Cadmium-Induced Pathologies: Where Is the Oxidative Balance Lost (or Not)?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
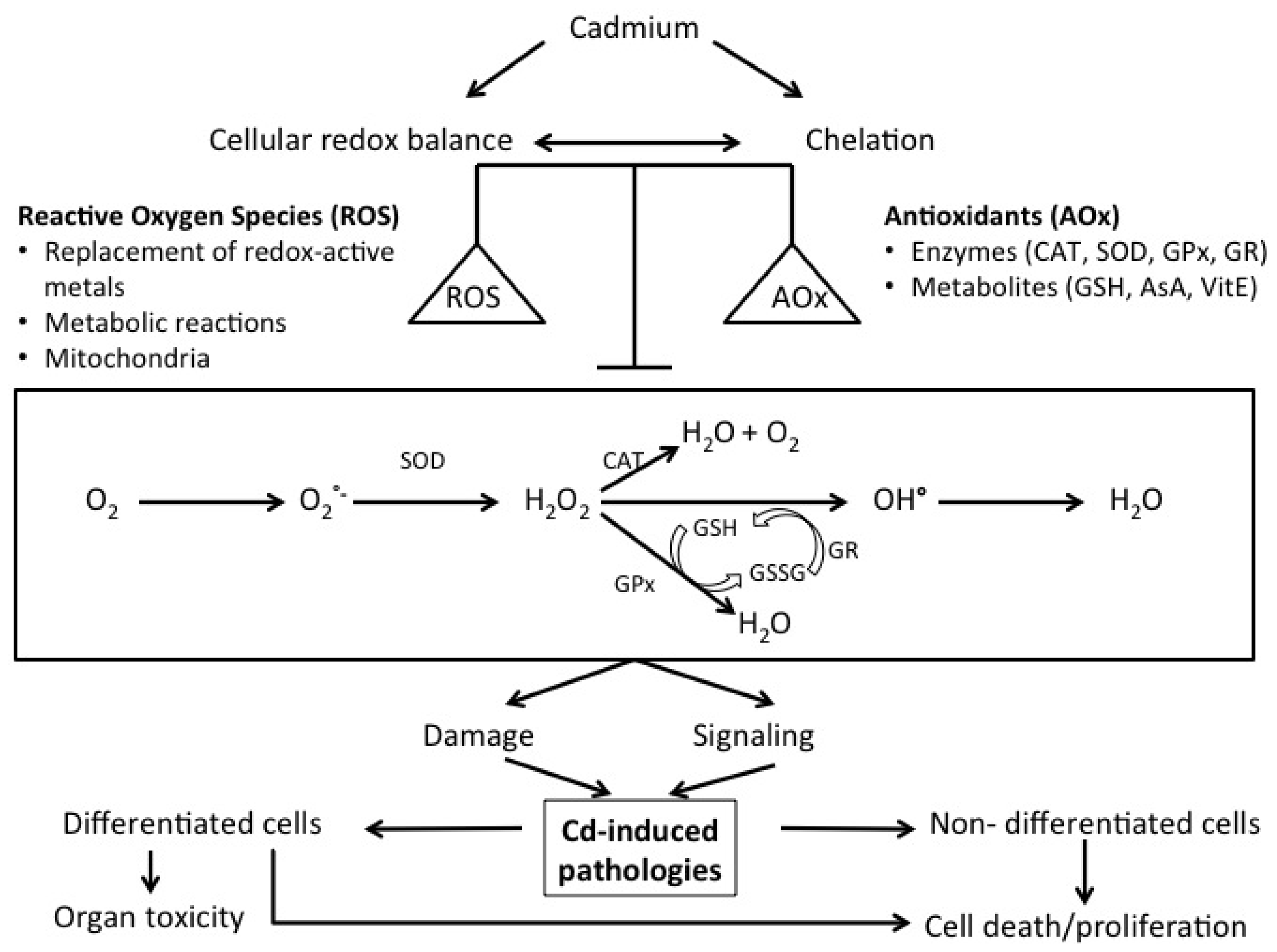
2. Cellular Mechanisms of Cd Toxicity: A Central Role for Oxidative Stress
3. Cd-Induced Pathologies: A Central Role for Oxidative Stress
3.1. Kidney
3.2. Liver
3.3. Bone
3.4. Lungs
3.5. Cardiovascular System
3.6. Brain
3.7. Testis
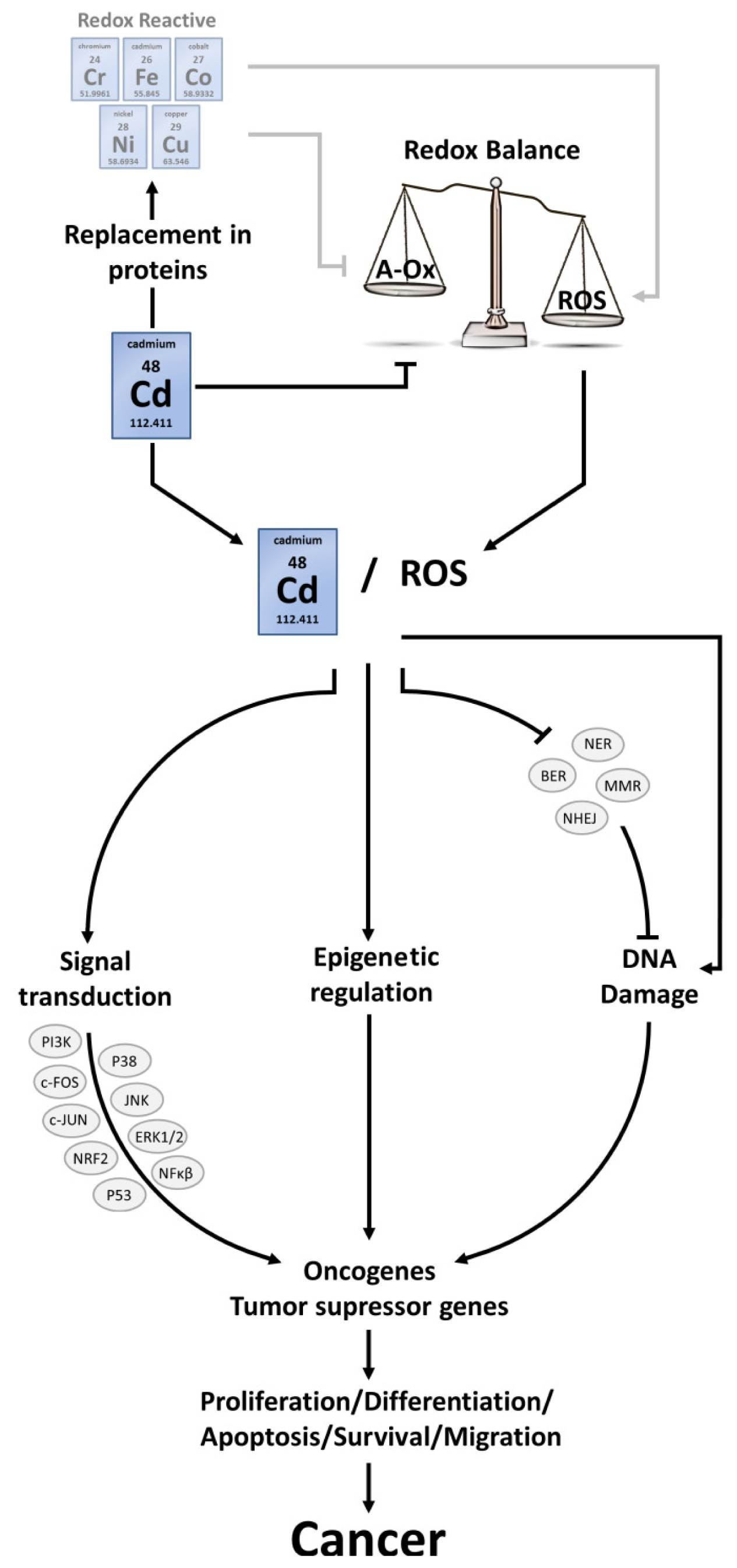
4. Cancer
4.1. ROS Interconnect with Signalling Pathways
4.2. ROS-Induced DNA Damage
4.3. ROS and Epigenetic Alterations
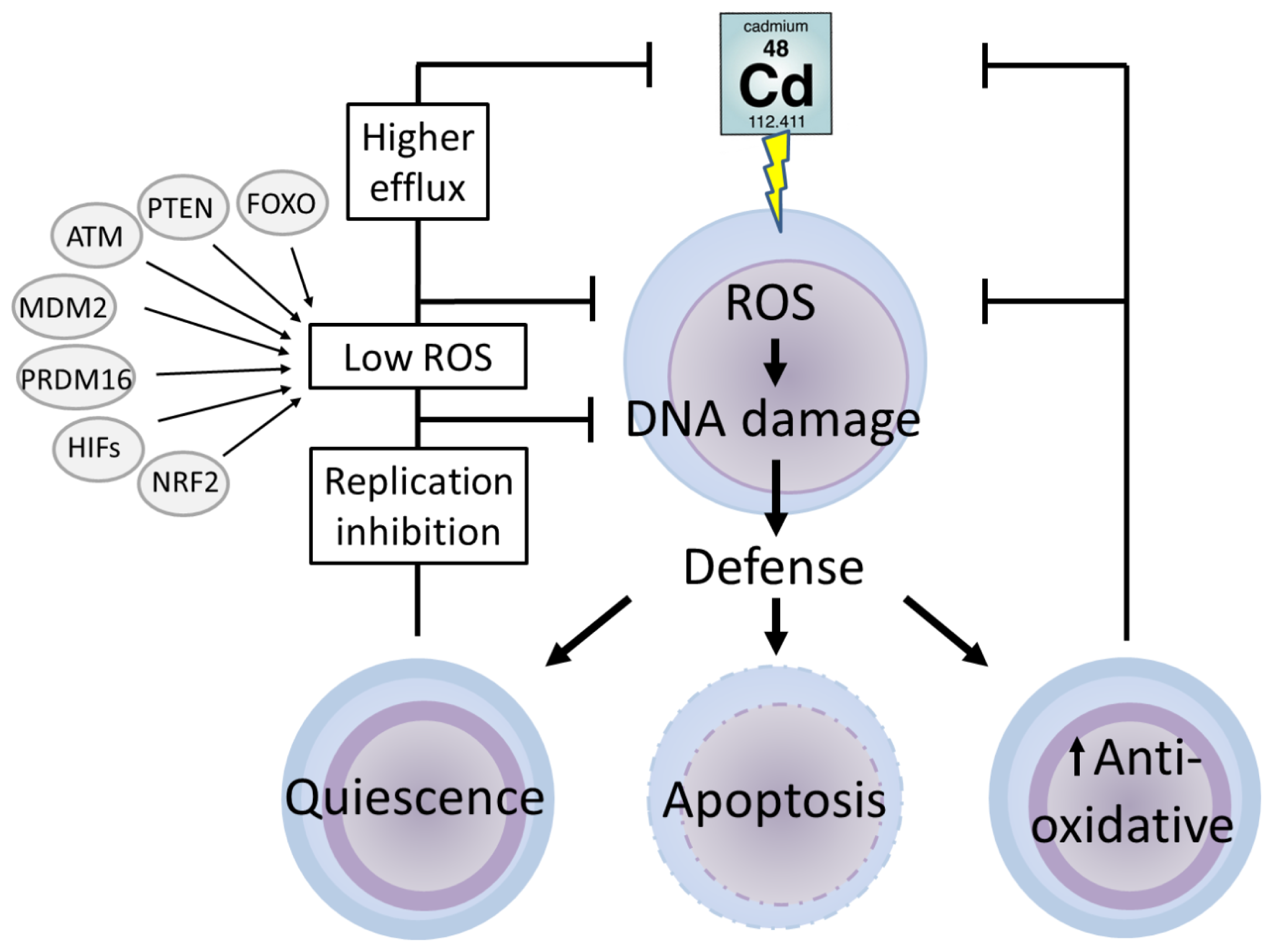
5. Stem Cells
5.1. Defence Mechanisms in Stem Cells
5.2. Cadmium and Stem Cells
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Satarug, S. Long-term exposure to cadmium in food and cigarette smoke, liver effects and hepatocellular carcinoma. Curr. Drug Metab 2012, 13, 257–271. [Google Scholar]
- Sarwar, N.; Malhi, S.S.; Zia, M.H.; Naeem, A.; Bibi, S.; Farid, G. Role of mineral nutrition in minimizing cadmium accumulation by plants. J. Sci. Food Agric 2010, 90, 925–937. [Google Scholar]
- Satarug, S.; Garrett, S.H.; Sens, M.A.; Sens, D.A. Cadmium, environmental exposure, and health outcomes. Environ. Health Perspect 2010, 118, 182–190. [Google Scholar]
- Satarug, S.; Haswell-Elkins, M.R.; Moore, M.R. Safe levels of cadmium intake to prevent renal toxicity in human subjects. Br. J. Nutr 2000, 84, 791–802. [Google Scholar]
- Hogervorst, J.; Plusquin, M.; Vangronsveld, J.; Nawrot, T.; Cuypers, A.; Van Hecke, E.; Roels, H.A.; Carleer, R.; Staessen, J.A. House dust as possible route of environmental exposure to cadmium and lead in the adult general population. Environ. Res 2007, 103, 30–37. [Google Scholar]
- Satarug, S.; Moore, M.R. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ. Health Perspect 2004, 112, 1099–1103. [Google Scholar]
- Jarup, L.; Akesson, A. Current status of cadmium as an environmental health problem. Toxicol. Appl. Pharmacol 2009, 238, 201–208. [Google Scholar]
- Thijssen, S.; Cuypers, A.; Maringwa, J.; Smeets, K.; Horemans, N.; Lambrichts, I.; Van Kerkhove, E. Low cadmium exposure triggers a biphasic oxidative stress response in mice kidneys. Toxicology 2007, 236, 29–41. [Google Scholar]
- Inaba, T.; Kobayashi, E.; Suwazono, Y.; Uetani, M.; Oishi, M.; Nakagawa, H.; Nogawa, K. Estimation of cumulative cadmium intake causing itai-itai disease. Toxicol. Lett 2005, 159, 192–201. [Google Scholar]
- Kirschvink, N.; Martin, N.; Fievez, L.; Smith, N.; Marlin, D.; Gustin, P. Airway inflammation in cadmium-exposed rats is associated with pulmonary oxidative stress and emphysema. Free Radic. Res 2006, 40, 241–250. [Google Scholar]
- Messner, B.; Bernhard, D. Cadmium and cardiovascular diseases: Cell biology, pathophysiology, and epidemiological relevance. Biometals 2010, 23, 811–822. [Google Scholar]
- Schwartz, G.G.; Il’yasova, D.; Ivanova, A. Urinary cadmium, impaired fasting glucose, and diabetes in the NHANES III. Diabetes Care 2003, 26, 468–470. [Google Scholar]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Roles of dioxins and heavy metals in cancer and neurological diseases using ros-mediated mechanisms. Free Radic. Biol. Med 2010, 49, 1328–1341. [Google Scholar]
- Waalkes, M.P. Cadmium carcinogenesis. Mutat. Res 2003, 533, 107–120. [Google Scholar]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium stress: An oxidative challenge. Biometals 2010, 23, 927–940. [Google Scholar]
- Moulis, J.M. Cellular mechanisms of cadmium toxicity related to the homeostasis of essential metals. Biometals 2010, 23, 877–896. [Google Scholar]
- Vesey, D.A. Transport pathways for cadmium in the intestine and kidney proximal tubule: Focus on the interaction with essential metals. Toxicol. Lett 2010, 198, 13–19. [Google Scholar]
