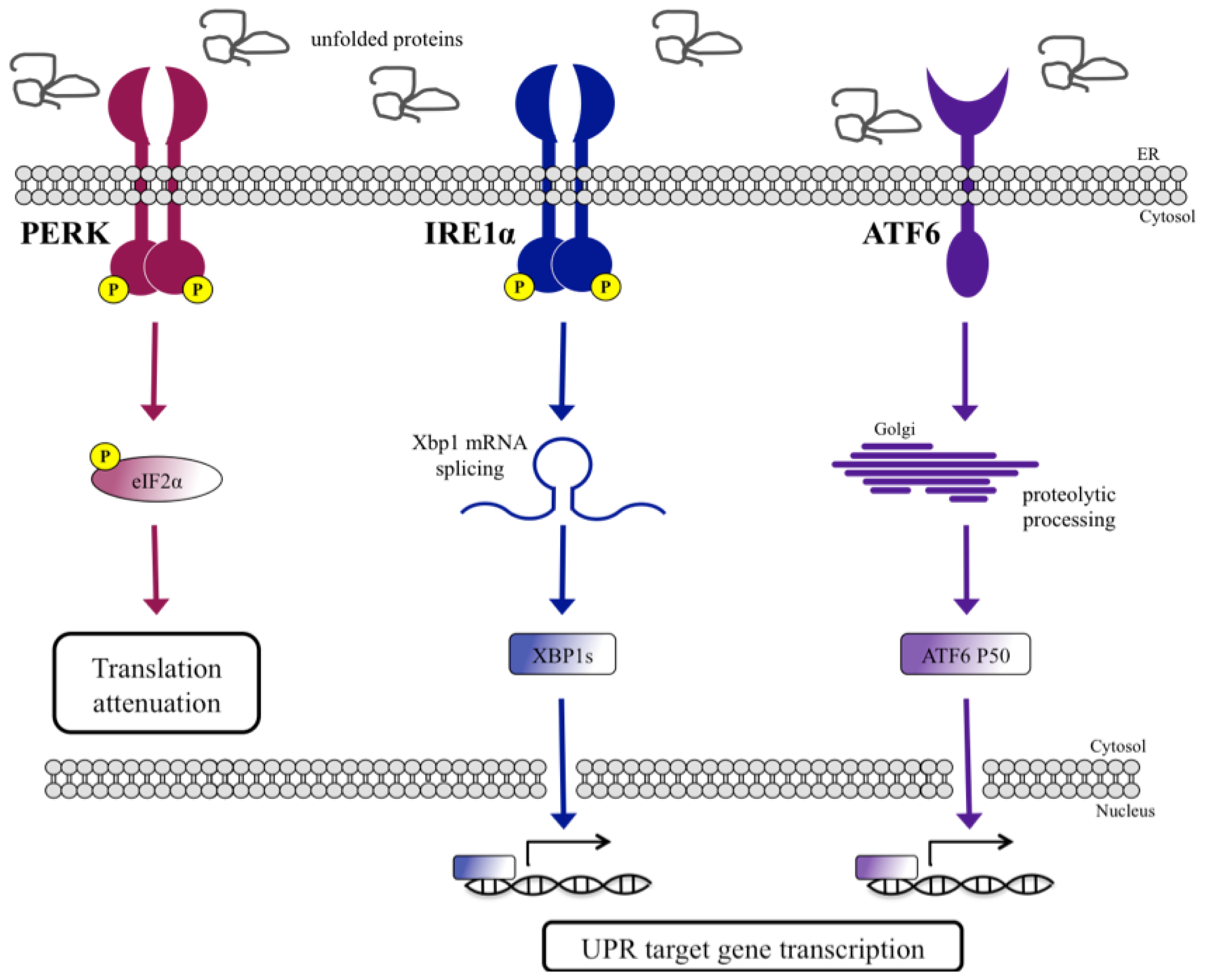
UPR Signal Activation by Luminal Sensor Domains

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
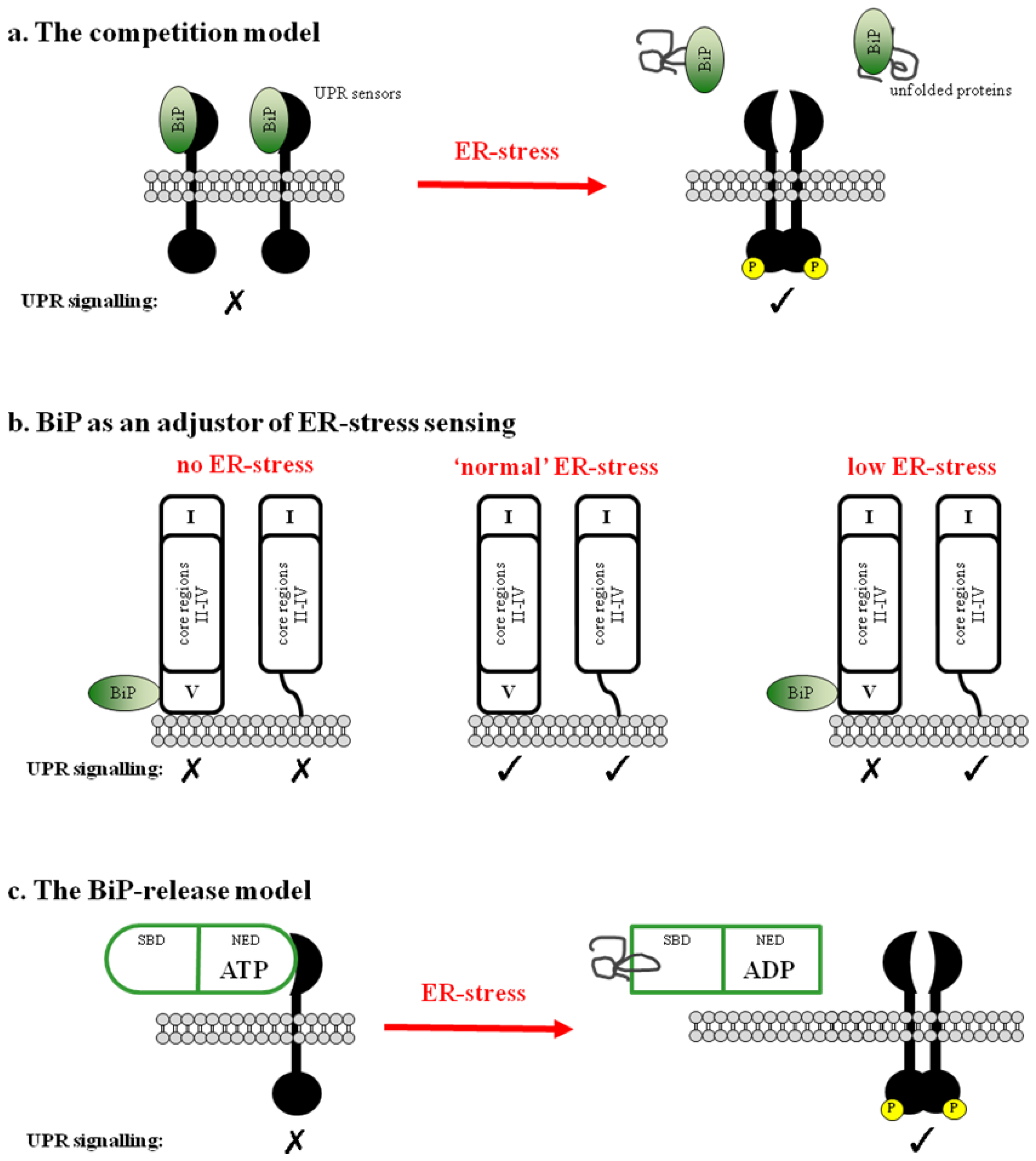
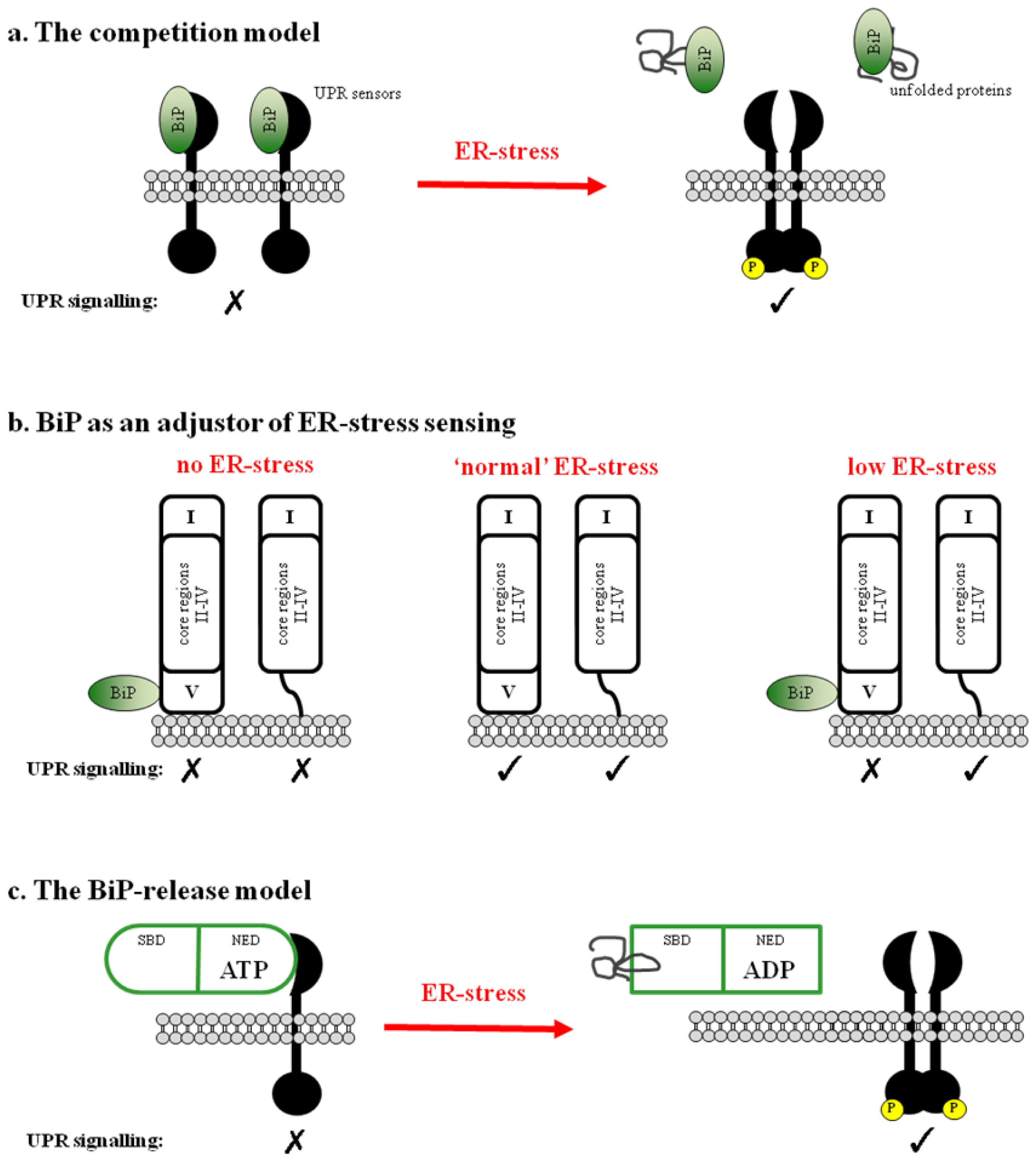
2. BiP-Dependent Models
2.1. BiP Acts as a Negative Regulator of UPR Signaling
2.2. BiP May Act to Fine Tune the UPR Signal
2.3. Regulation of BiP ATPase Activity and the BiP-Release Model
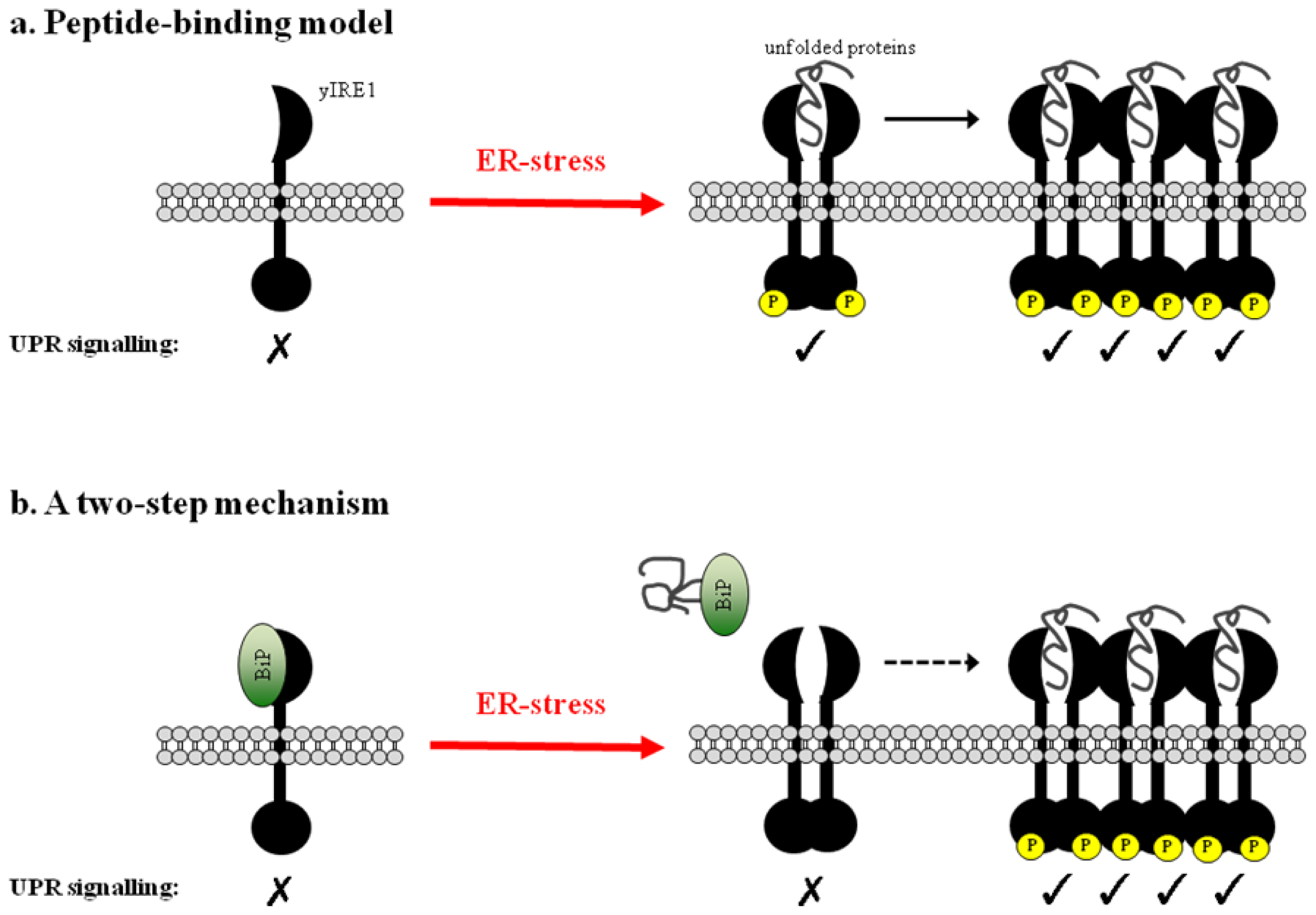
3. The Peptide-Binding Model
3.1. Direct Binding of Unfolded Proteins to the Luminal Domain of yIRE1
3.2. Clustering of Active yIRE1 on the ER Membrane
3.3. The Peptide-Binding Model in Mammals
4. A Two-Step Mechanism for UPR Activation
5. Alternative Mechanisms of Sensing ER-Stress
Acknowledgments
Conflict of Interest
References
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol 2003, 4, 181–191. [Google Scholar]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem 2006, 281, 30299–30304. [Google Scholar]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem 2005, 74, 739–789. [Google Scholar]
- Mori, K. Signalling pathways in the unfolded protein response: Development from yeast to mammals. J. Biochem 2009, 146, 743–750. [Google Scholar]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem 1998, 273, 33741–33749. [Google Scholar]
