Structure-Based Search for New Inhibitors of Cholinesterases

 ,
, 
Abstract
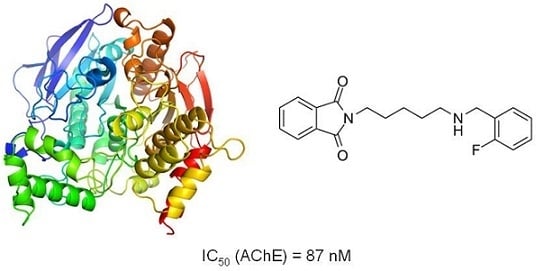
:
Abbreviations
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| BuChE | butyrylcholinesterase |
| hBuChE | human butyrylcholinesterase |
| NMDA | N-methyl-D-aspartate |
| PAS | peripheral anionic site |
| PDB | Protein Data Bank |
| TcAChE | Torpedo californica acetylcholinesterase |
1. Introduction
2. Results and Discussion
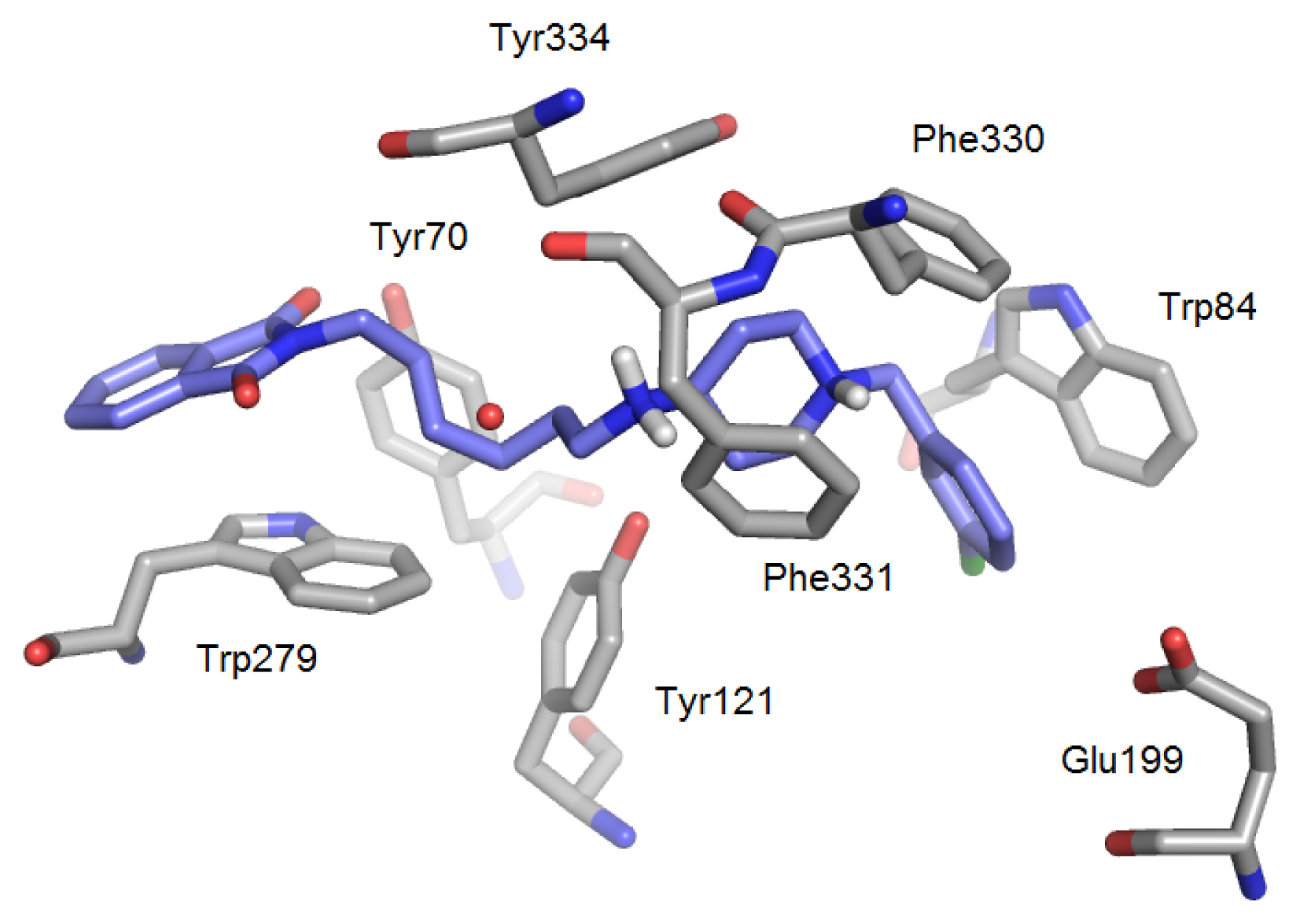
2.1. Analysis of Enzyme Structures
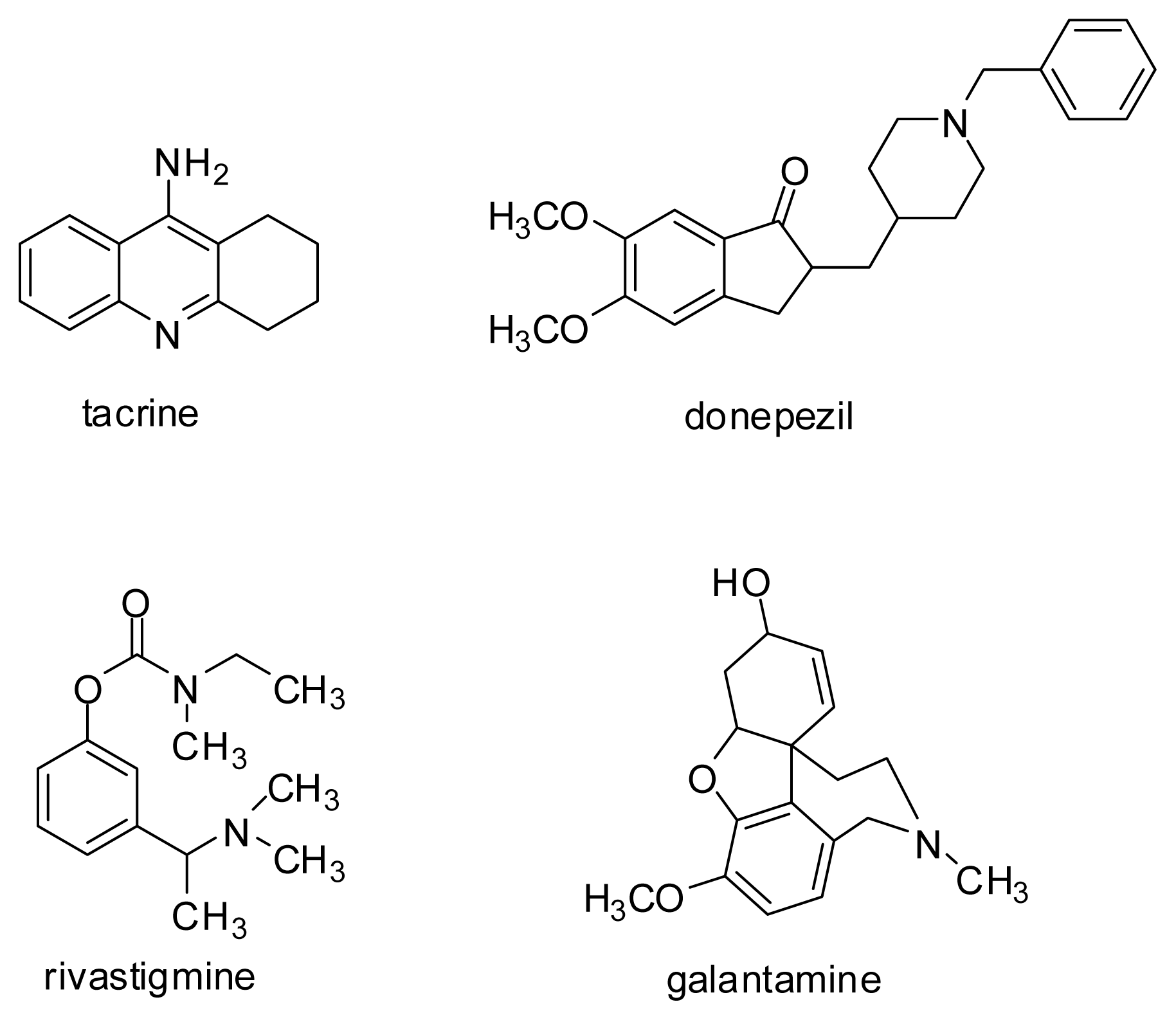
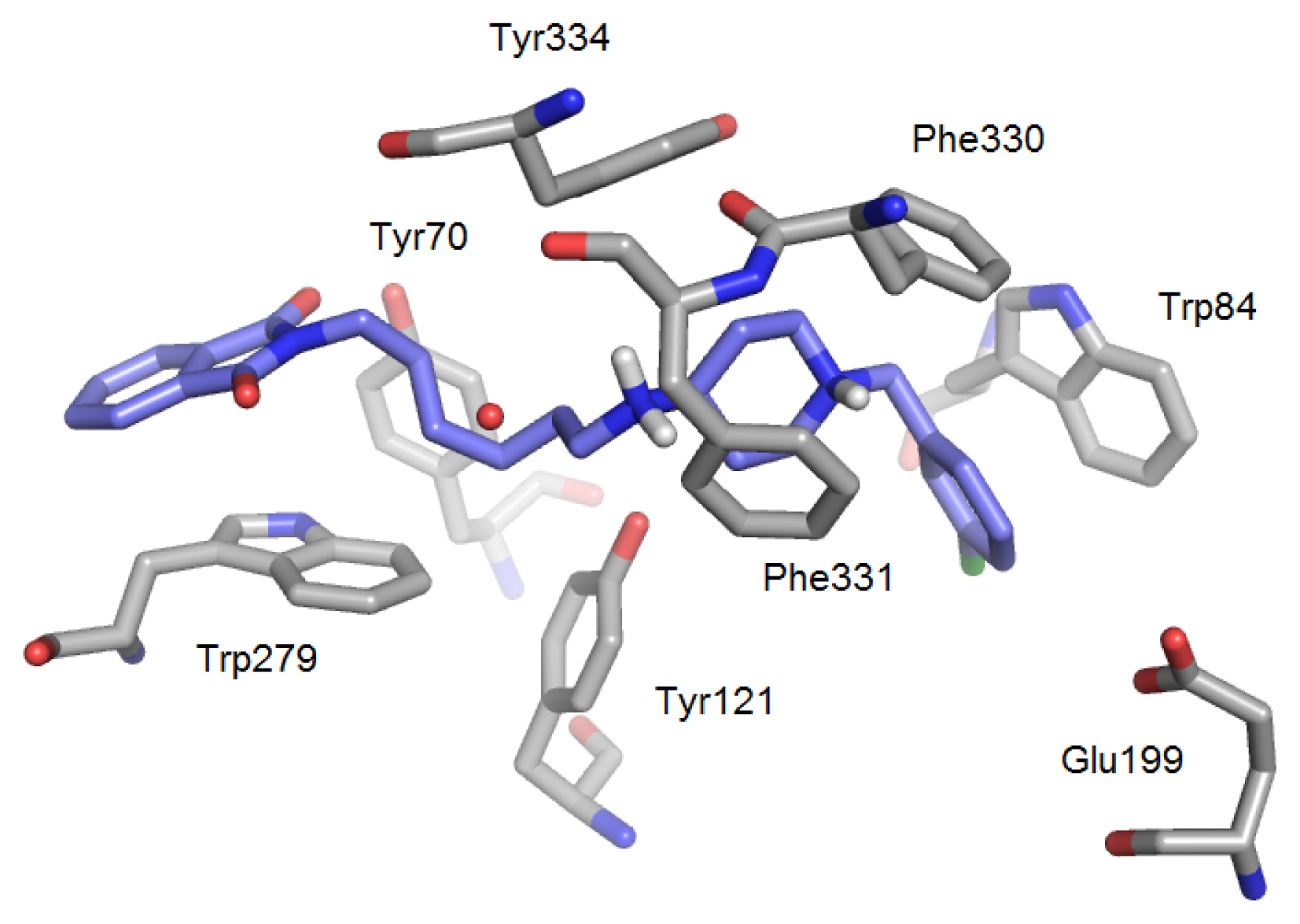
2.1.1. Acetylcholinesterase
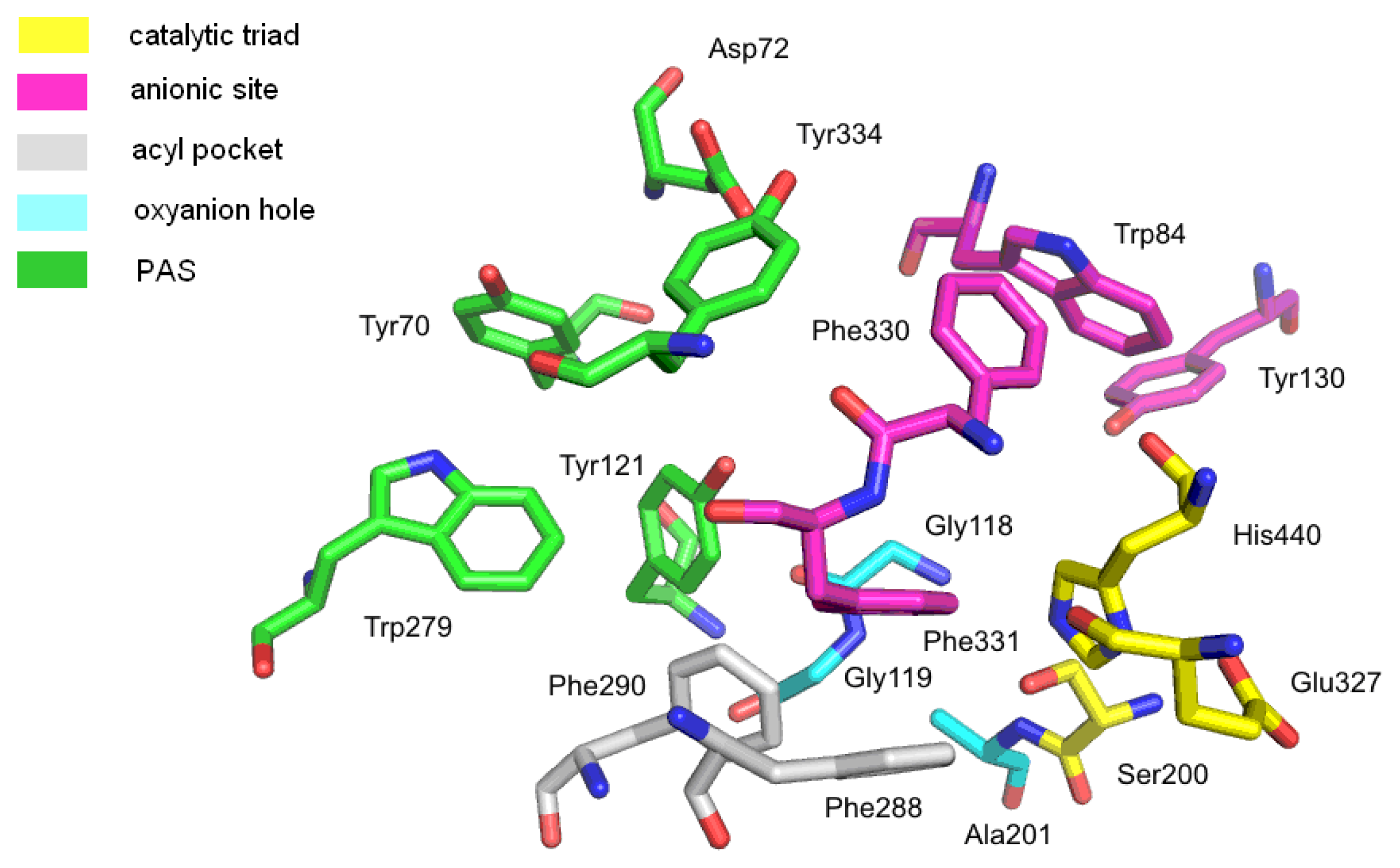
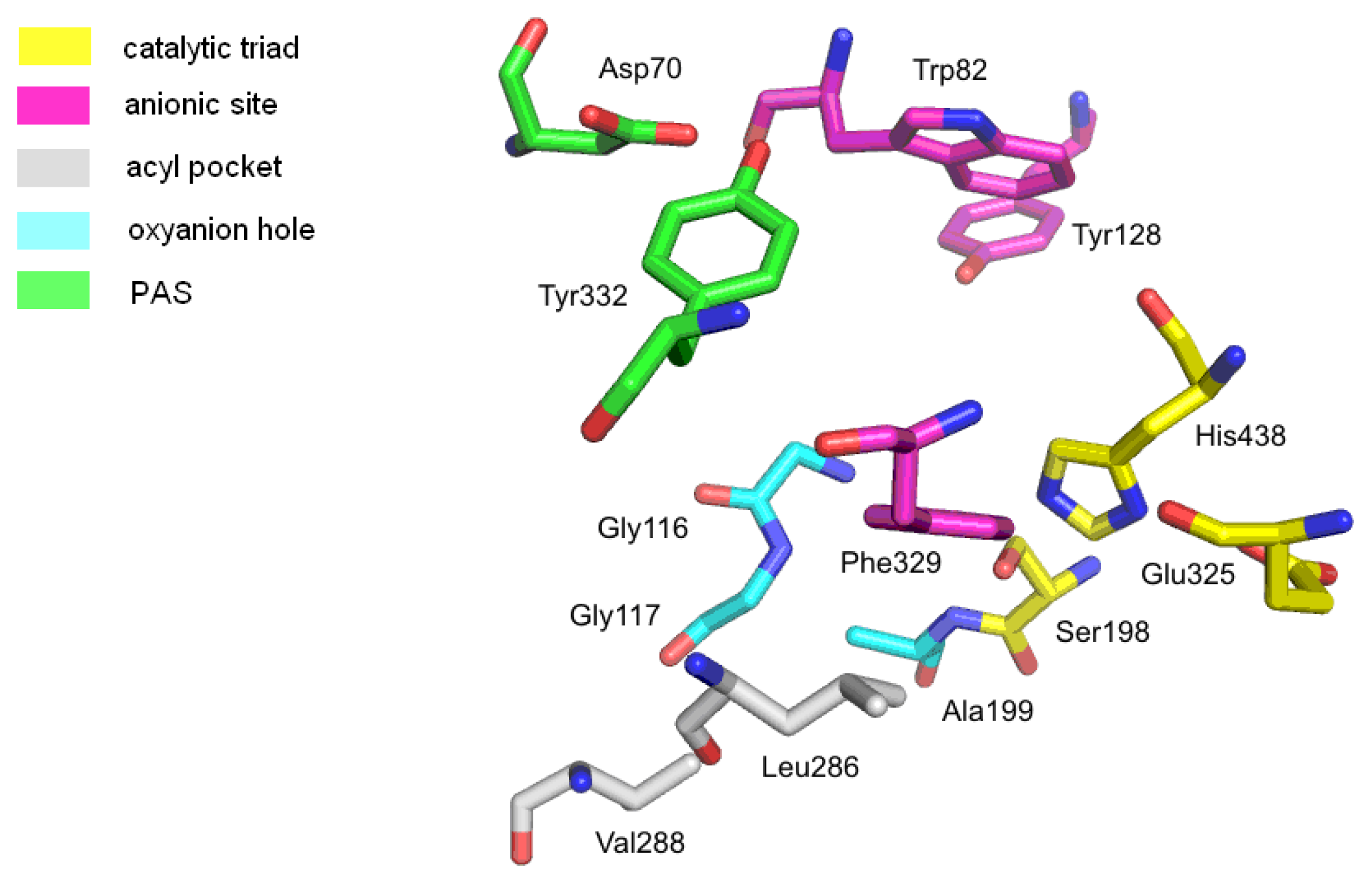
2.1.2. Butyrylcholinesterase
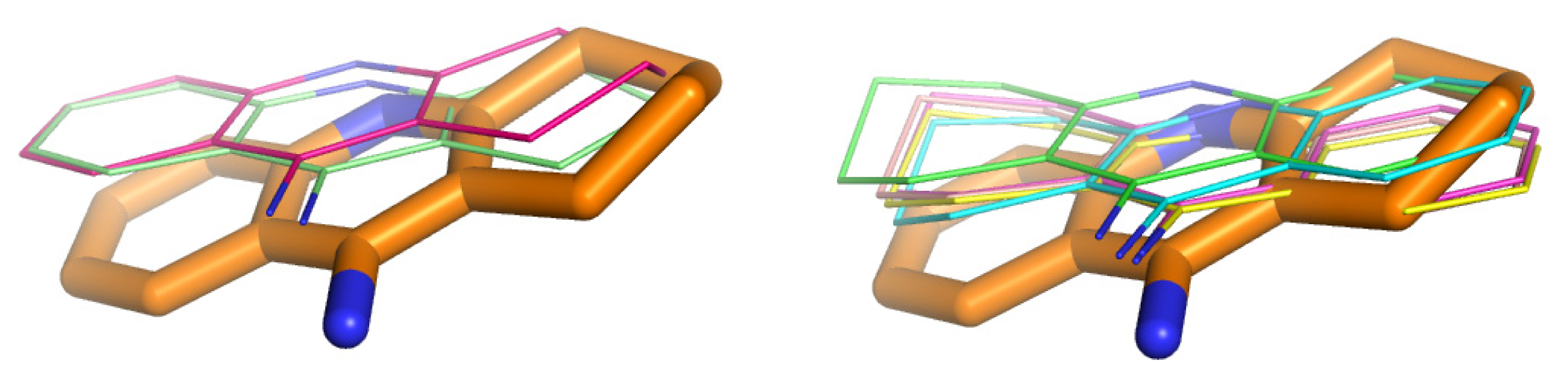
2.2. Validation of Docking with Gold Software
- rmsd ≤ 1.0 Å, good pose;
- Å < rmsd ≤ 2.0 Å, close pose;
- Å < rmsd ≤ 3.0 Å, pose with errors;
- rmsd > 3.0 Å, bad pose.
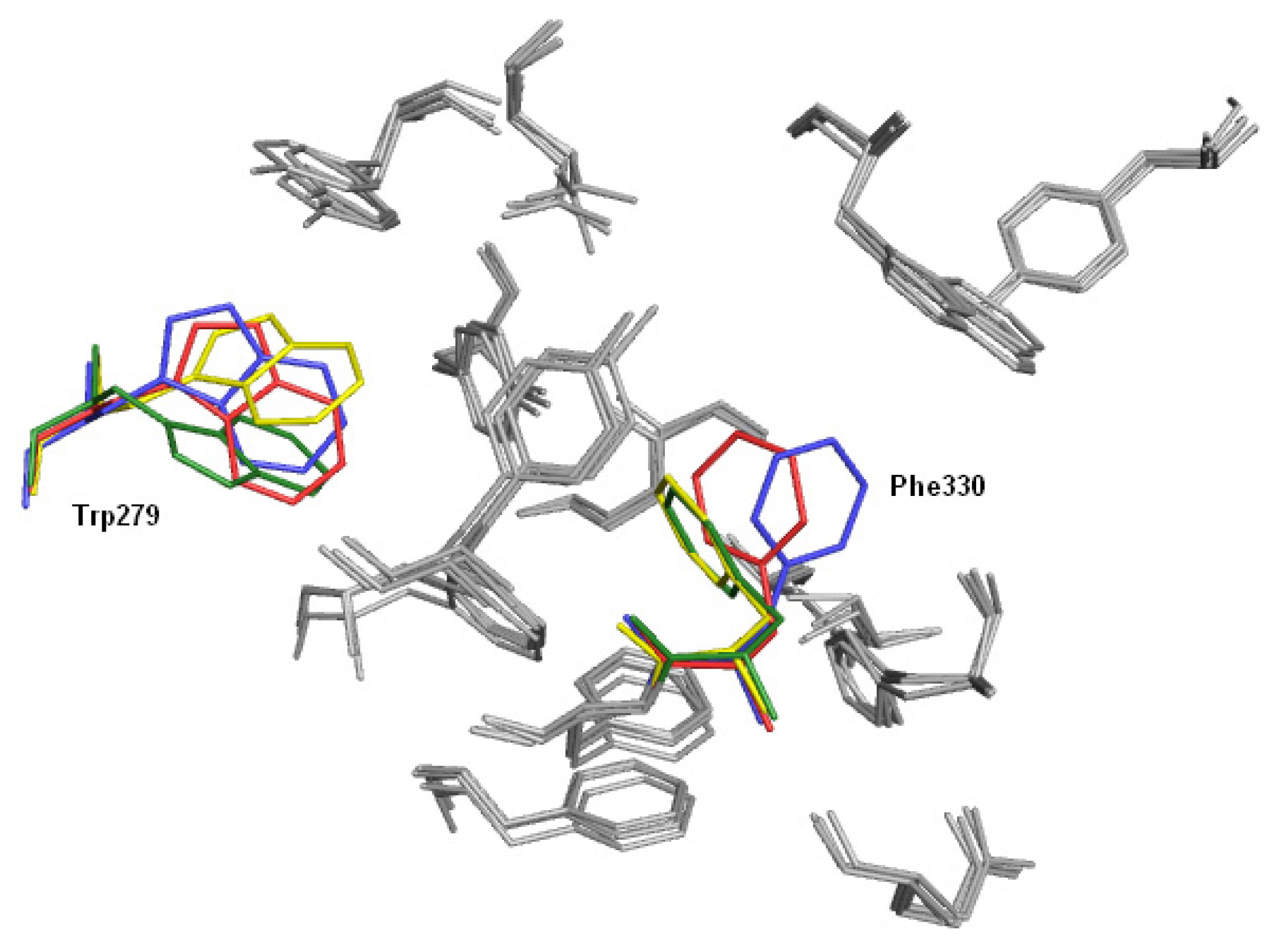
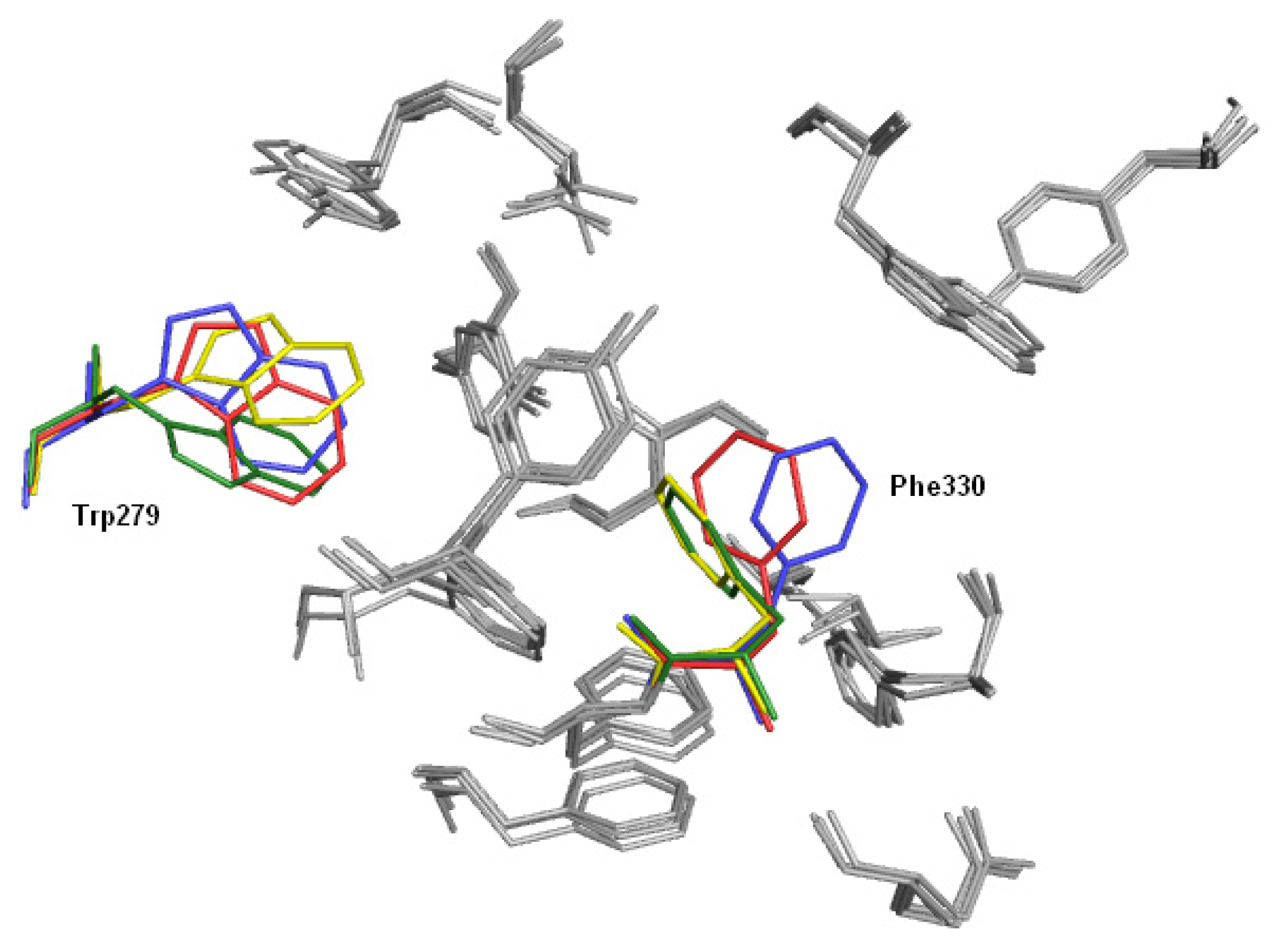
2.3. Modifications of Heterodimeric Structures from Our Library
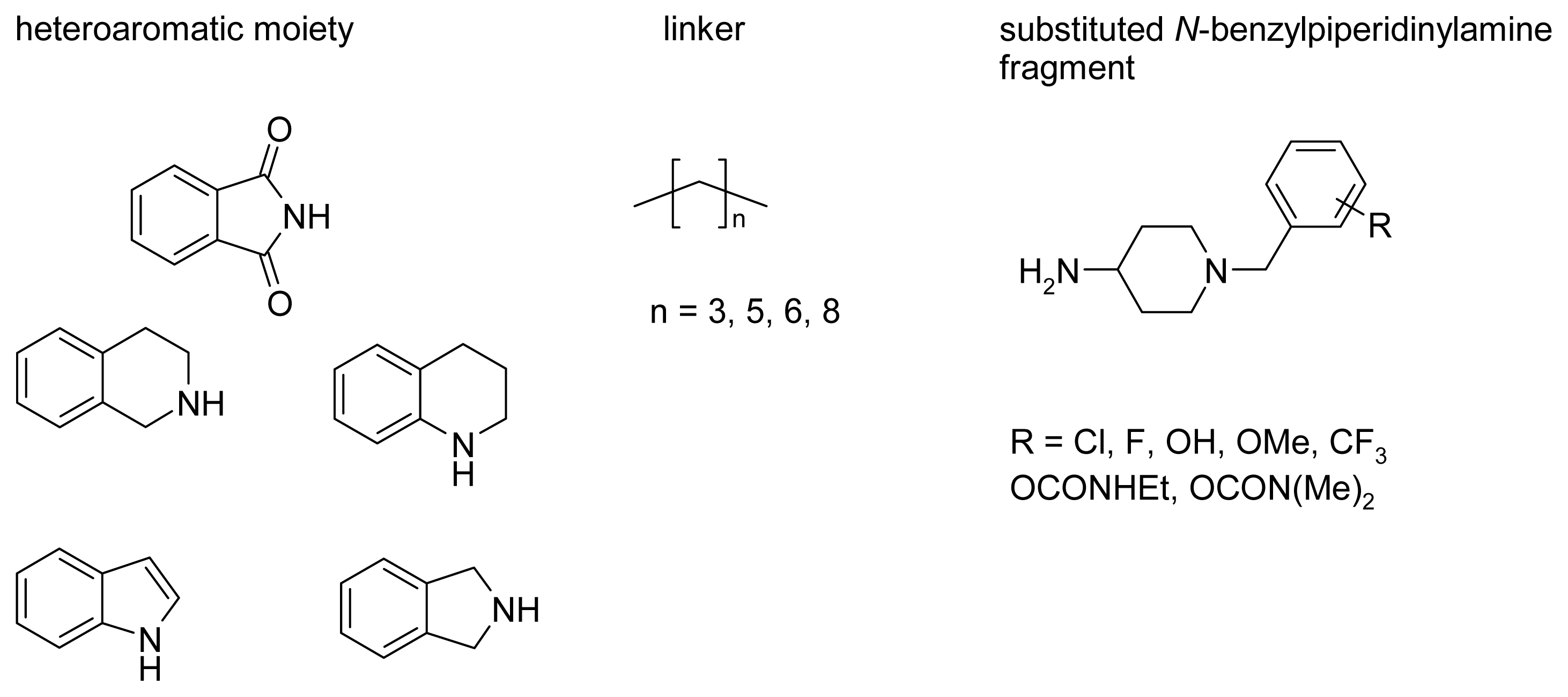
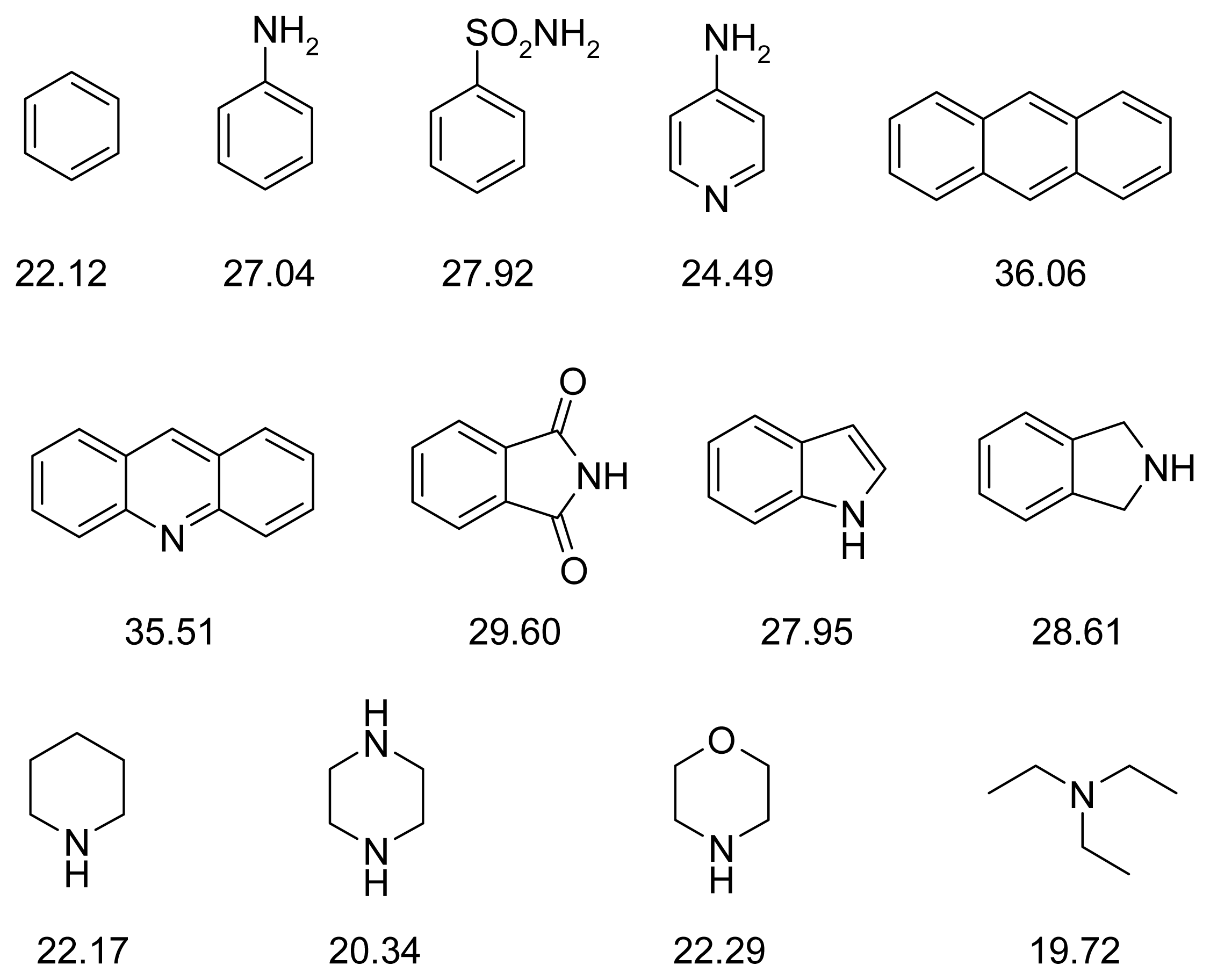