- Dalton, T.P.; He, L.; Wang, B.; Miller, M.L.; Jin, L.; Stringer, K.F.; Chang, X.; Baxter, C.S.; Nebert, D.W. Identification of mouse slc39a8 as the transporter responsible for cadmium-induced toxicity in the testis. Proc. Natl. Acad. Sci. USA 2005, 102, 3401–3406. [Google Scholar]
- Johri, N.; Jacquillet, G.; Unwin, R. Heavy metal poisoning: The effects of cadmium on the kidney. Biometals 2010, 23, 783–792. [Google Scholar]
- Abouhamed, M.; Wolff, N.A.; Lee, W.K.; Smith, C.P.; Thevenod, F. Knockdown of endosomal/lysosomal divalent metal transporter 1 by rna interference prevents cadmium-metallothionein-1 cytotoxicity in renal proximal tubule cells. Am. J. Physiol. Renal. Physiol 2007, 293, F705–F712. [Google Scholar]
- Thevenod, F. Cadmium and cellular signaling cascades: To be or not to be? Toxicol. Appl. Pharmacol 2009, 238, 221–239. [Google Scholar]
- Hart, B.A.; Potts, R.J.; Watkin, R.D. Cadmium adaptation in the lung—A double-edged sword? Toxicology 2001, 160, 65–70. [Google Scholar]
- Jimenez, I.; Gotteland, M.; Zarzuelo, A.; Uauy, R.; Speisky, H. Loss of the metal binding properties of metallothionein induced by hydrogen peroxide and free radicals. Toxicology 1997, 120, 37–46. [Google Scholar]
- Cannino, G.; Ferruggia, E.; Luparello, C.; Rinaldi, A.M. Cadmium and mitochondria. Mitochondrion 2009, 9, 377–384. [Google Scholar]
- Templeton, D.M.; Liu, Y. Multiple roles of cadmium in cell death and survival. Chem. Biol. Interact 2010, 188, 267–275. [Google Scholar]
- Luparello, C.; Sirchia, R.; Longo, A. Cadmium as a transcriptional modulator in human cells. Crit. Rev. Toxicol 2011, 41, 75–82. [Google Scholar]
- Casalino, E.; Sblano, C.; Landriscina, C. Enzyme activity alteration by cadmium administration to rats: The possibility of iron involvement in lipid peroxidation. Arch. Biochem. Biophys 1997, 346, 171–179. [Google Scholar]
- Koizumi, T.; Li, Z.G. Role of oxidative stress in single-dose, cadmium-induced testicular cancer. J. Toxicol. Environ. Health 1992, 37, 25–36. [Google Scholar]
- Ikediobi, C.O.; Badisa, V.L.; Ayuk-Takem, L.T.; Latinwo, L.M.; West, J. Response of antioxidant enzymes and redox metabolites to cadmium-induced oxidative stress in crl-1439 normal rat liver cells. Int. J. Mol. Med 2004, 14, 87–92. [Google Scholar]
- Waisberg, M.; Joseph, P.; Hale, B.; Beyersmann, D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 2003, 192, 95–117. [Google Scholar]
- Lopez, E.; Arce, C.; Oset-Gasque, M.J.; Canadas, S.; Gonzalez, M.P. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic. Biol. Med 2006, 40, 940–951. [Google Scholar]
- Wang, Y.; Fang, J.; Leonard, S.S.; Rao, K.M. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic. Biol. Med 2004, 36, 1434–1443. [Google Scholar]
- Hegedus, A.; Erdei, S.; Horvath, G. Comparative studies of H2O2 detoxifying enzymes in green and greening barley seedlings under cadmium stress. Plant Sci 2001, 160, 1085–1093. [Google Scholar]
- Pereira, G.J.G.; Molina, S.M.G.; Lea, P.J.; Azevedo, R.A. Activity of antioxidant enzymes in response to cadmium in crotalaria juncea. Plant Soil 2002, 239, 123–132. [Google Scholar]
- Shaikh, Z.A.; Vu, T.T.; Zaman, K. Oxidative stress as a mechanism of chronic cadmium-induced hepatotoxicity and renal toxicity and protection by antioxidants. Toxicol. Appl. Pharmacol 1999, 154, 256–263. [Google Scholar]
- Kamiya, T.; Izumi, M.; Hara, H.; Adachi, T. Propolis suppresses CdCl2-induced cytotoxicity of COS7 cells through the prevention of intracellular reactive oxygen species accumulation. Biol. Pharm. Bull 2012, 35, 1126–1131. [Google Scholar]
- Wang, L.; Cao, J.; Chen, D.; Liu, X.; Lu, H.; Liu, Z. Role of oxidative stress, apoptosis, and intracellular homeostasis in primary cultures of rat proximal tubular cells exposed to cadmium. Biol. Trace Elem. Res 2009, 127, 53–68. [Google Scholar]
