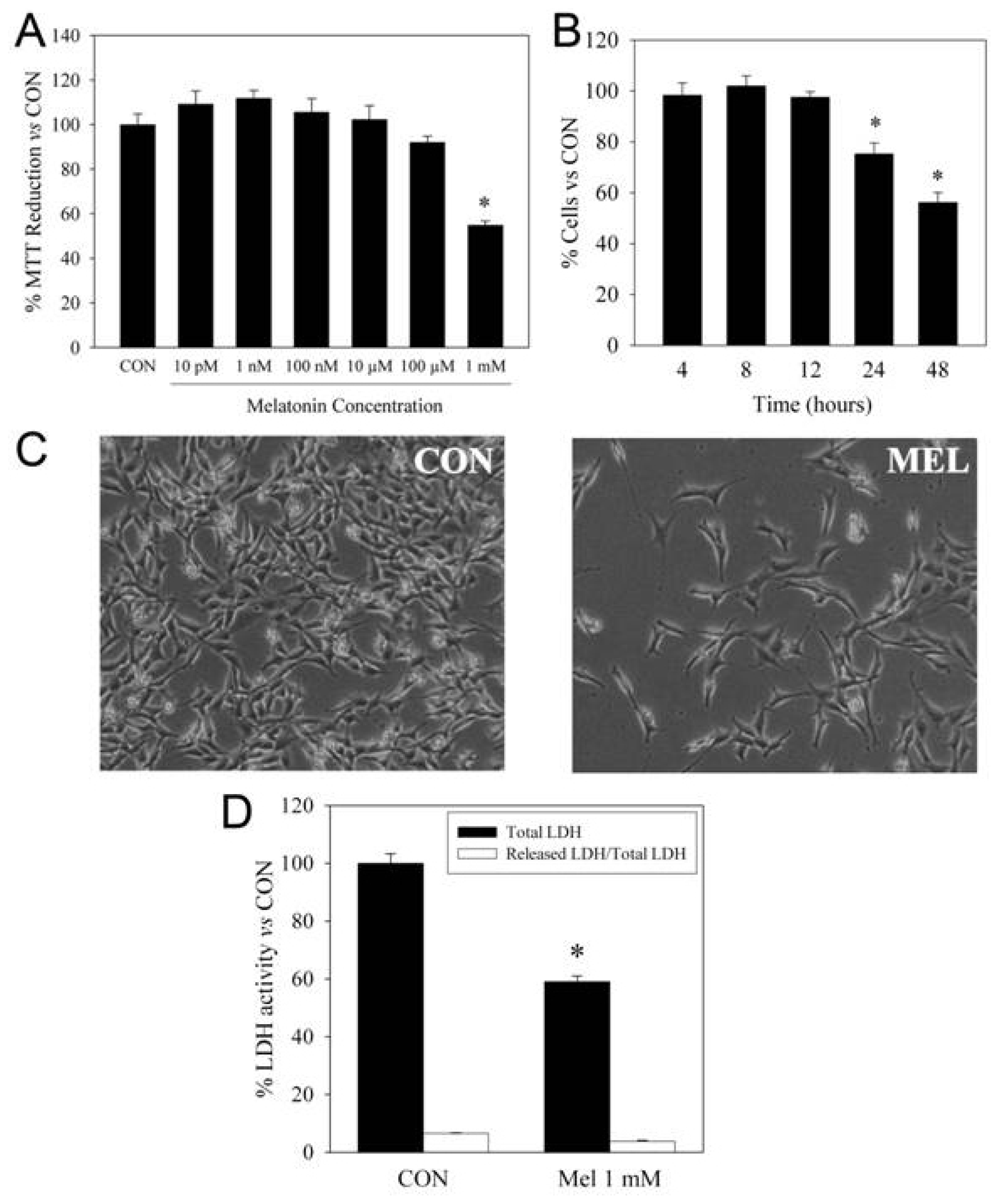
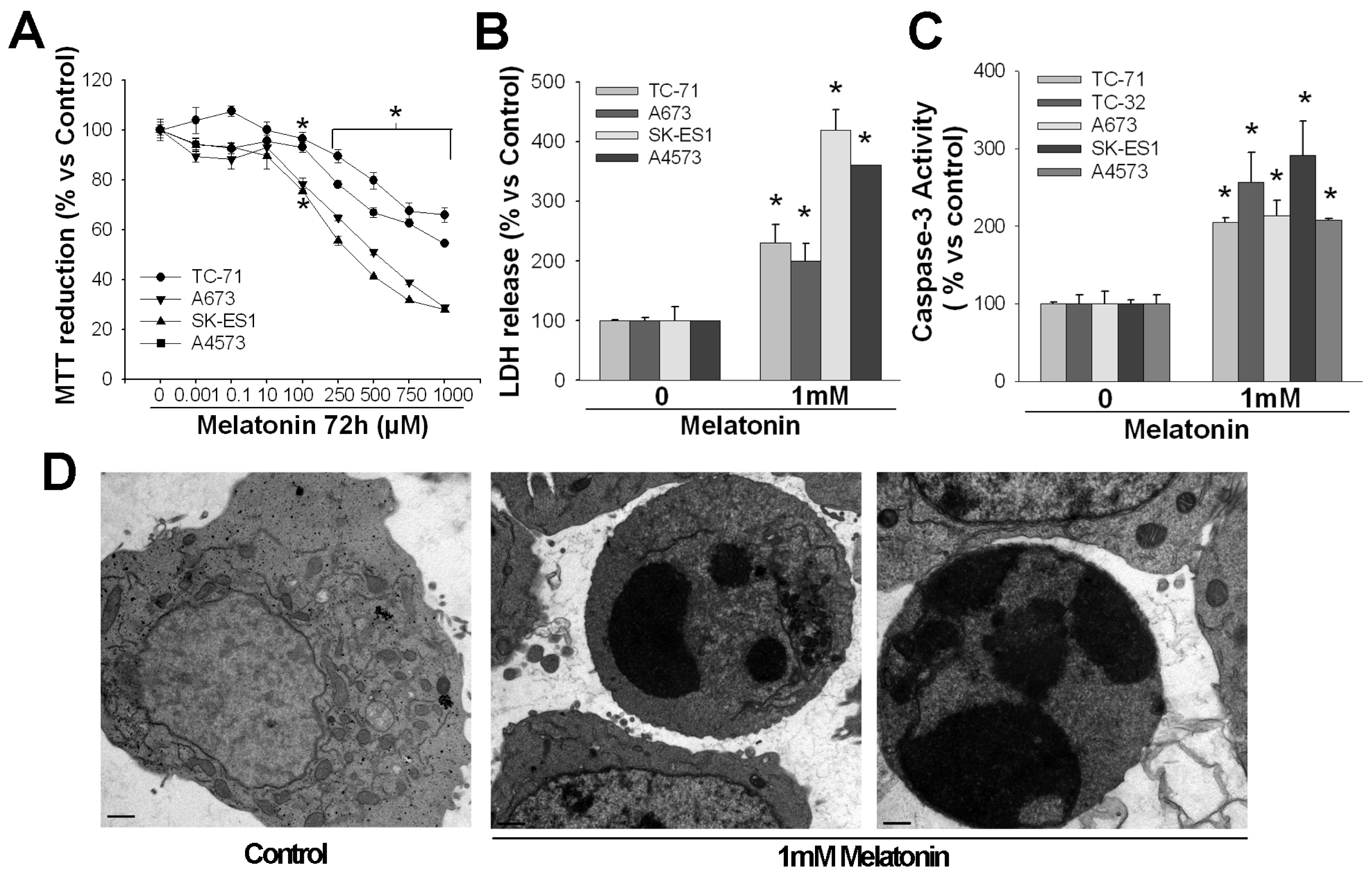
2. Pro-Apoptotic Effects of High Concentrations of Melatonin
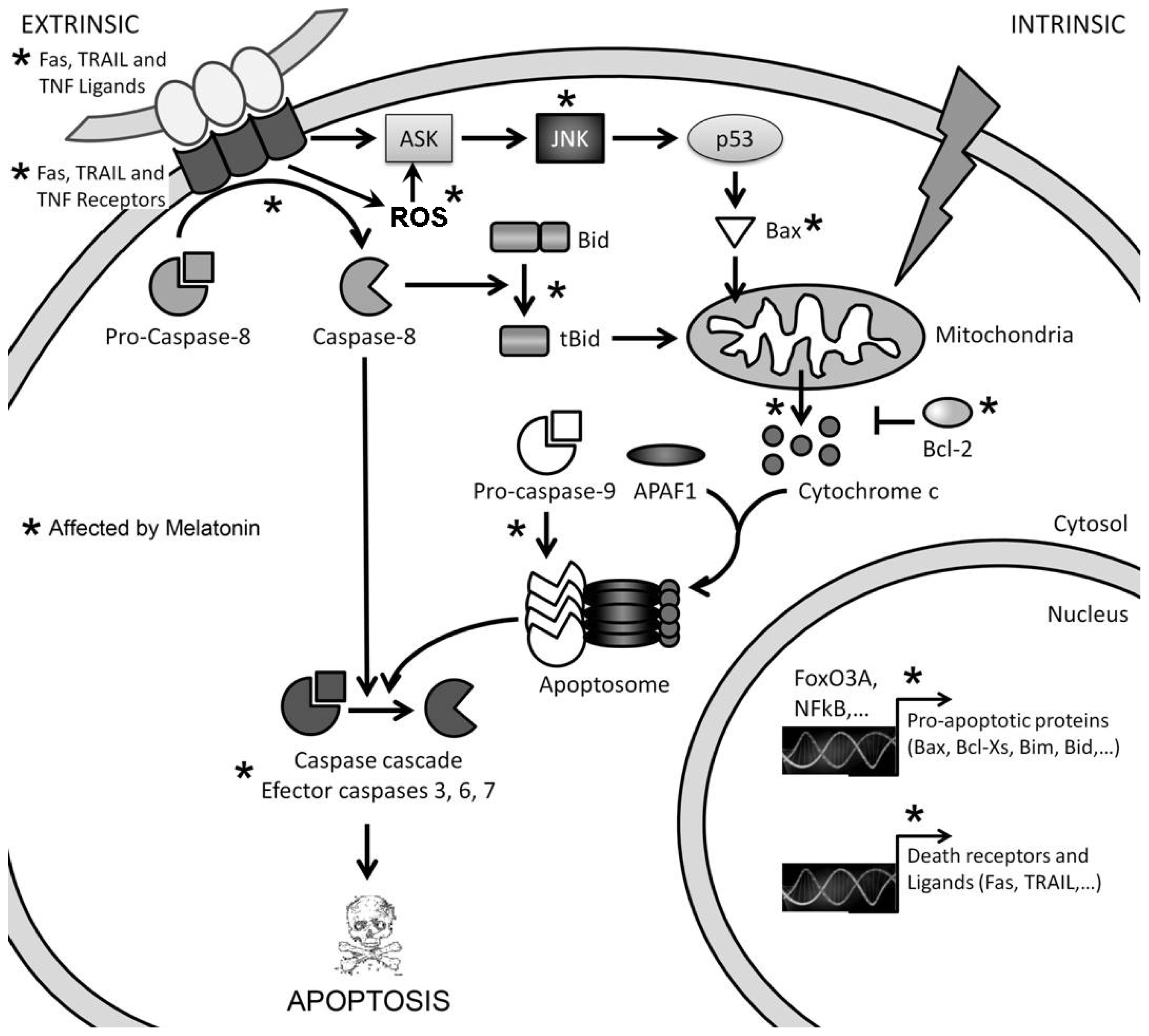
3. Mechanisms of Action Involved in the Pro-Apoptotic Effect of High Concentrations of Melatonin
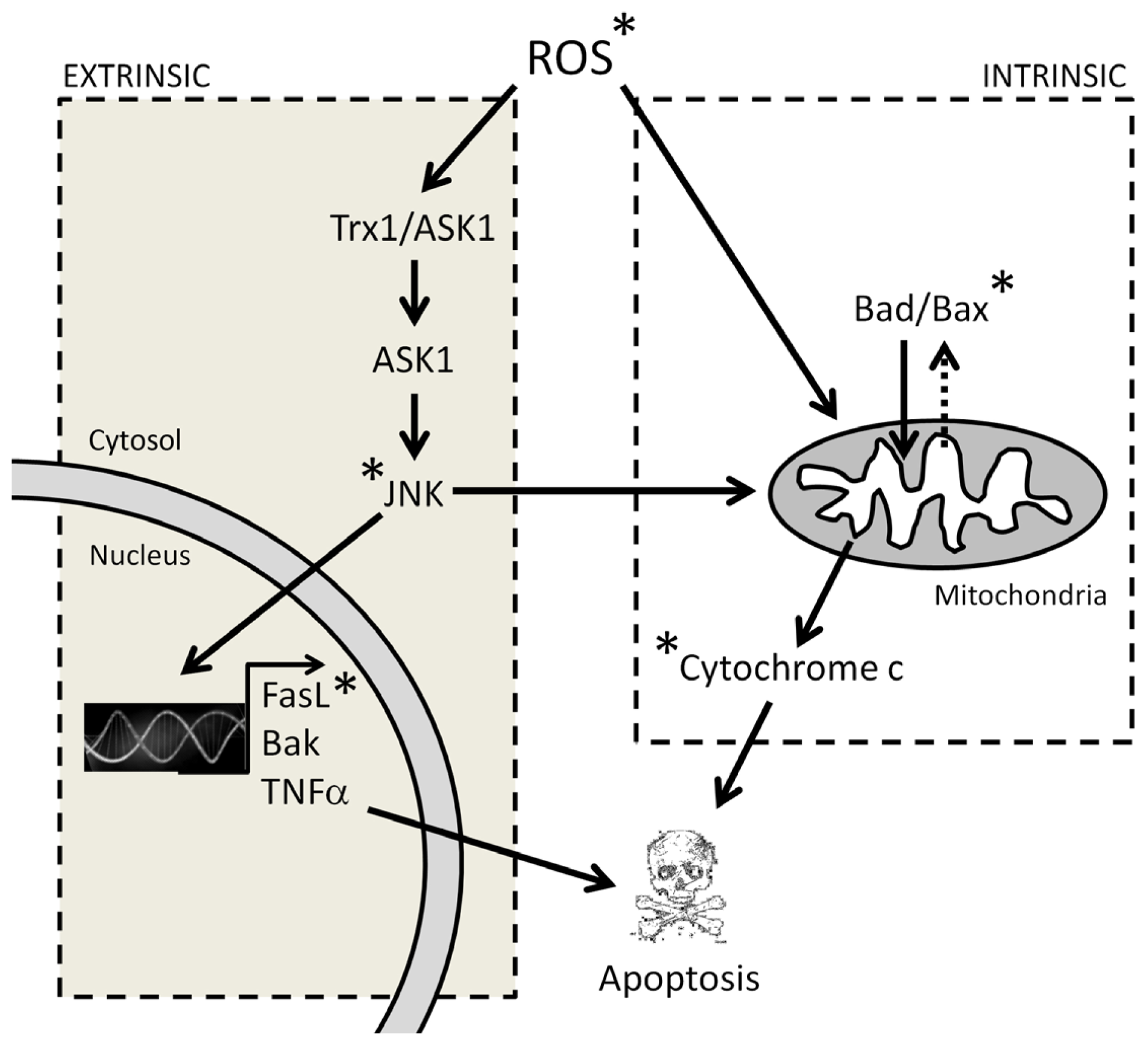
3.1. Melatonin-Induced Apoptosis Is Associated with an Early Increase of Reactive Oxygen Species (ROS)
3.2. The Early Increase of ROS Production Determines if Cancer Cells Die or Stop Proliferating in Response to Melatonin
3.3. The Reduction in Intracellular GSH Levels Could also Be Involved in the Pro-Apoptotic Effect of Melatonin
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Hill, S.M.; Blask, D.E. Effects of the pineal hormone melatonin on the proliferation and morphological characteristics of human breast cancer cells (MCF-7) in culture. Cancer Res 1988, 48, 6121–6126. [Google Scholar]
- Antolin, I.; Rodriguez, C.; Sainz, R.M.; Mayo, J.C.; Uría, H.; Kotler, M.L.; Rodriguez-Colunga, M.J.; Tolivia, D.; Menendez-Pelaez, A. Neurohormone melatonin prevents cell damage: Effect on gene expression for antioxidant enzymes. FASEB J 1996, 10, 882–890. [Google Scholar]
- Antolín, I.; Mayo, J.C.; Sainz, R.M.; del Brio, M.A.; Herrera, F.; Martin, V.; Rodriguez, C. Protective effect of melatonin in a chronic experimental model of Parkinson’s disease. Brain Res 2002, 943, 163–173. [Google Scholar]
- Vijayalaxmi; Thomas, C.R., Jr.; Reiter, R.J.; Herman, T.S. Melatonin: From basic research to cancer treatment clinics. J. Clin. Oncol. 2002, 20, 2575–2601. [Google Scholar]
- Reiter, R.J. Melatonin: Clinical relevance. Best Pract. Res. Clin. Endocrinol. Metab 2003, 2, 273–285. [Google Scholar]
- Herrera, F.; Martín, V.; García-Santos, G.; Rodriguez-Blanco, J.; Antolin, I.; Rodriguez, C. Melatonin prevents glutamate-induced oxytosis in the HT22 mouse hippocampal cell line through an antioxidant effect specifically targeting mitochondria. J. Neurochem 2007, 100, 736–746. [Google Scholar]
- Schernhammer, E.S.; Hankinson, S.E. Urinary melatonin levels and breast cancer risk. J. Natl. Cancer Inst 2005, 97, 1084–1087. [Google Scholar]
- Schernhammer, E.S.; Hankinson, S.E. Urinary melatonin levels and postmenopausal breast cancer risk in the Nurses’ Health Study cohort. Cancer Epidem. Biomar 2009, 18, 74–79. [Google Scholar]
- Srinivasan, V.; Spence, D.W.; Seithikurippu, M.A.; Pandi-Perumal, R.; Trakht, I.; Cardinali, D.P. Therapeutic actions of melatonin in cancer: Possible mechanisms. Integr. Cancer Ther 2008, 7, 189–203. [Google Scholar]