- Hampton, R.Y. IRE1: A role in UPREgulation of ER degradation. Dev. Cell 2003, 4, 144–146. [Google Scholar]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar]
- Groenendyk, J.; Michalak, M. Endoplasmic reticulum quality control and apoptosis. Acta Biochim. Pol 2005, 52, 381–395. [Google Scholar]
- Parmar, V.M.; Schröder, M. Sensing endoplasmic reticulum stress. Adv. Exp. Med. Biol 2012, 738, 153–168. [Google Scholar]
- Kimata, Y.; Kimata, Y.I.; Shimizu, Y.; Abe, H.; Farcasanu, I.C.; Takeuchi, M.; Rose, M.D.; Kohno, K. Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol. Biol. Cell 2003, 14, 2559–2569. [Google Scholar]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol 2000, 2, 326–332. [Google Scholar]
- Dorner, A.J.; Wasley, L.C.; Kaufman, R.J. Overexpression of GRP78 mitigates stress induction of glucose regulated proteins and blocks secretion of selective proteins in Chinese hamster ovary cells. EMBO J 1992, 11, 1563–1571. [Google Scholar]
- Kohno, K.; Normington, K.; Sambrook, J.; Gething, M.J.; Mori, K. The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol. Cell Biol 1993, 13, 877–890. [Google Scholar]