- Thevenod, F.; Friedmann, J.M. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of na+/k+-atpase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J 1999, 13, 1751–1761. [Google Scholar]
- Thevenod, F.; Friedmann, J.M.; Katsen, A.D.; Hauser, I.A. Up-regulation of multidrug resistance p-glycoprotein via nuclear factor-kappa b activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J. Biol. Chem 2000, 275, 1887–1896. [Google Scholar]
- Thevenod, F. Nephrotoxicity and the proximal tubule. Insights from cadmium. Nephron. Physiol 2003, 93, 87–93. [Google Scholar]
- Tang, W.; Shaikh, Z.A. Renal cortical mitochondrial dysfunction upon cadmium metallothionein administration to sprague-dawley rats. J. Toxicol. Environ. Health 2001, 63, 221–235. [Google Scholar]
- Bagchi, D.; Vuchetich, P.J.; Bagchi, M.; Hassoun, E.A.; Tran, M.X.; Tang, L.; Stohs, S.J. Induction of oxidative stress by chronic administration of sodium dichromate [chromium VI] and cadmium chloride [cadmium II] to rats. Free Radic. Biol. Med 1997, 22, 471–478. [Google Scholar]
- Beytut, E.; Yuce, A.; Kamiloglu, N.N.; Aksakal, M. Role of dietary vitamin E in cadmium-induced oxidative damage in rabbit’s blood, liver and kidneys. Int. J. Vitam. Nutr. Res 2003, 73, 351–355. [Google Scholar]
- Lawal, A.O.; Lawal, A.F.; Ologundudu, A.; Adeniran, O.Y.; Omonkhua, A.; Obi, F. Antioxidant effects of heated garlic juice on cadmium-induced liver damage in rats as compared to ascorbic acid. J. Toxicol. Sci 2011, 36, 549–557. [Google Scholar]
- Masso, E.L.; Corredor, L.; Antonio, M.T. Oxidative damage in liver after perinatal intoxication with lead and/or cadmium. J. Trace Elem. Med. Biol 2007, 21, 210–216. [Google Scholar]
- Nigam, D.; Shukla, G.S.; Agarwal, A.K. Glutathione depletion and oxidative damage in mitochondria following exposure to cadmium in rat liver and kidney. Toxicol. Lett 1999, 106, 151–157. [Google Scholar]
- Shukla, G.S.; Hussain, T.; Srivastava, R.S.; Chandra, S.V. Glutathione peroxidase and catalase in liver, kidney, testis and brain regions of rats following cadmium exposure and subsequent withdrawal. Ind. Health 1989, 27, 59–69. [Google Scholar]
- Latinwo, L.M.; Badisa, V.L.; Ikediobi, C.O.; Odewumi, C.O.; Lambert, A.T.; Badisa, R.B. Effect of cadmium-induced oxidative stress on antioxidative enzymes in mitochondria and cytoplasm of CRL-1439 rat liver cells. Int. J. Mol. Med 2006, 18, 477–481. [Google Scholar]
- Lemarie, A.; Lagadic-Gossmann, D.; Morzadec, C.; Allain, N.; Fardel, O.; Vernhet, L. Cadmium induces caspase-independent apoptosis in liver hep3b cells: Role for calcium in signaling oxidative stress-related impairment of mitochondria and relocation of endonuclease g and apoptosis-inducing factor. Free Radic. Biol. Med 2004, 36, 1517–1531. [Google Scholar]
- Liu, J.; Qu, W.; Kadiiska, M.B. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol. Appl. Pharmacol 2009, 238, 209–214. [Google Scholar]
- Liu, J.; Kadiiska, M.B.; Corton, J.C.; Qu, W.; Waalkes, M.P.; Mason, R.P.; Liu, Y.; Klaassen, C.D. Acute cadmium exposure induces stress-related gene expression in wild-type and metallothionein-I/II-null mice. Free Radic. Biol. Med 2002, 32, 525–535. [Google Scholar]
- Liu, J.; Qian, S.Y.; Guo, Q.; Jiang, J.; Waalkes, M.P.; Mason, R.P.; Kadiiska, M.B. Cadmium generates reactive oxygen- and carbon-centered radical species in rats: Insights from in vivo spin-trapping studies. Free Radic. Biol. Med 2008, 45, 475–481. [Google Scholar]
- Bhattacharyya, M.H. Cadmium osteotoxicity in experimental animals: Mechanisms and relationship to human exposures. Toxicol. Appl. Pharmacol 2009, 238, 258–265. [Google Scholar]