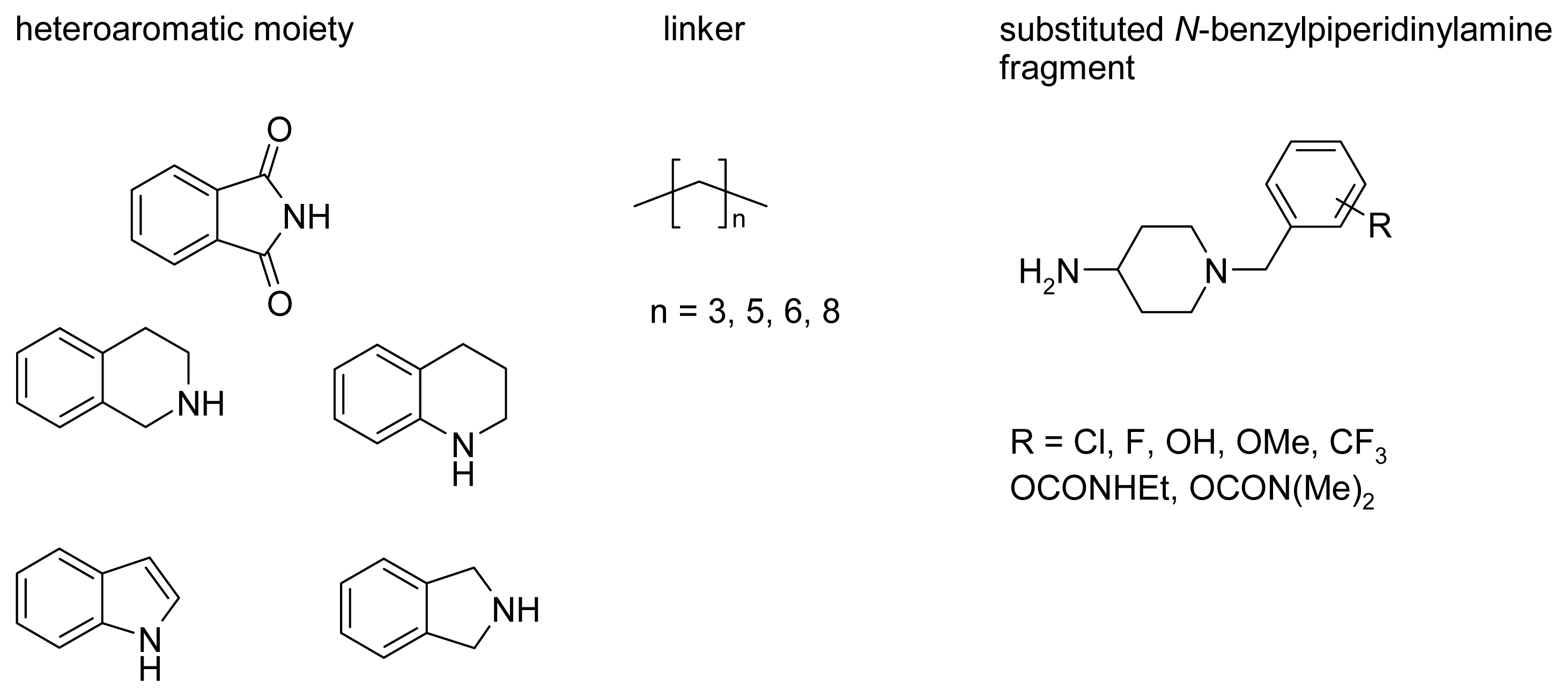
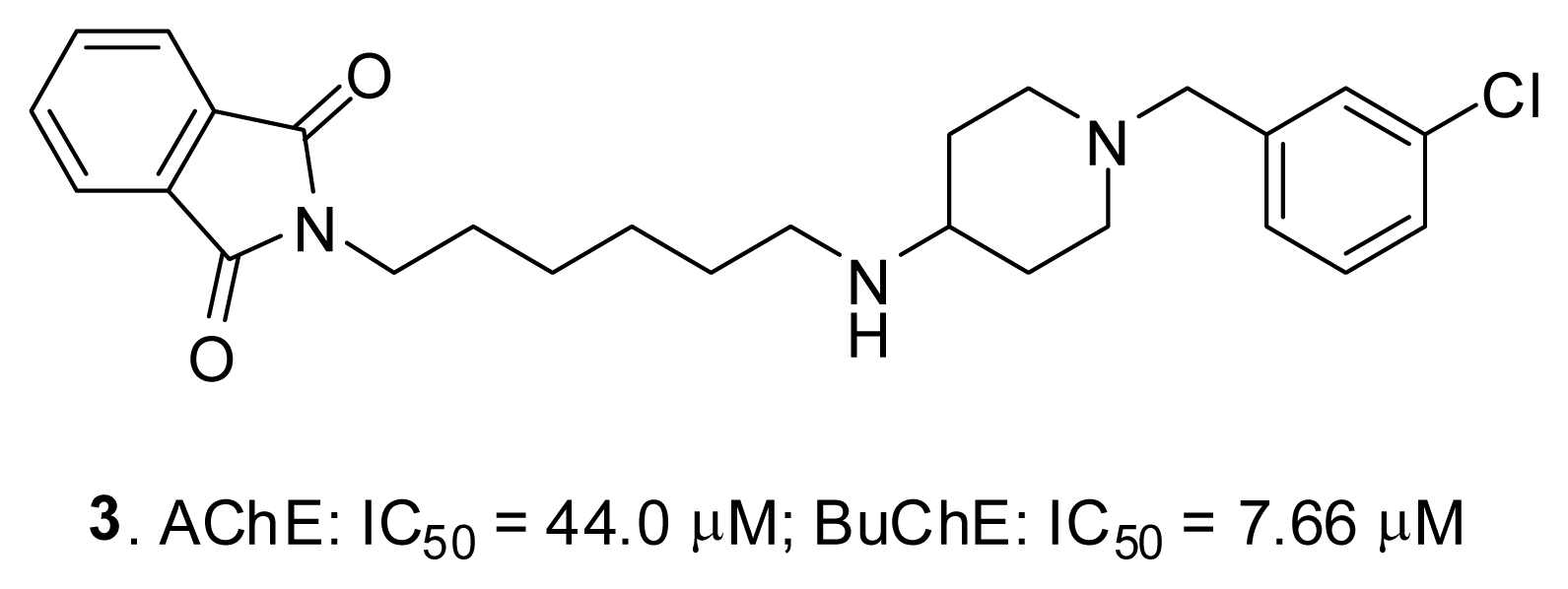
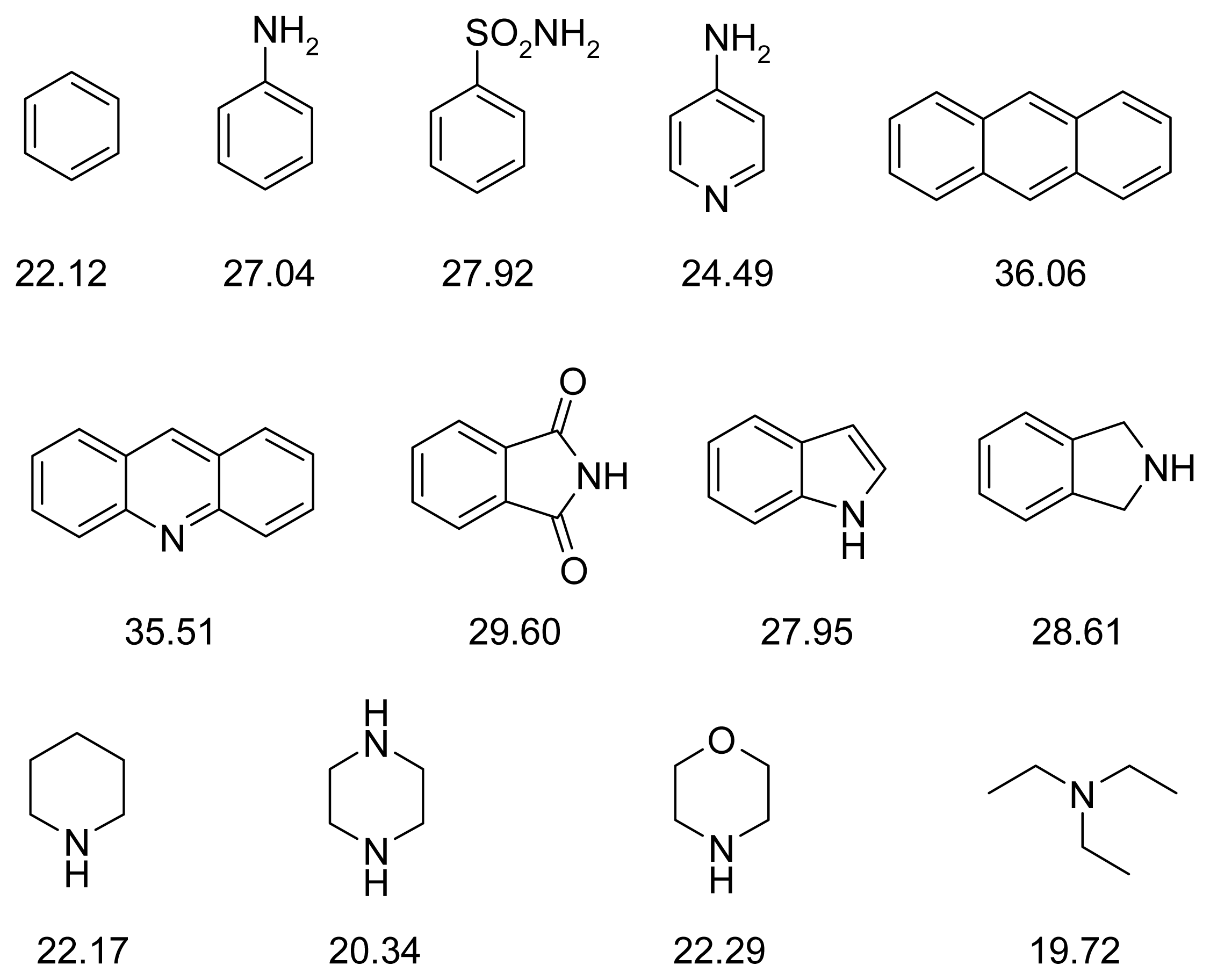
2.4. Fragment-Based Design of Cholinesterase Inhibitors
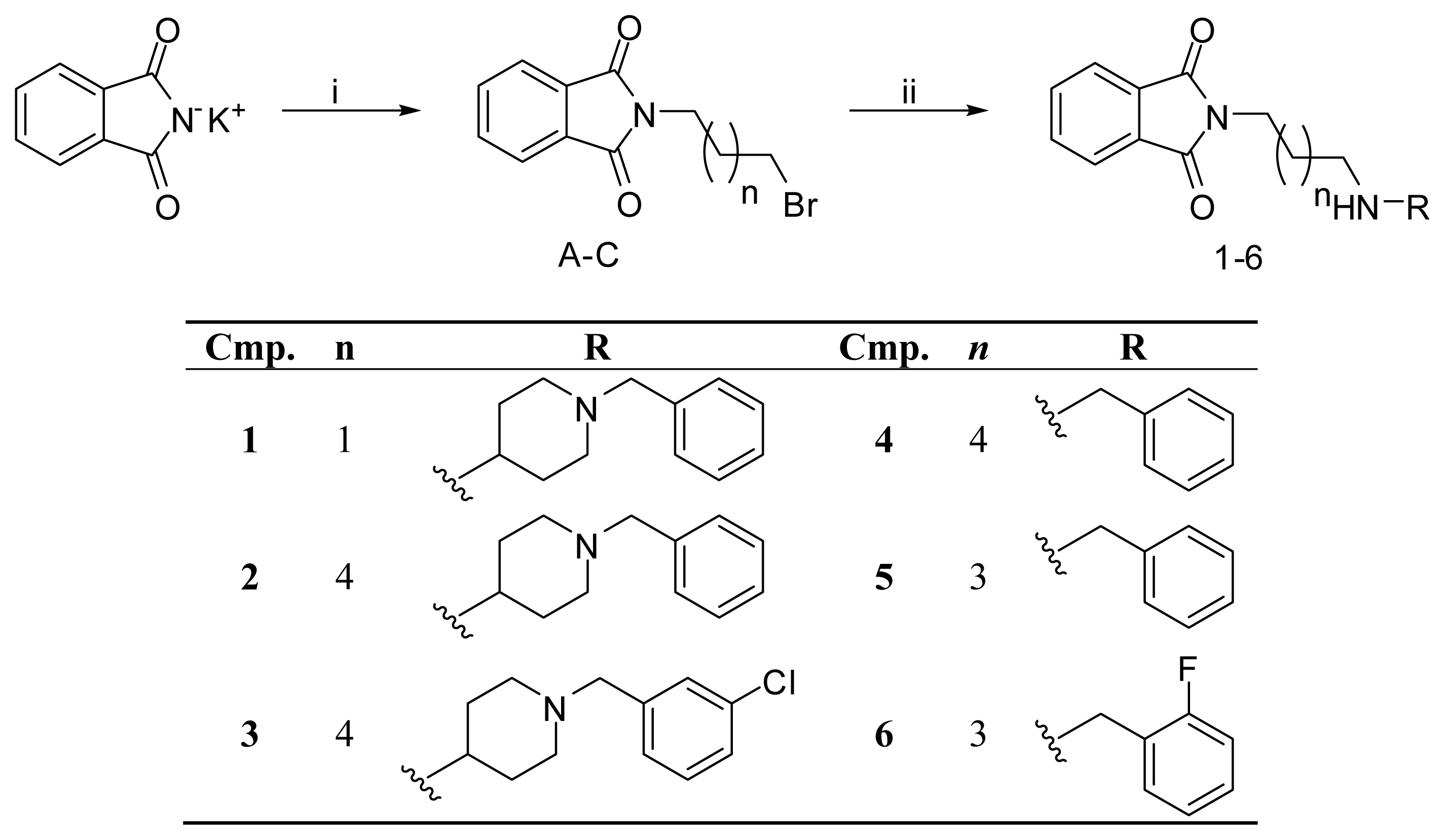
2.5. Chemistry
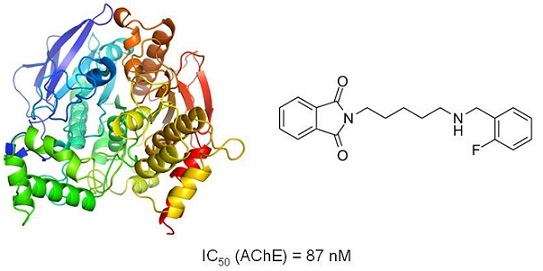
2.6. Biological Assays
3. Experimental Section
3.1. Docking
3.1.1. Ligands
3.1.2. Acetylcholinesterase
3.1.3. Butyrylcholinesterase
3.2. Synthesis and Biological Assay
3.2.1. General Information
3.2.1.1. Chemistry
3.2.1.2. Materials
3.2.1.3. Biological Activity
3.2.2. Experimental Details and Spectroscopic Data
3.2.3. Ellman’s Assay—AChE/BuChE Inhibition Activity
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar]
- Giacobini, E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res 2004, 50, 433–440. [Google Scholar]
- Melnikova, I. Therapies for Alzheimer’s disease. Nat. Rev. Drug Discov 2007, 6, 341–342. [Google Scholar]
- Ballenger, J.F. Progress in the history of Alzheimer’s disease: The importance of context. J. Alzheimers Dis 2006, 9, 5–13. [Google Scholar]
- Rountree, S.D.; Chan, W.; Pavlik, V.N.; Darby, E.J.; Siddiqui, S. Persistant treatment with cholinesterase inhibitors and/or memantine slows clinical progression of Alzheimer disease. Alzheimers Res. Ther 2009, 1, 1–7. [Google Scholar]
