Mitogen-Activated Protein (MAP) Kinase Scaffolding Proteins: A Recount


{kind=link}
{kind=link}
{kind=link}
Abstract
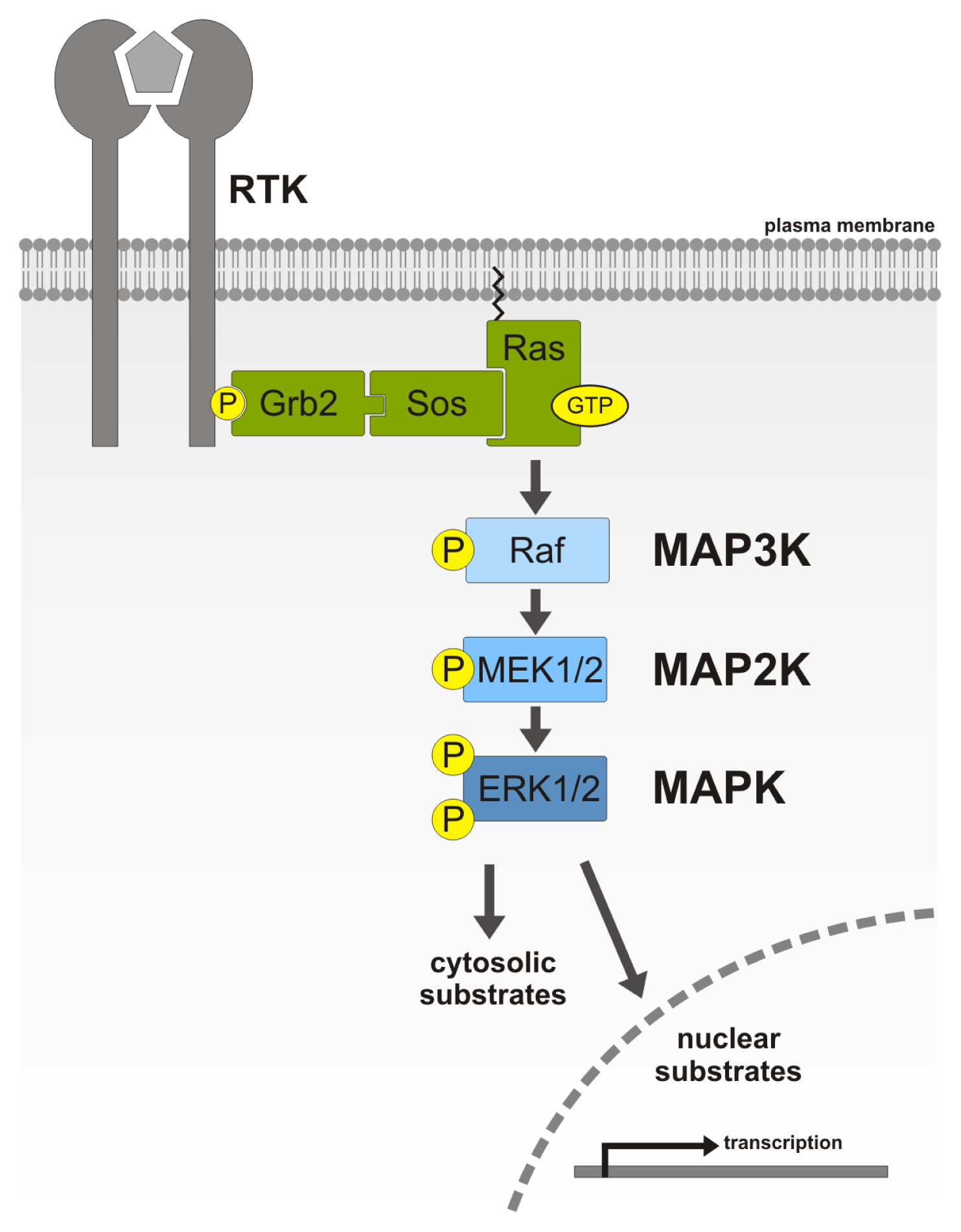
:1. MAP Kinase Signaling Cascade
1.1. Components of the MAPK/ERK Signaling Cascade
1.2. Function and Regulation of ERK
2. MAP Kinase Scaffolders
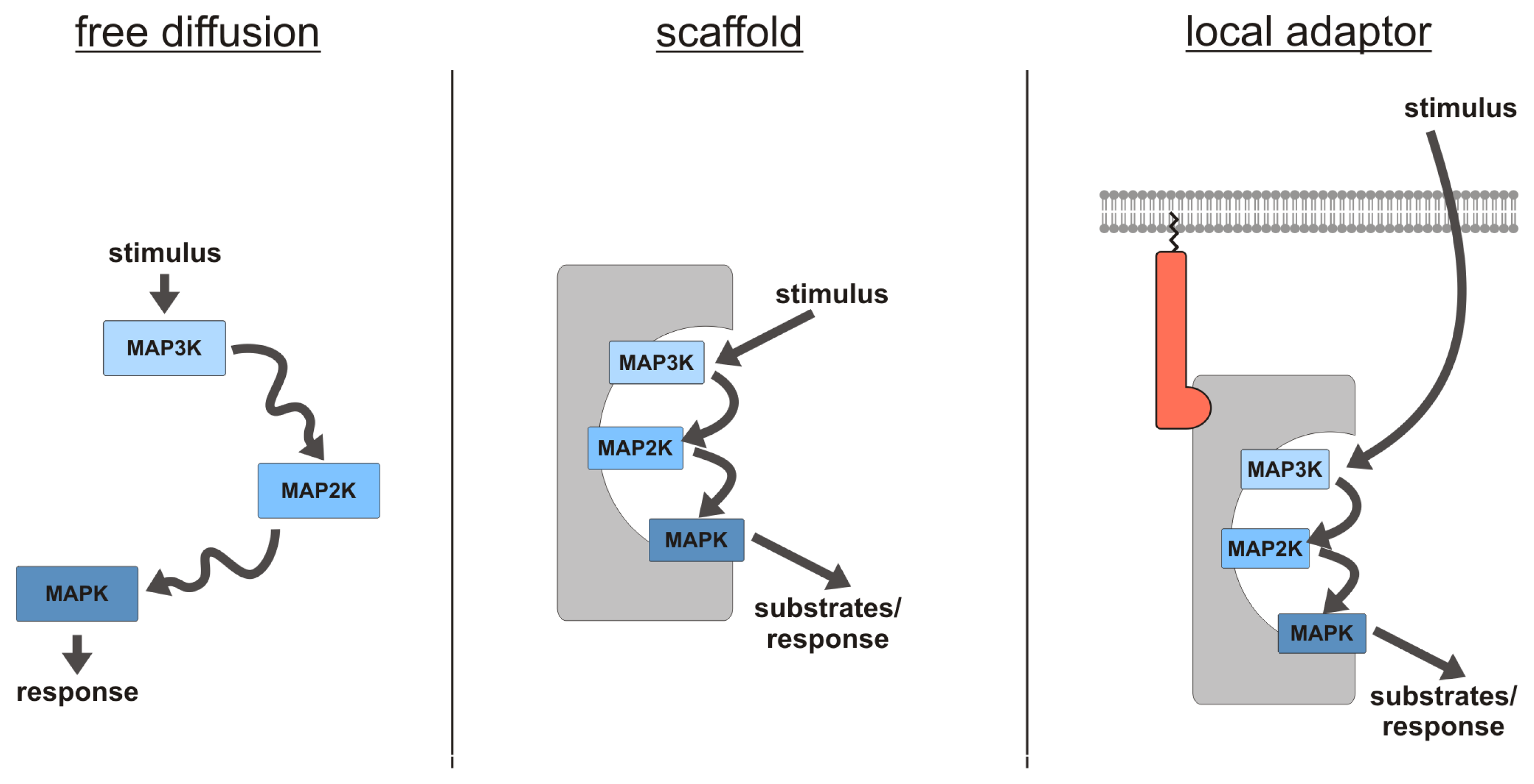
2.1. MAPK Scaffolding Proteins—An Introduction
2.2. Kinase Suppressor of Ras
2.3. MEK Partner 1/p14 Complex
2.4. β-Arrestins
2.5. Fibroblast Growth Factor Receptor Substrate 2
2.6. MAP Kinase Organizer 1
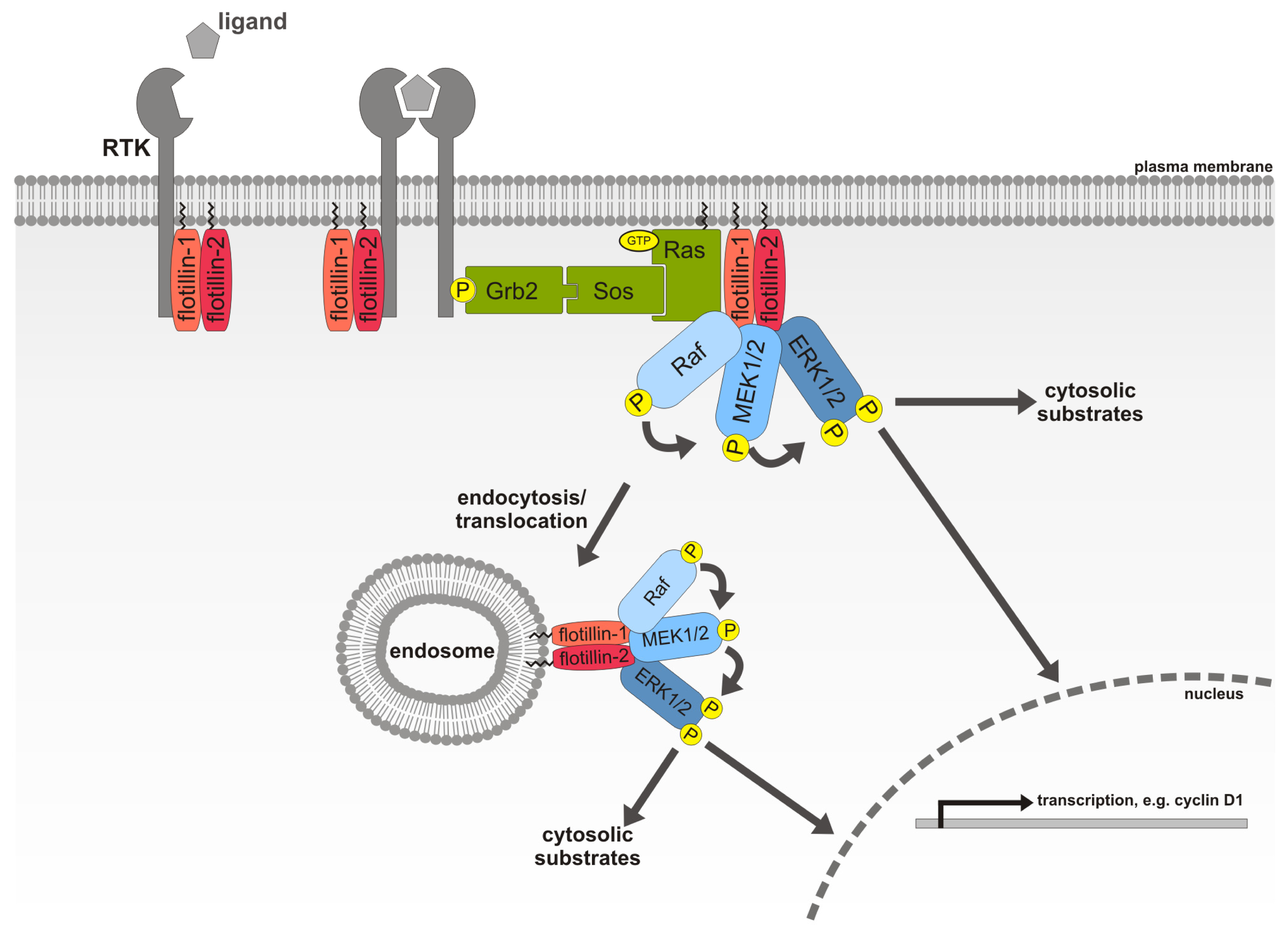
2.7. Flotillin-1, a Novel MAPK Scaffolding Protein
3. MAP Kinase Scaffolders: What Are They Good for?
Acknowledgments
Conflict of Interest
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar]
- Zhou, M.; Felder, S.; Rubinstein, M.; Hurwitz, D.R.; Ullrich, A.; Lax, I.; Schlessinger, J. Real-time measurements of kinetics of EGF binding to soluble EGF receptor monomers and dimers support the dimerization model for receptor activation. Biochemistry 1993, 32, 8193–8198. [Google Scholar]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar]
- Yarden, Y.; Schlessinger, J. Self-phosphorylation of epidermal growth factor receptor: Evidence for a model of intermolecular allosteric activation. Biochemistry 1987, 26, 1434–1442. [Google Scholar]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar]
- Egan, S.E.; Giddings, B.W.; Brooks, M.W.; Buday, L.; Sizeland, A.M.; Weinberg, R.A. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 1993, 363, 45–51. [Google Scholar]
- Li, N.; Batzer, A.; Daly, R.; Yajnik, V.; Skolnik, E.; Chardin, P.; Bar-Sagi, D.; Margolis, B.; Schlessinger, J. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 1993, 363, 85–88. [Google Scholar]
- Lowenstein, E.J.; Daly, R.J.; Batzer, A.G.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.Y.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar]
- Batzer, A.G.; Rotin, D.; Urena, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol 1994, 14, 5192–5201. [Google Scholar]
- Rozakis-Adcock, M.; Fernley, R.; Wade, J.; Pawson, T.; Bowtell, D. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 1993, 363, 83–85. [Google Scholar]
- Chardin, P.; Camonis, J.H.; Gale, N.W.; van Aelst, L.; Schlessinger, J.; Wigler, M.H.; Bar-Sagi, D. Human Sos1: A guanine nucleotide exchange factor for Ras that binds to GRB2. Science 1993, 260, 1338–1343. [Google Scholar]