- Okamura, K.; Kimata, Y.; Higashio, H.; Tsuru, A.; Kohno, K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun 2000, 279, 445–450. [Google Scholar]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar]
- Graham, K.S.; Le, A.; Sifers, R.N. Accumulation of the insoluble PiZ variant of human alpha 1-antitrypsin within the hepatic endoplasmic reticulum does not elevate the steady-state level of grp78/BiP. J. Biol. Chem 1990, 265, 20463–20468. [Google Scholar]
- Ng, D.T.; Watowich, S.S.; Lamb, R.A. Analysis in vivo of GRP78-BiP/substrate interactions and their role in induction of the GRP78-BiP gene. Mol. Biol. Cell 1992, 3, 143–155. [Google Scholar]
- Oikawa, D.; Kimata, Y.; Kohno, K.; Iwawaki, T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res 2009, 315, 2496–2504. [Google Scholar]
- Kimata, Y.; Kohno, K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol 2011, 23, 135–142. [Google Scholar]
- Ma, K.; Vattem, K.M.; Wek, R.C. Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J. Biol. Chem 2002, 277, 18728–18735. [Google Scholar]
- Kimata, Y.; Oikawa, D.; Shimizu, Y.; Ishiwata-Kimata, Y.; Kohno, K. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J. Cell Biol 2004, 167, 445–456. [Google Scholar]