- Nawrot, T.S.; Staessen, J.A.; Roels, H.A.; Munters, E.; Cuypers, A.; Richart, T.; Ruttens, A.; Smeets, K.; Clijsters, H.; Vangronsveld, J. Cadmium exposure in the population: From health risks to strategies of prevention. Biometals 2010, 23, 769–782. [Google Scholar]
- Ozgocmen, S.; Kaya, H.; Fadillioglu, E.; Aydogan, R.; Yilmaz, Z. Role of antioxidant systems, lipid peroxidation, and nitric oxide in postmenopausal osteoporosis. Mol. Cell Biochem 2007, 295, 45–52. [Google Scholar]
- Brzoska, M.M.; Rogalska, J.; Kupraszewicz, E. The involvement of oxidative stress in the mechanisms of damaging cadmium action in bone tissue: A study in a rat model of moderate and relatively high human exposure. Toxicol. Appl. Pharmacol 2011, 250, 327–335. [Google Scholar]
- Smith, S.S.; Reyes, J.R.; Arbon, K.S.; Harvey, W.A.; Hunt, L.M.; Heggland, S.J. Cadmium-induced decrease in runx2 mrna expression and recovery by the antioxidant n-acetylcysteine (nac) in the human osteoblast-like cell line, saos-2. Toxicol. In Vitro 2009, 23, 60–66. [Google Scholar]
- Lizotte, J.; Abed, E.; Signor, C.; Malu, D.T.; Cuevas, J.; Kevorkova, O.; Sanchez-Dardon, J.; Satoskar, A.; Scorza, T.; Jumarie, C.; et al. Expression of macrophage migration inhibitory factor by osteoblastic cells: Protection against cadmium toxicity. Toxicol. Lett 2012, 215, 167–173. [Google Scholar]
- Rennolds, J.; Butler, S.; Maloney, K.; Boyaka, P.N.; Davis, I.C.; Knoell, D.L.; Parinandi, N.L.; Cormet-Boyaka, E. Cadmium regulates the expression of the cftr chloride channel in human airway epithelial cells. Toxicol. Sci 2010, 116, 349–358. [Google Scholar]
- Childers, M.; Eckel, G.; Himmel, A.; Caldwell, J. A new model of cystic fibrosis pathology: Lack of transport of glutathione and its thiocyanate conjugates. Med. Hypotheses 2007, 68, 101–112. [Google Scholar]
- Kogan, I.; Ramjeesingh, M.; Li, C.; Kidd, J.F.; Wang, Y.; Leslie, E.M.; Cole, S.P.; Bear, C.E. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J 2003, 22, 1981–1989. [Google Scholar]
- Hart, B.A.; Lee, C.H.; Shukla, G.S.; Shukla, A.; Osier, M.; Eneman, J.D.; Chiu, J.F. Characterization of cadmium-induced apoptosis in rat lung epithelial cells: Evidence for the participation of oxidant stress. Toxicology 1999, 133, 43–58. [Google Scholar]
- Lag, M.; Westly, S.; Lerstad, T.; Bjornsrud, C.; Refsnes, M.; Schwarze, P.E. Cadmium-induced apoptosis of primary epithelial lung cells: Involvement of bax and p53, but not of oxidative stress. Cell Biol. Toxicol 2002, 18, 29–42. [Google Scholar]
- Lin, Y.S.; Caffrey, J.L.; Chang, M.H.; Dowling, N.; Lin, J.W. Cigarette smoking, cadmium exposure, and zinc intake on obstructive lung disorder. Respir. Res. 2010, 11, 53:1–53:8. [Google Scholar]
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Invest. 2012, 122, 2749–2755. [Google Scholar]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.J.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig 2004, 114, 1248–1259. [Google Scholar]
- Messner, B.; Knoflach, M.; Seubert, A.; Ritsch, A.; Pfaller, K.; Henderson, B.; Shen, Y.H.; Zeller, I.; Willeit, J.; Laufer, G.; et al. Cadmium is a novel and independent risk factor for early atherosclerosis mechanisms and in vivo relevance. Arterioscler. Thromb. Vasc. Biol 2009, 29, 1392–1398. [Google Scholar]
- Alissa, E.M.; Ferns, G.A. Heavy metal poisoning and cardiovascular disease. J. Toxicol. 2011, 2011. [Google Scholar] [CrossRef]
- Martynowicz, H.; Skoczynska, A.; Wojakowska, A.; Turczyn, B. Serum vasoactive agents in rats poisoned with cadmium. Int. J. Occup. Med. Environ. Health 2004, 17, 479–485. [Google Scholar]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Eng. J. Med. 1989, 320, 915–924. [Google Scholar]
- Donpunha, W.; Kukongviriyapan, U.; Sompamit, K.; Pakdeechote, P.; Kukongviriyapan, V.; Pannangpetch, P. Protective effect of ascorbic acid on cadmium-induced hypertension and vascular dysfunction in mice. Biometals 2011, 24, 105–115. [Google Scholar]