- Medema, R.H.; de Vries-Smits, A.M.; van der Zon, G.C.; Maassen, J.A.; Bos, J.L. Ras activation by insulin and epidermal growth factor through enhanced exchange of guanine nucleotides on p21ras. Mol. Cell. Biol 1993, 13, 155–162. [Google Scholar]
- Satoh, T.; Endo, M.; Nakafuku, M.; Akiyama, T.; Yamamoto, T.; Kaziro, Y. Accumulation of p21ras. GTP in response to stimulation with epidermal growth factor and oncogene products with tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 1990, 87, 7926–7929. [Google Scholar]
- Wood, K.W.; Sarnecki, C.; Roberts, T.M.; Blenis, J. Ras mediates nerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell 1992, 68, 1041–1050. [Google Scholar]
- Van Aelst, L.; Barr, M.; Marcus, S.; Polverino, A.; Wigler, M. Complex formation between Ras and Raf and other protein kinases. Proc. Natl. Acad. Sci. USA 1993, 90, 6213–6217. [Google Scholar]
- Vojtek, A.B.; Hollenberg, S.M.; Cooper, J.A. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 1993, 74, 205–214. [Google Scholar]
- Warne, P.H.; Viciana, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355. [Google Scholar]
- Zhang, X.F.; Settleman, J.; Kyriakis, J.M.; Takeuchi-Suzuki, E.; Elledge, S.J.; Marshall, M.S.; Bruder, J.T.; Rapp, U.R.; Avruch, J. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993, 364, 308–313. [Google Scholar]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras. GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar]
- Leevers, S.J.; Paterson, H.F.; Marshall, C.J. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 1994, 369, 411–414. [Google Scholar]
- Marais, R.; Light, Y.; Paterson, H.F.; Marshall, C.J. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J 1995, 14, 3136–3145. [Google Scholar]
- Stokoe, D.; Macdonald, S.G.; Cadwallader, K.; Symons, M.; Hancock, J.F. Activation of Raf as a result of recruitment to the plasma membrane. Science 1994, 264, 1463–1467. [Google Scholar]
- Clark, G.J.; Drugan, J.K.; Rossman, K.L.; Carpenter, J.W.; Rogers-Graham, K.; Fu, H.; Der, C.J.; Campbell, S.L. 14-3-3 zeta negatively regulates raf-1 activity by interactions with the Raf-1 cysteine-rich domain. J. Biol. Chem 1997, 272, 20990–20993. [Google Scholar]
- Roy, S.; McPherson, R.A.; Apolloni, A.; Yan, J.; Lane, A.; Clyde-Smith, J.; Hancock, J.F. 14-3-3 facilitates Ras-dependent Raf-1 activation in vitro and in vivo. Mol. Cell. Biol 1998, 18, 3947–3955. [Google Scholar]
- Ueda, Y.; Hirai, S.; Osada, S.; Suzuki, A.; Mizuno, K.; Ohno, S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J. Biol. Chem 1996, 271, 23512–23519. [Google Scholar]