- Blask, D.E.; Sauer, L.A.; Dauchy, R.T.; Holowachuk, E.W.; Ruhoff, M.S.; Kopff, H.S. Melatonin inhibition of cancer growth in vivo involves suppression of tumor fatty acid metabolism via melatonin receptor-mediated signal transduction events. Cancer Res 1999, 59, 4693–4701. [Google Scholar]
- Cos, S.; Gonzalez, A.; Martinez-Campa, C.; Mediavilla, M.D.; Alonso-Gonzalez, C.; Sanchez-Barceló, E.J. Estrogen-signaling pathway: A link between cancer and melatonin oncostatic actions. Cancer Detect. Prev 2006, 30, 118–128. [Google Scholar]
- Hill, S.M.; Frash, T.; Xiang, S.; Yuan, L.; Duplessis, T.; Mao, L. Molecular mechanisms of melatonin anticancer effects. Integr. Cancer Ther 2009, 8, 337–346. [Google Scholar]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Qi, W.; Hanes, M.A.; Farley, N.J. High physiological levels of melatonin in bile of mammals. Life Sci 1999, 65, 523–529. [Google Scholar]
- Shinner, D.C.; Malpaux, B. High melatonin concentrations in third ventricular cerebrospinal fluid are not due to Galen vein blood in recirculation through the choroids plexus. Endocrinology 1999, 140, 4399–4405. [Google Scholar]
- Sainz, R.M.; Mayo, J.C.; Tan, D.X.; León, J.; Manchester, L.; Reiter, R.J. Melatonin reduces prostate cancer cell growth leading to neuroendocrine differentiation via a receptor and PKA independent mechanism. Prostate 2005, 63, 29–43. [Google Scholar]
- Martín, V.; Herrera, F.; Carrera-Gonzalez, P.; García-Santos, G.; Antolín, I.; Rodriguez-Blanco, J.; Rodriguez, C. Intracellular signaling pathways involved in the cell growth inhibition of glioma cells by melatonin. Cancer Res 2006, 66, 1–8. [Google Scholar]
- Tan, D.X.; Poeggeler, B.; Reiter, J.F.; Chen, I.D.; Chen, S.; Manchester, L.C.; Barlow-Walden, L.R. The pineal hormone melatonin inhibits DNA adduct formation induced by chemical carcinogen safrole in vivo. Cancer Lett 1993, 70, 65–71. [Google Scholar]
- Reiter, R.J. Antioxidant actions of melatonin. Adv. Pharmacol 1997, 38, 103–117. [Google Scholar]
- Herrera, F.; Sainz, R.M.; Mayo, J.C.; Martin, V.; Antolin, I.; Rodriguez, C. Glutamate induces oxidative stress not mediated by glutamate receptors or cystine transporters: Protective effect of melatonin and other antioxidants. J. Pineal Res 2001, 31, 353–362. [Google Scholar]