- Musiał, A.; Bajda, M.; Malawska, B. Recent developments in cholinesterases inhibitors for Alzheimer’s disease treatment. Curr. Med. Chem 2007, 14, 2654–2679. [Google Scholar]
- Holzgrabe, U.; Kapkova, P.; Alptüzün, V.; Scheiber, J.; Kugelmann, E. Targeting acetylcholinesterase to treat neurodegeneration. Expert Opin. Ther. Targets 2007, 11, 161–179. [Google Scholar]
- Nachon, F.; Masson, P.; Nicolet, Y.; Lockridge, O.; Fontecilla-Camps, J.C. Comparison of the Structures of Butyrylcholinesterase and Acetylcholinesterase. In Butyrylcholinesterase: Its Function and Inhibitors, 1st ed; Giacobini, E., Ed.; Martin Dunitz: London, UK, 2003; pp. 39–54. [Google Scholar]
- Cokugras, A.N. Butyrylcholinesterase: Structure and physiological importance. Turk. J. Biochem 2003, 28, 54–61. [Google Scholar]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J.; et al. The α/β hydrolase fold. Protein Eng 1992, 5, 197–211. [Google Scholar]
- Cygler, M.; Schrag, J.D.; Sussman, J.L.; Harel, M.; Silman, I.; Gentry, M.K.; Doctor, B.P. Relationship between sequence conservation and three-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci 1993, 2, 366–382. [Google Scholar]