- Zou, Y.; Komuro, I.; Yamazaki, T.; Aikawa, R.; Kudoh, S.; Shiojima, I.; Hiroi, Y.; Mizuno, T.; Yazaki, Y. Protein kinase C, but not tyrosine kinases or Ras, plays a critical role in angiotensin II-induced activation of Raf-1 kinase and extracellular signal-regulated protein kinases in cardiac myocytes. J. Biol. Chem 1996, 271, 33592–33597. [Google Scholar]
- Schaap, D.; van der Wal, J.; Howe, L.R.; Marshall, C.J.; van Blitterswijk, W.J. A dominant-negative mutant of raf blocks mitogen-activated protein kinase activation by growth factors and oncogenic p21ras. J. Biol. Chem 1993, 268, 20232–20236. [Google Scholar]
- Ahn, N.G.; Krebs, E.G. Evidence for an epidermal growth factor-stimulated protein kinase cascade in Swiss 3T3 cells. Activation of serine peptide kinase activity by myelin basic protein kinases in vitro. J. Biol. Chem 1990, 265, 11495–11501. [Google Scholar]
- Alessi, D.R.; Saito, Y.; Campbell, D.G.; Cohen, P.; Sithanandam, G.; Rapp, U.; Ashworth, A.; Marshall, C.J.; Cowley, S. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J 1994, 13, 1610–1619. [Google Scholar]
- Gomez, N.; Cohen, P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 1991, 353, 170–173. [Google Scholar]
- Kyriakis, J.M.; App, H.; Zhang, X.F.; Banerjee, P.; Brautigan, D.L.; Rapp, U.R.; Avruch, J. Raf-1 activates MAP kinase-kinase. Nature 1992, 358, 417–421. [Google Scholar]
- Dhanasekaran, N.; Premkumar Reddy, E. Signaling by dual specificity kinases. Oncogene 1998, 17, 1447–1455. [Google Scholar]
- Ahn, N.G.; Seger, R.; Bratlien, R.L.; Diltz, C.D.; Tonks, N.K.; Krebs, E.G. Multiple components in an epidermal growth factor-stimulated protein kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J. Biol. Chem 1991, 266, 4220–4227. [Google Scholar]
- Crews, C.M.; Alessandrini, A.; Erikson, R.L. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science 1992, 258, 478–480. [Google Scholar]
- Payne, D.M.; Rossomando, A.J.; Martino, P.; Erickson, A.K.; Her, J.H.; Shabanowitz, J.; Hunt, D.F.; Weber, M.J.; Sturgill, T.W. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J 1991, 10, 885–892. [Google Scholar]
- Canagarajah, B.J.; Khokhlatchev, A.; Cobb, M.H.; Goldsmith, E.J. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 1997, 90, 859–869. [Google Scholar]
- Chen, R.H.; Sarnecki, C.; Blenis, J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell. Biol 1992, 12, 915–927. [Google Scholar]
- Khokhlatchev, A.V.; Canagarajah, B.; Wilsbacher, J.; Robinson, M.; Atkinson, M.; Goldsmith, E.; Cobb, M.H. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 1998, 93, 605–615. [Google Scholar]