- Szuster-Ciesielska, A.; Stachura, A.; Slotwinska, M.; Kaminska, T.; Sniezko, R.; Paduch, R.; Abramczyk, D.; Filar, J.; Kandefer-Szerszen, M. The inhibitory effect of zinc on cadmium-induced cell apoptosis and reactive oxygen species (ros) production in cell cultures. Toxicology 2000, 145, 159–171. [Google Scholar]
- Pal, R.; Nath, R.; Gill, K.D. Influence of ethanol on cadmium accumulation and its impact on lipid peroxidation and membrane bound functional enzymes (Na+, K(+)-ATPase and acetylcholinesterase) in various regions of adult rat brain. Neurochem. Int 1993, 23, 451–458. [Google Scholar]
- Sinha, M.; Manna, P.; Sil, P.C. Cadmium-induced neurological disorders: Prophylactic role of taurine. J. Appl. Toxicol 2008, 28, 974–986. [Google Scholar]
- Wong, K.L.; Klaassen, C.D. Neurotoxic effects of cadmium in young-rats. Toxicol. Appl. Pharm 1982, 63, 330–337. [Google Scholar]
- Gutierrez-Reyes, E.Y.; Albores, A.; Rios, C. Increase of striatal dopamine release by cadmium in nursing rats and its prevention by dexamethasone-induced metallothionein. Toxicology 1998, 131, 145–154. [Google Scholar]
- Bar-Sela, S.; Reingold, S.; Richter, E.D. Amyotrophic lateral sclerosis in a battery-factory worker exposed to cadmium. Int. J. Occup. Environ. Health 2001, 7, 109–112. [Google Scholar]
- Kumar, R.; Agarwal, A.K.; Seth, P.K. Oxidative stress-mediated neurotoxicity of cadmium. Toxicol Lett 1996, 89, 65–69. [Google Scholar]
- Shukla, G.S.; Srivastava, R.S.; Chandra, S.V. Glutathione status and cadmium neurotoxicity— Studies in discrete brain-regions of growing-rats. Fund Appl. Toxicol 1988, 11, 229–235. [Google Scholar]
- Figueiredo-Pereira, M.E.; Li, Z.; Jansen, M.; Rockwell, P. N-acetylcysteine and celecoxib lessen cadmium cytotoxicity which is associated with cyclooxygenase-2 up-regulation in mouse neuronal cells. J. Biol. Chem 2002, 277, 25283–25289. [Google Scholar]
- Chen, L.; Liu, L.; Huang, S.L. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic. Biol. Med 2008, 45, 1035–1044. [Google Scholar]
- Rockwell, P.; Martinez, J.; Papa, L.; Gomes, E. Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cell. Signal 2004, 16, 343–353. [Google Scholar]
- Yuan, Y.; Bian, J.C.; Liu, X.Z.; Zhang, Y.; Sun, Y.; Liu, Z.P. Oxidative stress and apoptotic changes of rat cerebral cortical neurons exposed to cadmium in vitro. Biomed. Environ. Sci 2012, 25, 172–181. [Google Scholar]
- Acharya, U.R.; Mishra, M.; Patro, J.; Panda, M.K. Effect of vitamins c and e on spermatogenesis in mice exposed to cadmium. Reprod Toxicol 2008, 25, 84–88. [Google Scholar]
- Sen Gupta, R.; Sen Gupta, E.; Dhakal, B.K.; Thakur, A.R.; Ahnn, J. Vitamin c and vitamin e protect the rat testes from cadmium-induced reactive oxygen species. Mol. Cells 2004, 17, 132–139. [Google Scholar]
- El-Missiry, M.A.; Shalaby, F. Role of beta-carotene in ameliorating the cadmium-induced oxidative stress in rat brain and testis. J. Biochem. Mol. Toxic 2000, 14, 238–243. [Google Scholar]
- Georgiou, M.; Perkins, L.M.; Payne, A.H. Steroid synthesis-dependent, oxygen-mediated damage of mitochondrial and microsomal cytochrome p-450 enzymes in rat leydig cell cultures. Endocrinology 1987, 121, 1390–1399. [Google Scholar]
- International Agency for Research on Cancer. Monographs on the Evaluation of Carcinogenic Risks to Humans. October 2004. Available online: http://monographs.iarc.fr/ENG/recentpub/mono89.pdf accessed on 7 March 2013.
- National Institute of Environmental Health Sciences; National Toxicology Program (U.S.). Cadmium and Cadmium Compounds, Report on Carcinogens, Twelfth Edition ed. 2011. Available online: http://ntp.niehs.nih.gov/ntp/roc/twelfth/profiles/Cadmium.pdf accessed on 7 March 2013.