- Pincus, D.; Chevalier, M.W.; Aragón, T.; van Anken, E.; Vidal, S.E.; El-Samad, H.; Walter, P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol 2010, 8, e1000415. [Google Scholar]
- Liu, C.Y.; Xu, Z.; Kaufman, R.J. Structure and intermolecular interactions of the luminal dimerization domain of human IRE1alpha. J. Biol. Chem 2003, 278, 17680–17687. [Google Scholar]
- Sou, S.N.; Ilieva, K.M.; Polizzi, K.M. Binding of human BiP to the ER stress transducers IRE1 and PERK requires ATP. Biochem. Biophys. Res. Commun 2012, 420, 473–478. [Google Scholar]
- Todd-Corlett, A.; Jones, E.; Seghers, C.; Gething, M.-J. Lobe IB of the ATPase domain of Kar2p/BiP interacts with Ire1p to negatively regulate the unfolded protein response in Saccharomyces cerevisiae. J. Mol. Biol 2007, 367, 770–787. [Google Scholar]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell Biol 2005, 25, 921–932. [Google Scholar]
- Steel, G.J.; Fullerton, D.M.; Tyson, J.R.; Stirling, C.J. Coordinated activation of Hsp70 chaperones. Science 2004, 303, 98–101. [Google Scholar]
- Shen, Y.; Meunier, L.; Hendershot, L.M. Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J. Biol. Chem 2002, 277, 15947–15956. [Google Scholar]