- García-Navarro, A.; González-Puga, C.; Escames, G.; Lopez, L.C.; López, A.; López-Cantarero, M.; Camacho, E.; Espinosa, A.; Gallo, M.A.; Acuña-Castroviejo, D. Cellular mechanisms involved in the melatonin inhibition of HT-29 human colon cancer cell proliferation in culture. J. Pineal Res. 2007, 43, 195–205. [Google Scholar]
- Hermann, R.; Podhajsky, S.; Jungnickel, S.; Lerchl, A. Potentiation of antiproliferative effects on tamoxifen and ethanol on mouse hepatoma cells by melatonin, possible involvement of mitogen-activated protein kinase and induction of apoptosis. J. Pineal Res 2002, 33, 8–13. [Google Scholar]
- Wenzel, U.; Nickel, A.; Daniel, H. Melatonin potentiates flavone-induced apoptosis in human colon cancer cells by increasing the level of glycolytic end products. Int. J. Cancer 2005, 116, 236–242. [Google Scholar]
- González-Puga, C.; García-Navarro, A.; Escames, G.; León, J.; Lopez-Cantarero, M.; Ros, E.; Acuña-Castroviejo, D. Selective CCK-A but not CCK-B receptor antagonists inhibit HT-29 cell proliferation: Synergism with pharmacological levels of melatonin. J. Pineal Res 2005, 39, 243–250. [Google Scholar]
- Sainz, R.M.; Reiter, R.J.; Tan, D.X.; Roldan, F.; Natarajan, M.; Quirós, I.; Hevia, D.; Rodriguez, C.; Mayo, J.C. Critical role of glutathione in melatonin enhancement of tumor necrosis factor and ionizing radiation-induced apoptosis in prostate cancer cells in vitro. J. Pineal Res 2008, 45, 258–270. [Google Scholar]
- Korkmaz, A.; Tamura, H.; Manchester, L.C.; Ogden, G.B.; Tan, D.X.; Reiter, R.J. Combination of melatonin and a peroxisome proliferator-activated receptor-gamma agonist induces apoptosis in a breast cancer cell line. J. Pineal Res 2009, 46, 115–116. [Google Scholar]
- Martin, V.; García-Santos, G.; Rodriguez-Blanco, J.; Casado-Zapico, S.; Sanchez-Sanchez, A.; Antolin, I.; Medina, M.; Rodriguez, C. Melatonin sensitizes human malignant glioma cells against TRAIL-induced cell death. Cancer Let 2010, 287, 216–223. [Google Scholar]
- Casado-Zapico, S.; Rodriguez-Blanco, J.; García-Santos, G.; Martin, V.; Sánchez-Sánchez, A.M.; Antolin, I.; Rodriguez, C. Synergistic antitumor effect of melatonin with several chemotherapeutic drugs on human Ewing sarcoma cancer cells. Potentiation of the extrinsic apoptotic pathway. J. Pineal Res 2010, 48, 72–80. [Google Scholar]
- Bejarano, I.; Espino, J.; Marchena, A.M.; Barriga, C.; Paredes, S.D.; Rodriguez, A.B.; Pariente, J.A. Melatonin enhances hydrogen peroxide-induced apoptosis in human promyelocytic leukemia HL-60 cells. Mol. Cell. Biochem 2011, 353, 167–176. [Google Scholar]