- Bachour, M.; Khaddour, H.H.; Alammori, M.; Al-Quobaili, F.A.; Kuatli, K. Comparison of smoking effects on blood cadmium levels between lung cancer patients and healthy volunteers (smokers and non-smokers) in syria. J. Thorac. Oncol 2012, 7, S28–S29. [Google Scholar]
- Nawrot, T.; Plusquin, M.; Hogervorst, J.; Roels, H.A.; Celis, H.; Thijs, L.; Vangronsveld, J.; Van Hecke, E.; Staessen, J.A. Environmental exposure to cadmium and risk of cancer: A prospective population-based study. Lancet Oncol 2006, 7, 119–126. [Google Scholar]
- Julin, B.; Wolk, A.; Johansson, J.E.; Andersson, S.O.; Andren, O.; Akesson, A. Dietary cadmium exposure and prostate cancer incidence: A population-based prospective cohort study. Br. J. Cancer 2012, 107, 895–900. [Google Scholar]
- Blanco, A.; Moyano, R.; Lopez, A.M.M.; Blanco, C.; Flores-Acuna, R.; Garcia-Flores, J.R.; Espada, M.; Monterde, J.G. Preneoplastic and neoplastic changes in the leydig cells population in mice exposed to low doses of cadmium. Toxicol. Ind. Health 2010, 26, 451–457. [Google Scholar]
- Vinceti, M.; Venturelli, M.; Sighinolfi, C.; Trerotoli, P.; Bonvicini, F.; Ferrari, A.; Bianchi, G.; Serio, G.; Bergomi, M.; Vivoli, G. Case-control study of toenail cadmium and prostate cancer risk in Italy. Sci. Total Environ 2007, 373, 77–81. [Google Scholar]
- Boffetta, P.; Fontana, L.; Stewart, P.; Zaridze, D.; Szeszenia-Dabrowska, N.; Janout, V.; Bencko, V.; Foretova, L.; Jinga, V.; Matveev, V.; et al. Occupational exposure to arsenic, cadmium, chromium, lead and nickel, and renal cell carcinoma: A case-control study from central and eastern europe. Occup. Environ. Med 2011, 68, 723–728. [Google Scholar]
- Il’yasova, D.; Schwartz, G.G. Cadmium and renal cancer. Toxicol. Appl. Pharmacol 2005, 207, 179–186. [Google Scholar]
- Gallagher, C.M.; Chen, J.J.; Kovach, J.S. Environmental cadmium and breast cancer risk. Aging 2010, 2, 804–814. [Google Scholar]
- Julin, B.; Wolk, A.; Bergkvist, L.; Bottai, M.; Akesson, A. Dietary cadmium exposure and risk of postmenopausal breast cancer: A population-based prospective cohort study. Cancer Res 2012, 72, 1459–1466. [Google Scholar]
- Akesson, A.; Julin, B.; Wolk, A. Long-term dietary cadmium intake and postmenopausal endometrial cancer incidence: A population-based prospective cohort study. Cancer Res 2008, 68, 6435–6441. [Google Scholar]
- Amaral, A.F.S.; Cymbron, T.; Gartner, F.; Lima, M.; Rodrigues, A.S. Trace metals and over-expression of metallothioneins in bladder tumoral lesions: A case-control study. BMC Vet. Res. 2009, 5, 24:1–24:6. [Google Scholar]
- Huff, J.; Lunn, R.M.; Waalkes, M.P.; Tomatis, L.; Infante, P.F. Cadmium-induced cancers in animals and in humans. Int. J. Occup. Environ. Health 2007, 13, 202–212. [Google Scholar]
- Kellen, E.; Zeegers, M.P.; Den Hond, E.; Buntinx, F. Blood cadmium may be associated with bladder carcinogenesis: The belgian case-control study on bladder cancer. Cancer Detect. Prev 2007, 31, 77–82. [Google Scholar]
- Wolf, C.; Strenziok, R.; Kyriakopoulos, A. Elevated metallothionein-bound cadmium concentrations in urine from bladder carcinoma patients, investigated by size exclusion chromatography-inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2009, 631, 218–222. [Google Scholar]
- Amaral, A.F.S.; Porta, M.; Silverman, D.T.; Milne, R.L.; Kogevinas, M.; Rothman, N.; Cantor, K.P.; Jackson, B.P.; Pumarega, J.A.; Lopez, T.; et al. Pancreatic cancer risk and levels of trace elements. Gut 2012, 61, 1583–1588. [Google Scholar]
- Kriegel, A.M.; Soliman, A.S.; Zhang, Q.; El-Ghawalby, N.; Ezzat, F.; Soultan, A.; Abdel-Wahab, M.; Fathy, O.; Ebidi, G.; Bassiouni, N.; et al. Serum cadmium levels in pancreatic cancer patients from the east nile delta region of egypt. Environ. Health Perspect. 2006, 114, 113–119. [Google Scholar]
- Schwartz, G.G.; Reis, I.M. Is cadmium a cause of human pancreatic cancer? Cancer Epidemiol. Biomarkers Prev 2000, 9, 139–145. [Google Scholar]
- Filipic, M. Mechanisms of cadmium induced genomic instability. Mutat. Res 2012, 733, 69–77. [Google Scholar]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar]
- Cheng, T.F.; Choudhuri, S.; Muldoon-Jacobs, K. Epigenetic targets of some toxicologically relevant metals: A review of the literature. J. Appl. Toxicol 2012, 32, 643–653. [Google Scholar]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Oxidative stress in apoptosis and cancer: An update. Arch. Toxicol 2012, 86, 1649–1665. [Google Scholar]
- Lee, J.C.; Son, Y.O.; Pratheeshkumar, P.; Shi, X. Oxidative stress and metal carcinogenesis. Free Radic. Biol. Med 2012, 53, 742–757. [Google Scholar]
- Hartwig, A. Metal interaction with redox regulation: An integrating concept in metal carcinogenesis? Free Radic. Biol. Med 2012, 55C, 63–72. [Google Scholar]
- Qu, W.; Diwan, B.A.; Reece, J.M.; Bortner, C.D.; Pi, J.; Liu, J.; Waalkes, M.P. Cadmium-induced malignant transformation in rat liver cells: Role of aberrant oncogene expression and minimal role of oxidative stress. Int. J. Cancer 2005, 114, 346–355. [Google Scholar]
- Shaulian, E.; Karin, M. Ap-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar]
- Joseph, P.; Muchnok, T.K.; Klishis, M.L.; Roberts, J.R.; Antonini, J.M.; Whong, W.Z.; Ong, T.M. Cadmium-induced cell transformation and tumorigenesis are associated with transcriptional activation of c-fos, c-jun, and c-myc proto-oncogenes: Role of cellular calcium and reactive oxygen species. Toxicol. Sci 2001, 61, 295–303. [Google Scholar]
- Qu, W.; Fuquay, R.; Sakurai, T.; Waalkes, M.P. Acquisition of apoptotic resistance in cadmium-induced malignant transformation: Specific perturbation of jnk signal transiduction pathway and associated metallothionein overexpression. Mol. Carcinog 2006, 45, 561–571. [Google Scholar]