- Kriegsheim, A.; Preisinger, C.; Kolch, W. Mapping of signaling pathways by functional interaction proteomics. Methods Mol. Biol 2008, 484, 177–192. [Google Scholar]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar]
- Rossomando, A.J.; Payne, D.M.; Weber, M.J.; Sturgill, T.W. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinase. Proc. Natl. Acad. Sci. USA 1989, 86, 6940–6943. [Google Scholar]
- Alvarez, E.; Northwood, I.C.; Gonzalez, F.A.; Latour, D.A.; Seth, A.; Abate, C.; Curran, T.; Davis, R.J. Pro-Leu-Ser/Thr-Pro is a consensus primary sequence for substrate protein phosphorylation. Characterization of the phosphorylation of c-myc and c-jun proteins by an epidermal growth factor receptor threonine 669 protein kinase. J. Biol. Chem 1991, 266, 15277–15285. [Google Scholar]
- Gonzalez, F.A.; Raden, D.L.; Davis, R.J. Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J. Biol. Chem 1991, 266, 22159–22163. [Google Scholar]
- Aebersold, D.M.; Shaul, Y.D.; Yung, Y.; Yarom, N.; Yao, Z.; Hanoch, T.; Seger, R. Extracellular signal-regulated kinase 1c (ERK1c), a novel 42-kilodalton ERK, demonstrates unique modes of regulation, localization, and function. Mol. Cell. Biol 2004, 24, 10000–10015. [Google Scholar]
- Yung, Y.; Yao, Z.; Hanoch, T.; Seger, R. ERK1b, a 46-kDa ERK isoform that is differentially regulated by MEK. J. Biol. Chem 2000, 275, 15799–15808. [Google Scholar]
- Fischer, A.M.; Katayama, C.D.; Pages, G.; Pouyssegur, J.; Hedrick, S.M. The role of erk1 and erk2 in multiple stages of T cell development. Immunity 2005, 23, 431–443. [Google Scholar]
- Lefloch, R.; Pouyssegur, J.; Lenormand, P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol. Cell. Biol 2008, 28, 511–527. [Google Scholar]
- Nekrasova, T.; Shive, C.; Gao, Y.; Kawamura, K.; Guardia, R.; Landreth, G.; Forsthuber, T.G. ERK1-deficient mice show normal T cell effector function and are highly susceptible to experimental autoimmune encephalomyelitis. J. Immunol 2005, 175, 2374–2380. [Google Scholar]
- Saba-El-Leil, M.K.; Vella, F.D.; Vernay, B.; Voisin, L.; Chen, L.; Labrecque, N.; Ang, S.L.; Meloche, S. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep 2003, 4, 964–968. [Google Scholar]
- Vantaggiato, C.; Formentini, I.; Bondanza, A.; Bonini, C.; Naldini, L.; Brambilla, R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J. Biol 2006, 5, 14. [Google Scholar]