- Alvarez, A.; Alarcon, R.; Opazo, C.; Campos, E.O.; Munoz, F.J.; Calderon, F.H.; Dajas, F.; Gentry, M.K.; Doctor, B.P.; de Mello, F.G.; Inestrosa, N.C. Stable complexes involving acetylcholinesterase and amyloid-beta peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer’s fibrils. J. Neurosci 1998, 18, 3213–3223. [Google Scholar]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: “Classical” and “non-classical” functions and pharmacology. Curr. Opin. Pharm 2005, 5, 293–302. [Google Scholar]
- Darvesh, S.; Cash, M.K.; Reid, G.A.; Martin, E.; Mitnitski, A.; Geula, C. Butyrylcholinesterase is associated with β-amyloid plaques in the transgenic APPSWE/PSEN1dE9 mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol 2012, 71, 2–14. [Google Scholar]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem 2003, 278, 41141–41147. [Google Scholar]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar]
- Masson, P.; Legrand, P.; Bartels, C.F.; Froment, M.T.; Schopfer, L.M.; Lockridge, O. Role of aspartate 70 and tryptophan 82 in binding of succinyldithiocholine to human butyrylcholinesterase. Biochemistry 1997, 36, 2266–2277. [Google Scholar]
- Nachon, F.; Ehret-Sabatier, L.; Loew, D.; Colas, C.; van Dorsselaer, A.; Goeldner, M. Trp82 and Tyr332 are involved in two quaternary ammonium binding domains of human butyrylcholinesterase as revealed by photoaffinity labeling with [3H]DDF. Biochemistry 1998, 37, 10507–10513. [Google Scholar]
- Xu, Y.; Colletier, J.P.; Weik, M.; Jiang, H.; Moult, J.; Silman, I.; Sussman, J.L. Flexibility of aromatic residues in the active-site gorge of acetylcholinesterase: X-ray versus molecular dynamics. Biophys. J 2008, 95, 2500–2511. [Google Scholar]
- Johnson, G.; Moore, S.W. The peripheral anionic site of acetylcholinesterase: Structure, functions and potential role in rational drug design. Curr. Pharm. Des 2006, 12, 217–225. [Google Scholar]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar]
- Koellner, G.; Kryger, G.; Millard, C.B.; Silman, I.; Sussman, J.L.; Steiner, T. Active-site gorge and buried water molecules in crystal structures of acetylcholinesterase from Torpedo californica. J. Mol. Biol 2000, 296, 713–735. [Google Scholar]
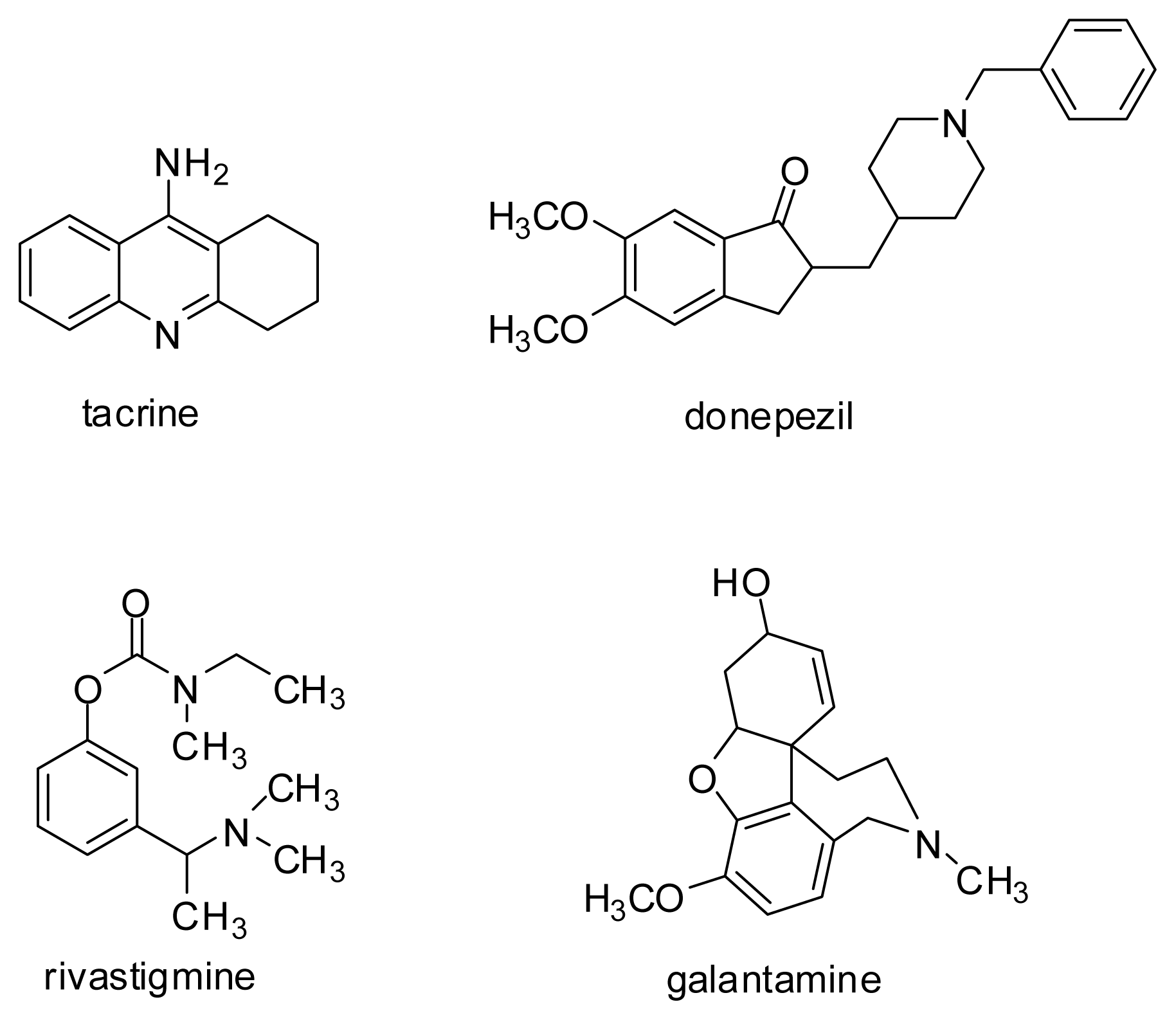