- Um, H.J.; Park, J.W.; Kwon, T.K. Melatonin sensitizes Caki renal cancer cells to kahweol-induced apoptosis through CHOP-mediated up-regulation of PUMA. J. Pineal Res 2011, 50, 359–366. [Google Scholar]
- Kim, J.H.; Jeong, S.J.; Kim, B.; Yun, S.M.; Choi, D.Y.; Kim, S.H. Melatonin synergistically enhances cisplatin-induced apoptosis via the dephosphorylation of ERK/p90 ribosomal S6 kinase/heat shock protein 27 in SK-OV-3 cells. J. Pineal Res 2012, 52, 244–252. [Google Scholar]
- Uguz, A.C.; Cig, B.; Espino, J.; Bejarano, I.; Naziroglu, M.; Rodriguez, A.B.; Pariente, J.A. Melatonin potentiates chemotherapy-induced cytotoxicity and apoptosis in rat pancreatic tumor cells. J. Pineal Res 2012, 53, 91–98. [Google Scholar]
- Anisimov, V.N.; Egormin, P.A.; Piskunova, T.S.; Popovich, I.G.; Tyndyk, M.L.; Yurova, M.N.; Zabezhinski, M.A.; Anikin, I.V.; Karkach, A.S.; Romanyukha, A.A. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle 2010, 9, 188–197. [Google Scholar]
- Padillo, F.J.; Ruiz-Rabelo, J.F.; Cruz, A.; Perea, M.D.; Tasset, I.; Montilla, P.; Tunez, I.; Muntané, J. Melatonin and celecoxib improve the outcomes in hamsters with experimental pancreatic cancer. J. Pineal Res 2010, 49, 264–270. [Google Scholar]
- Cos, S.; Gonzalez, A.; Guezmes, A.; Mediavilla, M.D.; Martinez-Campa, C.; Alonso-Gonzalez, C.; Sanchez-Barceló, E.J. Melatonin inhibits the growth of DMBA-induced mammary tumors by decreasing the local biosynthesis of estrogens through the modulation of aromatase activity. Int. J. Cancer 2006, 118, 274–278. [Google Scholar]
- Farriol, M.; Venereo, Y.; Orta, X.; Castellanos, J.M.; Segovia-Silvestre, T. In vitro effects of melatonin on cell proliferation in a colon adenocarcinoma line. J. Appl. Toxicol 2000, 20, 21–24. [Google Scholar]
- Sainz, R.M.; Mayo, J.C.; Tan, D.X.; Lopez-Burillo, S.; Natarajan, M.; Reiter, R.J. Antioxidant activity of melatonin in Chinese hamster ovarian cells: Changes in cellular proliferation and differentiation. Biochem. Bioph. Res. Commun 2003, 302, 625–634. [Google Scholar]
- Sánchez-Sánchez, A.M.; Martín, V.; García-Santos, G.; Rodriguez-Blanco, J.; Casado-Zapico, S.; Suarez-Garnacho, S.; Antolin, I.; Rodriguez, C. Intracellular redox state as determinant for melatonin antiproliferative vs. cytotoxic effects in cancer cells. Free Radic. Res 2011, 45, 1333–1341. [Google Scholar]
- Fu, J.; Zhao, S.D.; Liu, H.J.; Yuan, Q.H.; Liu, S.M.; Zhang, Y.M.; Ling, E.A.; Hao, A.J. Melatonin promotes proliferation and differentiation of neural stem cells subjected to hypoxia in vitro. J. Pineal Res 2011, 51, 104–112. [Google Scholar]