- Voisin, L.; Saba-El-Leil, M.K.; Julien, C.; Fremin, C.; Meloche, S. Genetic demonstration of a redundant role of extracellular signal-regulated kinase 1 (ERK1) and ERK2 mitogen-activated protein kinases in promoting fibroblast proliferation. Mol. Cell. Biol 2010, 30, 2918–2932. [Google Scholar]
- Von Thun, A.; Birtwistle, M.; Kalna, G.; Grindlay, J.; Strachan, D.; Kolch, W.; von Kriegsheim, A.; Norman, J.C. ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-beta2. J. Cell Sci 2012, 125, 1465–1477. [Google Scholar]
- Pages, G.; Guerin, S.; Grall, D.; Bonino, F.; Smith, A.; Anjuere, F.; Auberger, P.; Pouyssegur, J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 1999, 286, 1374–1377. [Google Scholar]
- Yao, Y.; Li, W.; Wu, J.; Germann, U.A.; Su, M.S.; Kuida, K.; Boucher, D.M. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 12759–12764. [Google Scholar]
- Brunet, A.; Roux, D.; Lenormand, P.; Dowd, S.; Keyse, S.; Pouyssegur, J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J 1999, 18, 664–674. [Google Scholar]
- Casar, B.; Pinto, A.; Crespo, P. Essential role of ERK dimers in the activation of cytoplasmic but not nuclear substrates by ERK-scaffold complexes. Mol. Cell 2008, 31, 708–721. [Google Scholar]
- Wolf, I.; Rubinfeld, H.; Yoon, S.; Marmor, G.; Hanoch, T.; Seger, R. Involvement of the activation loop of ERK in the detachment from cytosolic anchoring. J. Biol. Chem 2001, 276, 24490–24497. [Google Scholar]
- Adachi, M.; Fukuda, M.; Nishida, E. Two co-existing mechanisms for nuclear import of MAP kinase: Passive diffusion of a monomer and active transport of a dimer. EMBO J 1999, 18, 5347–5358. [Google Scholar]
- Chuderland, D.; Konson, A.; Seger, R. Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol. Cell 2008, 31, 850–861. [Google Scholar]
- Kosako, H.; Yamaguchi, N.; Aranami, C.; Ushiyama, M.; Kose, S.; Imamoto, N.; Taniguchi, H.; Nishida, E.; Hattori, S. Phosphoproteomics reveals new ERK MAP kinase targets and links ERK to nucleoporin-mediated nuclear transport. Nat. Structr. Mol. Biol 2009, 16, 1026–1035. [Google Scholar]
- Lorenzen, J.A.; Baker, S.E.; Denhez, F.; Melnick, M.B.; Brower, D.L.; Perkins, L.A. Nuclear import of activated D-ERK by DIM-7, an importin family member encoded by the gene moleskin. Development 2001, 128, 1403–1414. [Google Scholar]
- Plotnikov, A.; Chuderland, D.; Karamansha, Y.; Livnah, O.; Seger, R. Nuclear extracellular signal-regulated kinase 1 and 2 translocation is mediated by casein kinase 2 and accelerated by autophosphorylation. Mol. Cell. Biol 2011, 31, 3515–3530. [Google Scholar]