- Son, Y.O.; Wang, L.; Poyil, P.; Budhraja, A.; Hitron, J.A.; Zhang, Z.; Lee, J.C.; Shi, X.L. Cadmium induces carcinogenesis in BEAS-2B cells through ROS-dependent activation of PI3K/AKT/GSK-3β/β-catenin signaling. Toxicol. Appl. Pharmacol 2012, 264, 153–160. [Google Scholar]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive oxygen species in cancer stem cells. Antioxid. Redox. Signal 2012, 16, 1215–1228. [Google Scholar]
- Beyersmann, D.; Hartwig, A. Carcinogenic metal compounds: Recent insight into molecular and cellular mechanisms. Arch. Toxicol 2008, 82, 493–512. [Google Scholar]
- Bravard, A.; Vacher, M.; Gouget, B.; Coutant, A.; de Boisferon, F.H.; Marsin, S.; Chevillard, S.; Radicella, J.P. Redox regulation of human ogg1 activity in response to cellular oxidative stress. Mol. Cell. Biol 2006, 26, 7430–7436. [Google Scholar]
- Bravard, A.; Campalans, A.; Vacher, M.; Gouget, B.; Levalois, C.; Chevillard, S.; Radicella, J.P. Inactivation by oxidation and recruitment into stress granules of hOGG1 but not APE1 in human cells exposed to sub-lethal concentrations of cadmium. Mutat. Res 2010, 685, 61–69. [Google Scholar]
- Broedbaek, K.; Weimann, A.; Stovgaard, E.S.; Poulsen, H.E. Urinary 8-oxo-7,8-dihydro-2′- deoxyguanosine as a biomarker in type 2 diabetes. Free Radic. Biol. Med 2011, 51, 1473–1479. [Google Scholar]
- Nzengue, Y.; Steiman, R.; Rachidi, W.; Favier, A.; Guiraud, P. Oxidative stress induced by cadmium in the c6 cell line: Role of copper and zinc. Biol. Trace Elem. Res 2012, 146, 410–419. [Google Scholar]
- Lin, T.S.; Wu, C.C.; Wu, J.D.; Wei, C.H. Oxidative DNA damage estimated by urinary 8-hydroxy-2′-deoxyguanosine and arsenic in glass production workers. Toxicol. Ind. Health 2012, 28, 513–521. [Google Scholar]
- Diakowska, D.; Lewandowski, A.; Kopec, W.; Diakowski, W.; Chrzanowska, T. Oxidative DNA damage and total antioxidant status in serum of patients with esophageal squamous cell carcinoma. Hepatogastroenterology 2007, 54, 1701–1704. [Google Scholar]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′-deoxyguanosine (8-ohdg): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health Pt. C 2009, 27, 120–139. [Google Scholar]
- Muguruma, M.; Unami, A.; Kanki, M.; Kuroiwa, Y.; Nishimura, J.; Dewa, Y.; Umemura, T.; Oishi, Y.; Mitsumori, K. Possible involvement of oxidative stress in piperonyl butoxide induced hepatocarcinogenesis in rats. Toxicology 2007, 236, 61–75. [Google Scholar]
- Tost, J. Epigenetics; Caister Academic: Wymondham, UK, 2008. [Google Scholar]
- Fragou, D.; Fragou, A.; Kouidou, S.; Njau, S.; Kovatsi, L. Epigenetic mechanisms in metal toxicity. Toxicol. Mech. Methods 2011, 21, 343–352. [Google Scholar]
- Huang, D.; Zhang, Y.; Qi, Y.; Chen, C.; Ji, W. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicol. Lett 2008, 179, 43–47. [Google Scholar]
- Poirier, L.A.; Vlasova, T.I. The prospective role of abnormal methyl metabolism in cadmium toxicity. Environ. Health Perspect 2002, 110, 793–795. [Google Scholar]
- Benbrahim-Tallaa, L.; Waterlandz, R.A.; Dill, A.L.; Webber, M.M.; Waalkes, M.P. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexprression of de novo DNA methyltransferase. Environ. Health Perspect 2007, 115, 1454–1459. [Google Scholar]
- Zhou, Z.H.; Lei, Y.X.; Wang, C.X. Analysis of aberrant methylation in DNA repair genes during malignant transformation of human bronchial epithelial cells induced by cadmium. Toxicol. Sci 2012, 125, 412–417. [Google Scholar]
- Lim, S.O.; Gu, J.M.; Kim, M.S.; Kim, H.S.; Park, Y.N.; Park, C.K.; Cho, J.W.; Park, Y.M.; Jung, G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: Methylation of the e-cadherin promoter. Gastroenterology 2008, 135, 2128–2140. [Google Scholar]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive oxygen species (ROS)-induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res 2011, 711, 167–173. [Google Scholar]
- Shafritz, D.A.; Dabeva, M.D. Liver stem cells and model systems for liver repopulation. J. Hepatol 2002, 36, 552–564. [Google Scholar]
- Kobayashi, C.I.; Suda, T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J. Cell. Physiol 2012, 227, 421–430. [Google Scholar]
- Cheng, T.; Rodrigues, N.; Shen, H.M.; Yang, Y.G.; Dombkowski, D.; Sykes, M.; Scadden, D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000, 287, 1804–1808. [Google Scholar]
- Arai, F.; Suda, T. Quiescent Stem Cells in the Niche; Stembook: Cambridge, MA, USA, 2008. [Google Scholar]
- Blanpain, C.; Mohrin, M.; Sotiropoulou, P.A.; Passegue, E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011, 8, 16–29. [Google Scholar]
- Challen, G.A.; Little, M.H. A side order of stem cells: The sp phenotype. Stem Cells 2006, 24, 3–12. [Google Scholar]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. Mtor activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after pten deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar]
- Yilmaz, O.H.; Valdez, R.; Ferguson, D.O.; Morrison, S.J. Pten dependence distinguishes hematopoietic stem cells from leukemia-initiating cells. Dev. Biol 2006, 295, 359–359. [Google Scholar]
- Cosentino, C.; Grieco, D.; Costanzo, V. Atm activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J 2011, 30, 546–555. [Google Scholar]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Mak, T.W.; Suda, T. Regulation of oxidative stress by atm is required for the self-renewal of haematopoietic stem cells. Blood 2004, 104, 109A–109A. [Google Scholar]
- Abbas, H.A.; Maccio, D.R.; Coskun, S.; Jackson, J.G.; Hazen, A.L.; Sills, T.M.; You, M.J.; Hirschi, K.K.; Lozano, G. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ros-induced p53 activity. Cell Stem Cell 2010, 7, 606–617. [Google Scholar]
- Chuikov, S.; Levi, B.P.; Smith, M.L.; Morrison, S.J. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat. Cell. Biol 2010, 12, 999–1006. [Google Scholar]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the hif-1 alpha level is essential for hematopoietic stem cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamato, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 2002, 62, 5196–5203. [Google Scholar]