- Cui, P.; Yu, M.; Luo, Z.; Dai, M.; Han, J.; Xiu, R.; Yang, Z. Intracellular signaling pathways involved in cell growth inhibition of human umbilical vein endothelial cells by melatonin. J. Pineal Res 2008, 44, 107–114. [Google Scholar]
- Kojima, T.; Mochizuki, C.; Mitaka, T.; Mochizuki, Y. Effects of melatonin on proliferation, oxidative stress and Cx32 gap junction protein expression in primary cultures of adult rat hepatocytes. Cell Struct. Funct 1997, 22, 347–356. [Google Scholar]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Trubiani, O.; Recchioni, R.; Moroni, F.; Pizzicannella, J.; Caputi, S.; di Primio, R. Melatonin provokes cell death in human B-lymphoma cells by mitochondrial-dependent apoptotic pathway activation. J. Pineal Res 2005, 39, 425–431. [Google Scholar]
- Rubio, S.; Estévez, F.; Cabrera, J.; Reiter, R.J.; Loro, J.; Quintana, J. Inhibition of proliferation and induction of apoptosis by melatonin in human myeloid HL-60 cells. J. Pineal Res 2007, 42, 131–138. [Google Scholar]
- Bejarano, I.; Redondo, P.C.; Espino, J.; Rosado, J.A.; Paredes, S.D.; Barriga, C.; Reiter, R.J.; Pariente, J.A.; Rodriguez, A.B. Melatonin induces mitochondrial-mediated apoptosis in human myeloid HL-60 cells. J. Pineal Res 2009, 46, 392–400. [Google Scholar]
- Bejarano, I.; Espino, J.; Barriga, C.; Reiter, R.J.; Pariente, J.A.; Rodriguez, A.B. Pro-oxidant effect of melatonin in tumor leucocytes: Relation with its cytotoxic and pro-apoptotic effects. Basic Clin. Pharmacol. Toxicol 2010, 108, 14–20. [Google Scholar]
- Casado-Zapico, S.; Martín, V.; García-Santos, G.; Rodriguez-Blanco, J.; Sanchez-Sanchez, A.M.; Luño, E.; Suarez, C.; García-Pedrero, J.M.; Menendez, S.T.; Antolín, I.; et al. Regulation of the expression of death receptors and their ligands by melatonin in hematological cancer cell lines and in leukemia cells from patients. J. Pineal Res 2011, 50, 345–355. [Google Scholar]
- Sánchez-Hidalgo, M.L.; de la Lastra, C.A.; Guerrero, J.M.; Packham, G. Melatonin inhibits cell proliferation and induces caspase activation and apoptosis in human malignant lymphoid cell lines. J. Pineal Res 2012, 53, 366–373. [Google Scholar]
- García-Santos, G.; Antolin, I.; Herrera, F.; Martín, V.; Rodriguez-Blanco, J.; Carrera, M.P.; Rodriguez, C. Melatonin induces apoptosis in human neuroblastoma cancer cells. J. Pineal Res 2006, 41, 130–135. [Google Scholar]