- Eblen, S.T.; Kumar, N.V.; Shah, K.; Henderson, M.J.; Watts, C.K.; Shokat, K.M.; Weber, M.J. Identification of novel ERK2 substrates through use of an engineered kinase and ATP analogs. J. Biol. Chem 2003, 278, 14926–14935. [Google Scholar]
- Vomastek, T.; Iwanicki, M.P.; Burack, W.R.; Tiwari, D.; Kumar, D.; Parsons, J.T.; Weber, M.J.; Nandicoori, V.K. Extracellular signal-regulated kinase 2 (ERK2) phosphorylation sites and docking domain on the nuclear pore complex protein Tpr cooperatively regulate ERK2-Tpr interaction. Mol. Cell. Biol 2008, 28, 6954–6966. [Google Scholar]
- Cruzalegui, F.H.; Cano, E.; Treisman, R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene 1999, 18, 7948–7957. [Google Scholar]
- Gille, H.; Sharrocks, A.D.; Shaw, P.E. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature 1992, 358, 414–417. [Google Scholar]
- Marais, R.; Wynne, J.; Treisman, R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 1993, 73, 381–393. [Google Scholar]
- Rao, V.N.; Reddy, E.S. Elk-1 proteins interact with MAP kinases. Oncogene 1994, 9, 1855–1860. [Google Scholar]
- Chen, R.H.; Abate, C.; Blenis, J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 1993, 90, 10952–10956. [Google Scholar]
- Chen, R.H.; Juo, P.C.; Curran, T.; Blenis, J. Phosphorylation of c-Fos at the C-terminus enhances its transforming activity. Oncogene 1996, 12, 1493–1502. [Google Scholar]
- Ekerot, M.; Stavridis, M.P.; Delavaine, L.; Mitchell, M.P.; Staples, C.; Owens, D.M.; Keenan, I.D.; Dickinson, R.J.; Storey, K.G.; Keyse, S.M. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem. J 2008, 412, 287–298. [Google Scholar]
- Muda, M.; Boschert, U.; Dickinson, R.; Martinou, J.C.; Martinou, I.; Camps, M.; Schlegel, W.; Arkinstall, S. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J. Biol. Chem 1996, 271, 4319–4326. [Google Scholar]
- Zhang, Z.; Kobayashi, S.; Borczuk, A.C.; Leidner, R.S.; Laframboise, T.; Levine, A.D.; Halmos, B. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis 2010, 31, 577–586. [Google Scholar]
- Sgouras, D.N.; Athanasiou, M.A.; Beal, G.J., Jr; Fisher, R.J.; Blair, D.G.; Mavrothalassitis, G.J. ERF: An ETS domain protein with strong transcriptional repressor activity, can suppress ets-associated tumorigenesis and is regulated by phosphorylation during cell cycle and mitogenic stimulation. EMBO J. 1995, 14, 4781–4793. [Google Scholar]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar]