- Tumiatti, V.; Minarini, A.; Bolognesi, M.L.; Milelli, A.; Rosini, M.; Melchiorre, C. Tacrine derivatives and Alzheimer’s disease. Curr. Med. Chem 2010, 17, 1825–1838. [Google Scholar]
- Yan, J.W.; Li, Y.P.; Ye, W.J.; Chen, S.B.; Hou, J.Q.; Tan, J.H.; Ou, T.M.; Li, D.; Gu, L.Q.; Huang, Z.S. Design, synthesis and evaluation of isaindigotone derivatives as dual inhibitors for acetylcholinesterases and amyloid beta aggregation. Bioorg. Med. Chem 2012, 20, 2527–2534. [Google Scholar]
- Chen, X.; Tikhonova, I.G.; Decker, M. Probing the mid-gorge of cholinesterases with spacer-modified bivalent quinazolinimines leads to highly potent and selective butyrylcholinesterase inhibitors. Bioorg. Med. Chem 2011, 19, 1222–1235. [Google Scholar]
- Li, Y.P.; Ning, F.X.; Yang, M.B.; Li, Y.C.; Nie, M.H.; Ou, T.M.; Tan, J.H.; Huang, S.L.; Li, D.; Gu, L.Q.; Huang, Z.S. Syntheses and characterization of novel oxoisoaporphine derivatives as dual inhibitors for cholinesterases and amyloid beta aggregation. Eur. J. Med. Chem 2011, 46, 1572–1581. [Google Scholar]
- Du, D.M.; Carlier, P.R. Development of bivalent acetylcholinesterase inhibitors as potential therapeutic drugs for Alzheimer’s disease. Curr. Pharm. Des 2004, 10, 3141–3156. [Google Scholar]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem 2008, 51, 347–372. [Google Scholar]