- García-Santos, G.; Martin, V.; Rodriguez-Blanco, J.; Herrera, F.; Casado-Zapico, S.; Sanchez-Sanchez, A.M.; Antolin, I.; Rodriguez, C. Fas/Fas ligand regulation mediates cell death in human Ewing’s sarcoma cells treated with melatonin. Br. J. Cancer 2012, 106, 1288–1296. [Google Scholar]
- Winczyk, K.; Pawlikowski, M.; Karasek, M. Melatonin and RZR/ROR receptor ligand CGP 52608 induce apoptosis in the murine colonic cancer. J. Pineal Res 2001, 31, 179–182. [Google Scholar]
- Ozdemir, F.; Deniz, O.; Kaynar, K.; Arslan, M.; Kavgaci, H.; Yildiz, B.; Aydin, F. The effects of melatonin on human hepatoma (Hep G2) cell line. Bratisl. Lek. Listy 2009, 110, 276–279. [Google Scholar]
- Martín-Renedo, J.; Mauriz, J.L.; Jorquera, F.; Ruiz-Andrés, O.; González, P.; González-Gallego, J. Melatonin induces cell cycle arrest and apoptosis in hepatocarcinoma HepG2 cell line. J. Pineal Res 2008, 45, 532–540. [Google Scholar]
- Carbajo-Pescador, S.; Steinmetz, C.; Kashyap, A.; Lorenz, S.; Mauriz, J.L.; Heise, M.; Galle, P.R.; González-Gallego, J.; Strand, S. Melatonin induces transcriptional regulation of Bim by Fox03a in HepG2 cells. Br. J. Cancer 2012, 108, 442–449. [Google Scholar]
- Joo, S.S.; Yoo, Y.M. Melatonin induces apoptotic death in LNCaP cells via p38 and JNK pathways: Therapeutic implications for prostate cancer. J. Pineal Res 2009, 47, 8–14. [Google Scholar]
- Kim, C.H.; Yoo, Y.M. Melatonin induces apoptotic cell death via p53 in LNCaP cells. Korean J. Physiol. Pharmacol 2010, 14, 365–369. [Google Scholar]
- González, A.; del Castillo-Vaquero, A.; Miro-Moran, A.; Tapia, J.A.; Salido, G.M. Melatonin reduces pancreatic tumor cell viability by altering mitochondrial physiology. J. Pineal Res 2011, 50, 250–260. [Google Scholar]
- Osseni, R.A.; Rat, P.; Bogdan, A.; Warnet, J.M.; Touitou, Y. Evidence of prooxidant and antioxidant action of melatonin on human liver cell line HepG2. Life Sci 2000, 68, 387–399. [Google Scholar]
- Wolfler, A.; Caluba, H.C.; Abuja, P.M.; Dohr, G.; Schauenstein, K.; Liebmann, P.K. Prooxidant activity of melatonin promotes fas-induced cell death in human leukemic Jurkat cells. FEBS Lett 2001, 602, 127–131. [Google Scholar]
- Buyukavci, M.; Ozdemir, O.; Buck, S.; Stout, M.; Ravindranath, Y.; Savasan, S. Melatonin cytotoxicity in human leukemia cells: Relation with its pro-oxidant effect. Fund. Clin. Pharmacol 2006, 20, 73–79. [Google Scholar]
- Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. Intracellular redox status and oxidative stress: Implications for cell proliferation, apoptosis and carcinogenesis. Arch. Toxicol 2008, 82, 273–299. [Google Scholar]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems and apoptosis. Free Radic. Biol. Med 2010, 48, 749–762. [Google Scholar]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar]