- Yu, M.; Haslam, R.H.; Haslam, D.B. HEDJ, an Hsp40 co-chaperone localized to the endoplasmic reticulum of human cells. J. Biol. Chem 2000, 275, 24984–24992. [Google Scholar]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar]
- Kimata, Y.; Ishiwata-Kimata, Y.; Ito, T.; Hirata, A.; Suzuki, T.; Oikawa, D.; Takeuchi, M.; Kohno, K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol 2007, 179, 75–86. [Google Scholar]
- Gardner, B.M.; Walter, P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar]
- Aragón, T.; van Anken, E.; Pincus, D.; Serafimova, I.M.; Korennykh, A.V.; Rubio, C.A.; Walter, P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 2009, 457, 736–740. [Google Scholar]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar]
- Oikawa, D.; Kitamura, A.; Kinjo, M.; Iwawaki, T. Direct Association of Unfolded Proteins with Mammalian ER Stress Sensor, IRE1β. PLoS One 2012, 7, e51290. [Google Scholar]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol 2012, 28, 251–277. [Google Scholar]
- Wiseman, R.L.; Zhang, Y.; Lee, K.P.K.; Harding, H.P.; Haynes, C.M.; Price, J.; Sicheri, F.; Ron, D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol. Cell 2010, 38, 291–304. [Google Scholar]