- Madak-Erdogan, Z.; Lupien, M.; Stossi, F.; Brown, M.; Katzenellenbogen, B.S. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol. Cell. Biol 2011, 31, 226–236. [Google Scholar]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 1996, 274, 2100–2103. [Google Scholar]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: A link to histone acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar]
- Drobic, B.; Perez-Cadahia, B.; Yu, J.; Kung, S.K.; Davie, J.R. Promoter chromatin remodeling of immediate-early genes is mediated through H3 phosphorylation at either serine 28 or 10 by the MSK1 multi-protein complex. Nucleic Acids Res 2010, 38, 3196–3208. [Google Scholar]
- Soloaga, A.; Thomson, S.; Wiggin, G.R.; Rampersaud, N.; Dyson, M.H.; Hazzalin, C.A.; Mahadevan, L.C.; Arthur, J.S. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J 2003, 22, 2788–2797. [Google Scholar]
- Vicent, G.P.; Ballare, C.; Nacht, A.S.; Clausell, J.; Subtil-Rodriguez, A.; Quiles, I.; Jordan, A.; Beato, M. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol. Cell 2006, 24, 367–381. [Google Scholar]
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharm. Sci 2007, 28, 556–560. [Google Scholar]
- Nada, S.; Hondo, A.; Kasai, A.; Koike, M.; Saito, K.; Uchiyama, Y.; Okada, M. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J 2009, 28, 477–489. [Google Scholar]
- Schaeffer, H.J.; Catling, A.D.; Eblen, S.T.; Collier, L.S.; Krauss, A.; Weber, M.J. MP1: A MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 1998, 281, 1668–1671. [Google Scholar]
- Wunderlich, W.; Fialka, I.; Teis, D.; Alpi, A.; Pfeifer, A.; Parton, R.G.; Lottspeich, F.; Huber, L.A. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J. Cell Biol 2001, 152, 765–776. [Google Scholar]
- Alonso, M.; Melani, M.; Converso, D.; Jaitovich, A.; Paz, C.; Carreras, M.C.; Medina, J.H.; Poderoso, J.J. Mitochondrial extracellular signal-regulated kinases 1/2 (ERK1/2) are modulated during brain development. J. Neurochem 2004, 89, 248–256. [Google Scholar]
- Galli, S.; Jahn, O.; Hitt, R.; Hesse, D.; Opitz, L.; Plessmann, U.; Urlaub, H.; Poderoso, J.J.; Jares-Erijman, E.A.; Jovin, T.M. A new paradigm for MAPK: Structural interactions of hERK1 with mitochondria in HeLa cells. PLoS One 2009, 4, e7541. [Google Scholar]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar]
- Asano, E.; Maeda, M.; Hasegawa, H.; Ito, S.; Hyodo, T.; Yuan, H.; Takahashi, M.; Hamaguchi, M.; Senga, T. Role of palladin phosphorylation by extracellular signal-regulated kinase in cell migration. PLoS One 2011, 6, e29338. [Google Scholar]