- Luo, W.; Li, J.P.; He, Y.; Huang, S.L.; Tan, J.H.; Ou, T.M.; Li, D.; Gu, L.Q.; Huang, Z.S. Design, synthesis and evaluation of novel tacrine-multialkoxybenzene hybrids as dual inhibitors for cholinesterases and amyloid beta aggregation. Bioorg. Med. Chem 2011, 19, 763–770. [Google Scholar]
- Camps, P.; Formosa, X.; Galdeano, C.; Gomez, T.; Munoz-Torrero, D.; Ramirez, L.; Viayna, E.; Gomez, E.; Isambert, N.; Lavilla, R.; et al. Tacrine-based dual binding site acetylcholinesterase inhibitors as potential disease-modifying anti-Alzheimer drug candidates. Chem. Biol. Interact 2010, 187, 411–415. [Google Scholar]
- Więckowska, A. Novel Cholinesterases Inhibitors with Carbamoyloxyphenyl and N-Benzylpiperidine moieties. In Ph.D. thesis; Jagiellonian University: Kraków, Poland, 2008. [Google Scholar]
- Więckowska, A.; Bajda, M.; Guzior, N.; Malawska, B. Novel alkyl- and arylcarbamate derivatives with N-benzylpiperidine and N-benzylpiperazine moieties as cholinesterases inhibitors. Eur. J. Med. Chem 2010, 45, 5602–5611. [Google Scholar]
- Protein Data Bank. Available online: http://www.pdb.org accessed on 3 January 2009.