- Cook, S.A.; Sugden, P.H.; Clerk, A. Regulation of bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: Association with changes in mitochondrial membrane potential. Circ. Res 1999, 85, 940–949. [Google Scholar]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 1998, 17, 2596–2606. [Google Scholar]
- Fujino, G.; Noguchi, T.; Takeda, K.; Ichijo, H. Thioredoxin and protein kinases in redox signaling. Semin. Cancer Biol 2006, 16, 427–435. [Google Scholar]
- Fan, M.; Goodwin, M.E.; Birrer, M.J.; Chambers, T.C. The c-Jun NH(2)-terminal protein kinase/AP-1 pathway is required for efficient apoptosis induced by vinblastine. Cancer Res 2001, 61, 4450–4458. [Google Scholar]
- Kharbanda, S.; Saxena, S.; Yoshida, K.; Pandey, P.; Kaneki, M.; Wang, Q.; Chen, Y.N.; Campbell, A.; Sudha, T.; Yuan, Z.M.; et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(l) in response to DNA damage. J. Biol. Chem 2000, 275, 322–327. [Google Scholar]
- Franco, R.; Panayiotidis, M.I.; Cidlowski, J.A. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J. Biol. Chem 2007, 282, 30452–30465. [Google Scholar]
- Circu, M.L.; Stringer, S.; Rhoads, C.A.; Moyer, M.P.; Aw, T.Y. The role of GSH efflux in staurosporine-induced apoptosis in colonic epithelial cells. Biochem. Pharmacol 2009, 77, 76–85. [Google Scholar]
- Marengo, B.; de Ciucis, C.; Verzola, D.; Pistoia, V.; Raffaghello, L.; Patriarca, S.; Balbis, E.; Traverso, N.; Cottalasso, D.; Pronzato, M.A.; et al. Mechanisms of BSO (l-buthionine-SR-sulfoximine)-induced cytotoxic effects in neuroblastoma. Free Radic. Biol. Med 2008, 44, 474–482. [Google Scholar]
- Meurette, O.; Lefeuvre-Orfila, L.; Rebillard, A.; Lagadic-Gossmann, D.; Dimanche-Boirel, M.T. Role of intracellular glutathione in cell sensitivity to the apoptosis induced by tumor necrosis factor (alpha)-related apoptosis-inducing ligand/anticancer drug combinations. Clin. Cancer Res 2005, 11, 3075–3083. [Google Scholar]
- Albertini, M.C.; Radogna, F.; Accorsi, A.; Uguccioni, F.; Paternoster, L.; Cerella, C.; de Nicola, M.; D’Alessio, M.; Bergamaschi, A.; Magrini, A.; et al. Intracellular pro-oxidant activity of melatonin deprives U937 cells of reduced glutathione without affecting glutathione peroxidase activity. Ann. N. Y. Acad. Sci 2006, 1091, 10–16. [Google Scholar]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F.; Limson, J.; Weintraub, S.T.; Qi, W. Melatonin directly scavenges hydrogen peroxide: A new metabolic pathway. Free Radic. Biol. Med 2000, 29, 1177–1185. [Google Scholar]