- Vollrath, V.; Wielandt, A.M.; Iruretagoyena, M.; Chianale, J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J 2006, 395, 599–609. [Google Scholar]
- Huang, J.J.; Okuka, M.; McLean, M.; Keefe, D.L.; Liu, L. Effects of cigarette smoke on fertilization and embryo development in vivo. Fertil. Steril 2009, 92, 1456–1465. [Google Scholar]
- Uhal, B.D. Cell cycle kinetics in the alveolar epithelium. Am. J. Physiol 1997, 272, L1031–L1045. [Google Scholar]
- Hart, B.A.; Eneman, J.D.; Gong, Q.; DurieuxLu, C.C. Increased oxidant resistance of alveolar epithelial type ii cells. Isolated from rats following repeated exposure to cadmium aerosols. Toxicol. Lett 1995, 81, 131–139. [Google Scholar]
- Plusquin, M.; Stevens, A.S.; van Belleghem, F.; Degheselle, O.; van Roten, A.; Vroonen, J.; Blust, R.; Cuypers, A.; Artois, T.; Smeets, K. Physiological and molecular characterisation of cadmium stress in schmidtea mediterranea. Int. J. Dev. Biol 2012, 56, 183–191. [Google Scholar]
- Piersma, A.H.; Roelen, B.; Roest, P.; Haakmathoesenie, A.S.; Vanachterberg, T.A.E.; Mummery, C.L. Cadmium-induced inhibition of proliferation and differentiation of embryonal carcinoma-cells and mechanistic aspects of protection by zinc. Teratology 1993, 48, 335–341. [Google Scholar]
- Gadhia, S.R.; Calabro, A.R.; Barile, F.A. Trace metals alter DNA repair and histone modification pathways concurrently in mouse embryonic stem cells. Toxicol. Lett 2012, 212, 169–179. [Google Scholar]
- Jiang, G.F.; Xu, L.; Zhang, B.; Wu, L. Effects of cadmium on proliferation and self-renewal activity of prostate stem/progenitor cells. Environ. Toxicol. Pharmacol 2011, 32, 275–284. [Google Scholar]
- Kalafatic, M.; Kopjar, N.; Besendorfer, V. The impairments of neoblast division in regenerating planarian polycelis felina (daly.) caused by in vitro treatment with cadmium sulfate. Toxicol. in Vitro 2004, 18, 99–107. [Google Scholar]
- Gulisano, M.; Pacini, S.; Punzi, T.; Morucci, G.; Quagliata, S.; Delfino, G.; Sarchielli, E.; Marini, M.; Vannelli, G.B. Cadmium modulates proliferation and differentiation of human neuroblasts. J. Neurosci. Res 2009, 87, 228–237. [Google Scholar]
- Alvarado, A.S. Regeneration and the need for simpler model organisms. Philos. Trans. R. Soc. Lond. B 2004, 359, 759–763. [Google Scholar]
- Reddien, P.W.; Alvarado, A.S. Fundamentals of planarian regeneration. Annu. Rev. Cell. Dev. Biol 2004, 20, 725–757. [Google Scholar]
- Jiang, G.F.; Duan, W.X.; Xu, L.; Song, S.Z.; Zhu, C.C.; Wu, L. Biphasic effect of cadmium on cell proliferation in human embryo lung fibroblast cells and its molecular mechanism. Toxicol. in Vitro 2009, 23, 973–978. [Google Scholar]
- Fulladosa, E.; Murat, J.C.; Villaescusa, I. Effect of cadmium(II), chromium(VI), and arsenic(V) on long-term viability- and growth-inhibition assays using Vibrio fischeri marine bacteria. Arch. Environ. Contam. Toxicol 2005, 49, 299–306. [Google Scholar]
- Cao, X.J.; Chen, R.; Li, A.P.; Zhou, J.W. JWA gene is involved in cadmium-induced growth inhibition and apoptosis in HEK-293T cells. J. Toxicol. Environ. Health A 2007, 70, 931–937. [Google Scholar]
- Vonzglinicki, T.; Edwall, C.; Ostlund, E.; Lind, B.; Nordberg, M.; Ringertz, N.R.; Wroblewski, J. Very low cadmium concentrations stimulate DNA-synthesis and cell-growth. J. Cell. Sci. 1992, 103, 1073–1081. [Google Scholar]
- Misra, U.K.; Gawdi, G.; Akabani, G.; Pizzo, S.V. Cadmium-induced DNA synthesis and cell proliferation in macrophages: The role of intracellular calcium and signal transduction mechanisms. Cell. Signal 2002, 14, 327–340. [Google Scholar]
- Hussein, A.M.; Hasan, S. Cadmium affects viability of bone marrow mesenchymal stem cells through membrane impairment, intracellular calcium elevation and DNA breakage. Indian J. Med. Sci 2010, 64, 177–186. [Google Scholar]
- Huang, J.; Okuka, M.; Lu, W.; Tsibris, J.C.M.; McLean, M.P.; Keefe, D.L.; Liu, L. Telomere shortening and DNA damage of embryonic stem cells induced by cigarette smoke. Reprod. Toxicol 2013, 35, 89–95. [Google Scholar]
- Matsuoka, M.; Igisu, H.; Nakagawa, K.; Katada, T.; Nishina, H. Requirement of MKK4 and MKK7 for CdCl2- or HgCl2-induced activation of c-Jun NH2-terminal kinase in mouse embryonic stem cells. Toxicol. Lett 2004, 152, 175–181. [Google Scholar]
- Yamada, H.; Uenishi, R.; Suzuki, K.; Koizumi, S. Cadmium-induced alterations of gene expression in human cells. Environ. Toxicol. Pharmacol 2009, 28, 61–69. [Google Scholar]
- Hu, W.Y.; Shi, G.B.; Hu, D.P.; Nelles, J.L.; Prins, G.S. Actions of estrogens and endocrine disrupting chemicals on human prostate stem/progenitor cells and prostate cancer risk. Mol. Cell. Endocrinol 2012, 354, 63–73. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nair, A.R.; DeGheselle, O.; Smeets, K.; Van Kerkhove, E.; Cuypers, A. Cadmium-Induced Pathologies: Where Is the Oxidative Balance Lost (or Not)? Int. J. Mol. Sci. 2013, 14, 6116-6143. https://doi.org/10.3390/ijms14036116
Nair AR, DeGheselle O, Smeets K, Van Kerkhove E, Cuypers A. Cadmium-Induced Pathologies: Where Is the Oxidative Balance Lost (or Not)? International Journal of Molecular Sciences. 2013; 14(3):6116-6143. https://doi.org/10.3390/ijms14036116
Chicago/Turabian StyleNair, Ambily Ravindran, Olivier DeGheselle, Karen Smeets, Emmy Van Kerkhove, and Ann Cuypers. 2013. "Cadmium-Induced Pathologies: Where Is the Oxidative Balance Lost (or Not)?" International Journal of Molecular Sciences 14, no. 3: 6116-6143. https://doi.org/10.3390/ijms14036116
APA StyleNair, A. R., DeGheselle, O., Smeets, K., Van Kerkhove, E., & Cuypers, A. (2013). Cadmium-Induced Pathologies: Where Is the Oxidative Balance Lost (or Not)? International Journal of Molecular Sciences, 14(3), 6116-6143. https://doi.org/10.3390/ijms14036116