- PyMOL 0.99rc6; DeLano Scientific LLC: Palo Alto, CA, USA, 2006.
- Gold 4.0; The Cambridge Crystallographic Data Centre: Cambridge, UK, 2009.
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol 1961, 7, 88–95. [Google Scholar]
- Schneider, G.; Baringhaus, K.H. Molecular Design: Concepts and Applications, 1st ed.; Wiley-VCH: Weinheim, Germany; p. 2008.
- Ignasik, M.; Bajda, M.; Guzior, N.; Prinz, M.; Holzgrabe, U.; Malawska, B. Design, synthesis and evaluation of novel 2-(aminoalkyl)-isoindoline-1,3-dione derivatives as dual binding site acetylcholinesterase inhibitors. Arch. Pharm. Chem. Life Sci 2012, 345, 509–516. [Google Scholar]
- Quici, S.; Manfredi, A.; Pozzi, G.; Cavazzini, M.; Rozzoni, A. Ditopic receptors capable of hydrogen bonding: Synthesis and complexation behaviour of diaza crown-ethers having melamine sidearms. Tetrahedron 1999, 55, 10487–10496. [Google Scholar]
- Kong, X.; He, Z.; Zhang, Y.; Mu, L.; Liang, C.; Chen, B.; Jing, X.; Cammidge, A.A. Mesogenic triphenylene-perylene-triphenylene triad. Org. Lett 2011, 13, 764–767. [Google Scholar]
- Shum, P.W.; Peet, N.P.; Weintraub, P.M.; Le, T.B.; Zhao, Z.; Barbone, F.; Cashman, B.; Tsay, J.; Dwyer, S.; Loos, P.C.; et al. The design and synthesis of purine inhibitors of CDK2. III. Nucleosides Nucleotides Nucleic Acids 2001, 20, 1067–1078. [Google Scholar]
- Online Demo-Corina. Available online: http://www.molecular-networks.com/online_demos/corina_demo accessed on 3 January 2009.
- Sybyl 8.0; Tripos; St. Louis, MO, USA, 2007.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | 1ACJ | 1ACL | 1AX9 | 1DX6 | 1EVE | 1GQR | 1VOT | 2ACE | 2BAG | 2CKM | 2VQ6 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1ACJ | 0 | 1.062 | 0.784 | 0.915 | 0.907 | 1.030 | 0.803 | 0.830 | 0.907 | 1.059 | 0.751 |
| 1ACL | 0 | 0.728 | 0.732 | 0.691 | 0.850 | 0.706 | 0.757 | 0.663 | 1.144 | 0.852 | |
| 1AX9 | 0 | 0.722 | 0.714 | 0.760 | 0.274 | 0.617 | 0.692 | 0.878 | 0.536 | ||
| 1DX6 | 0 | 0.366 | 0.711 | 0.720 | 0.511 | 0.318 | 0.934 | 0.614 | |||
| 1EVE | 0 | 0.634 | 0.710 | 0.488 | 0.236 | 0.975 | 0.641 | ||||
| 1GQR | 0 | 0.781 | 0.625 | 0.579 | 1.040 | 0.743 | |||||
| 1VOT | 0 | 0.594 | 0.692 | 0.915 | 0.581 | ||||||
| 2ACE | 0 | 0.443 | 0810 | 0.573 | |||||||
| 2BAG | 0 | 0.958 | 0.627 | ||||||||
| 2CKM | 0 | 0.844 | |||||||||
| 2VQ6 | 0 |
| Complex | Binding site-radius (Å) | Water molecules | Scoring function | Value of scoring function | rmsd (Å) |
|---|---|---|---|---|---|
| 1EVE | 10 | 1159, 1249, 1254 (toggle *) | ChemScore | 49.48 | 0.8 |
| 1ACJ | 12 | 616, 634, 643 (toggle) | GoldScore | 67.95 | 0.4 |
| 2CKM | 10 | None | GoldScore | 80.72 | 1.3 |
| 1DX6 | 12 | None | GoldScore | 59.34 | 0.6 |
| 1ACL | 10 | None | GoldScore | 40.45 | 1.6 |
| 1AX9 | 12 | None | ChemScore | 26.36 | 1.4 |
| 1VOT | 12 | 619, 680 (toggle) | ChemScore | 41.60 | 0.9 |
| 1EVE | 1ACJ | 2CKM | 1DX6 | 1ACL | 1AX9 | 1VOT | |
|---|---|---|---|---|---|---|---|
| donepezil | 0.8 | 9.3 | 10.3 | 2.6 | 3.4 | 2.9 | 9.6 |
| tacrine | 7.6 | 0.4 | 2.9 | 6.0 | 5.3 | 9.6 | 6.1 |
| bis-7-tacrine | 4.3 | 5.7 | 1.3 | 5.1 | 5.1 | 4.8 | 5.0 |
| galantamine | 1.1 | 3.7 | 0.8 | 0.6 | 6.2 | 8.7 | 4.9 |
| decamethonium | 1.4 | 3.2 | 2.3 | 2.5 | 1.6 | 3.2 | 2.4 |
| edrophonium | 4.5 | 2.8 | 2.2 | 2.3 | 2.4 | 1.4 | 2.0 |
| huperzine A | 5.0 | 5.0 | 3.5 | 3.6 | 5.0 | 3.7 | 0.9 |
| Compound | ChemScore AChE | IC50 AChE (nM) |
|---|---|---|
| 4 | 45.29 | 330 |
| 5 | 41.83 | 170 |
| 6 | 41.91 | 87.2 |
| 7 | 40.54 | 920 |
| 8 | 41.35 | 1070 |
| 9 | 38.42 | 1230 |
| donepezil | 49.48 | 31.2 |
| Compound | AChE | AChE | BuChE | BuChE |
|---|---|---|---|---|
| IC50 (μM) | 95% CI (μM) | IC50 (μM) | 95% CI (μM) | |
| 1 | 18.2 | 14.2–23.3 | 22.8 | 13.1–39.8 |
| 2 | 53.7 | 40.6–71.1 | 24.5 | 15.9–37.7 |
| 3 | 44.0 | 40.2–48.3 | 7.66 | 5.92–9.91 |
| 4 | 0.330 | 0.313–0.348 | n.a. | n.a. |
| 5 | 0.170 | 0.153–0.188 | n.a. | n.a. |
| 6 | 0.0872 | 0.0777–0.0980 | n.a. | n.a. |
| 7 | 0.922 | 0.772–1.100 | 67.5 | 59.9–76.1 |
| 8 | 1.07 | 0.88–1.30 | n.a | n.a. |
| 9 | 1.23 | 1.06–1.42 | n.a | n.a. |
| Donepezil | 0.0312 | 0.0274–0.0355 | 2.84 | 2.49–3.24 |
| Tacrine | 0.0503 | 0.0465–0.0543 | 0.00536 | 0.00439–0.00655 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bajda, M.; Więckowska, A.; Hebda, M.; Guzior, N.; Sotriffer, C.A.; Malawska, B. Structure-Based Search for New Inhibitors of Cholinesterases. Int. J. Mol. Sci. 2013, 14, 5608-5632. https://doi.org/10.3390/ijms14035608
Bajda M, Więckowska A, Hebda M, Guzior N, Sotriffer CA, Malawska B. Structure-Based Search for New Inhibitors of Cholinesterases. International Journal of Molecular Sciences. 2013; 14(3):5608-5632. https://doi.org/10.3390/ijms14035608
Chicago/Turabian StyleBajda, Marek, Anna Więckowska, Michalina Hebda, Natalia Guzior, Christoph A. Sotriffer, and Barbara Malawska. 2013. "Structure-Based Search for New Inhibitors of Cholinesterases" International Journal of Molecular Sciences 14, no. 3: 5608-5632. https://doi.org/10.3390/ijms14035608