- Ressmeyer, A.R.; Mayo, J.C.; Zelosko, V.; Sainz, R.M.; Tan, D.X.; Poeggeler, B.; Antolín, I.; Zsizsik, B.K.; Reiter, R.J.; Hardeland, R. Antioxidant properties of the melatonin metabolite N1-acetyl-5-methoxykynuramine (AMK): Scavenging of free radicals and prevention of protein destruction. Redox Res 2003, 8, 205–213. [Google Scholar]
- Hardeland, R.; Backhaus, C.; Fadavi, A. Reactions of the NO redox forms NO+, *NO and HNO (protonated NO−) with the melatonin metabolite N1-acetyl-5-methoxykynuramine. J. Pineal Res 2007, 43, 382–388. [Google Scholar]
- Reiter, R.J.; Tan, D.X.; Terron, M.P.; Flores, L.J.; Czarnocki, Z. Melatonin and its metabolites: New findings regarding their production and their radical scavenging actions. Acta Biochim. Pol 2007, 54, 1–9. [Google Scholar]
- Schaefer, M.; Hardeland, R. The melatonin metabolite N-acetyl-5-methoxykynuramine is a potent singlet oxygen scavenger. J. Pineal Res 2009, 46, 49–52. [Google Scholar]
- Hardeland, R.; Tan, D.X.; Reiter, R.J. Kynuramines, metabolites of melatonin and other indoles: The resurrection of an almost forgotten class of biogenic amines. J. Pineal Res 2009, 47, 109–126. [Google Scholar]



© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rodriguez, C.; Martín, V.; Herrera, F.; García-Santos, G.; Rodriguez-Blanco, J.; Casado-Zapico, S.; Sánchez-Sánchez, A.M.; Suárez, S.; Puente-Moncada, N.; Anítua, M.J.; et al. Mechanisms Involved in the Pro-Apoptotic Effect of Melatonin in Cancer Cells. Int. J. Mol. Sci. 2013, 14, 6597-6613. https://doi.org/10.3390/ijms14046597
Rodriguez C, Martín V, Herrera F, García-Santos G, Rodriguez-Blanco J, Casado-Zapico S, Sánchez-Sánchez AM, Suárez S, Puente-Moncada N, Anítua MJ, et al. Mechanisms Involved in the Pro-Apoptotic Effect of Melatonin in Cancer Cells. International Journal of Molecular Sciences. 2013; 14(4):6597-6613. https://doi.org/10.3390/ijms14046597
Chicago/Turabian StyleRodriguez, Carmen, Vanesa Martín, Federico Herrera, Guillermo García-Santos, Jezabel Rodriguez-Blanco, Sara Casado-Zapico, Ana María Sánchez-Sánchez, Santos Suárez, Noelia Puente-Moncada, María José Anítua, and et al. 2013. "Mechanisms Involved in the Pro-Apoptotic Effect of Melatonin in Cancer Cells" International Journal of Molecular Sciences 14, no. 4: 6597-6613. https://doi.org/10.3390/ijms14046597