- Ishiwata-Kimata, Y.; Promlek, T.; Shido, M.; Sakuramoto, M.; Kohno, K.; Kimata, Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum-stress sensor Ire1 by different manners. Mol. Biol. Cell 2011, 22, 3520–3532. [Google Scholar]
- Jwa, M.; Chang, P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1α-mediated unfolded protein response. Nat. Cell Biol 2012, 14, 1223–1230. [Google Scholar]
- Hong, M.; Luo, S.; Baumeister, P.; Huang, J.-M.; Gogia, R.K.; Li, M.; Lee, A.S. Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J. Biol. Chem 2004, 279, 11354–11363. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carrara, M.; Prischi, F.; Ali, M.M.U. UPR Signal Activation by Luminal Sensor Domains. Int. J. Mol. Sci. 2013, 14, 6454-6466. https://doi.org/10.3390/ijms14036454
Carrara M, Prischi F, Ali MMU. UPR Signal Activation by Luminal Sensor Domains. International Journal of Molecular Sciences. 2013; 14(3):6454-6466. https://doi.org/10.3390/ijms14036454
Chicago/Turabian StyleCarrara, Marta, Filippo Prischi, and Maruf M. U. Ali. 2013. "UPR Signal Activation by Luminal Sensor Domains" International Journal of Molecular Sciences 14, no. 3: 6454-6466. https://doi.org/10.3390/ijms14036454