- Liu, Z.X.; Yu, C.F.; Nickel, C.; Thomas, S.; Cantley, L.G. Hepatocyte growth factor induces ERK-dependent paxillin phosphorylation and regulates paxillin-focal adhesion kinase association. J. Biol. Chem 2002, 277, 10452–10458. [Google Scholar]
- Romeo, Y.; Zhang, X.; Roux, P.P. Regulation and function of the RSK family of protein kinases. Biochem. J 2012, 441, 553–569. [Google Scholar]
- Sturgill, T.W.; Ray, L.B.; Erikson, E.; Maller, J.L. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature 1988, 334, 715–718. [Google Scholar]
- Brunet, A.; Pages, G.; Pouyssegur, J. Growth factor-stimulated MAP kinase induces rapid retrophosphorylation and inhibition of MAP kinase kinase (MEK1). FEBS Lett 1994, 346, 299–303. [Google Scholar]
- Mansour, S.J.; Resing, K.A.; Candi, J.M.; Hermann, A.S.; Gloor, J.W.; Herskind, K.R.; Wartmann, M.; Davis, R.J.; Ahn, N.G. Mitogen-activated protein (MAP) kinase phosphorylation of MAP kinase kinase: Determination of phosphorylation sites by mass spectrometry and site-directed mutagenesis. J. Biochem 1994, 116, 304–314. [Google Scholar]
- Eblen, S.T.; Slack-Davis, J.K.; Tarcsafalvi, A.; Parsons, J.T.; Weber, M.J.; Catling, A.D. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol. Cell. Biol 2004, 24, 2308–2317. [Google Scholar]
- Catalanotti, F.; Reyes, G.; Jesenberger, V.; Galabova-Kovacs, G.; de Matos Simoes, R.; Carugo, O.; Baccarini, M. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat. Structr. Mol. Biol 2009, 16, 294–303. [Google Scholar]
- Corbalan-Garcia, S.; Yang, S.S.; Degenhardt, K.R.; Bar-Sagi, D. Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol. Cell. Biol 1996, 16, 5674–5682. [Google Scholar]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar]
- Brondello, J.M.; Brunet, A.; Pouyssegur, J.; McKenzie, F.R. The dual specificity mitogen-activated protein kinase phosphatase-1 and −2 are induced by the p42/p44MAPK cascade. J. Biol. Chem 1997, 272, 1368–1376. [Google Scholar]
- Brondello, J.M.; Pouyssegur, J.; McKenzie, F.R. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 1999, 286, 2514–2517. [Google Scholar]
- Marchetti, S.; Gimond, C.; Chambard, J.C.; Touboul, T.; Roux, D.; Pouyssegur, J.; Pages, G. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell. Biol 2005, 25, 854–864. [Google Scholar]
- Furthauer, M.; Lin, W.; Ang, S.L.; Thisse, B.; Thisse, C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell. Biol 2002, 4, 170–174. [Google Scholar]
- Torii, S.; Kusakabe, M.; Yamamoto, T.; Maekawa, M.; Nishida, E. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev. Cell 2004, 7, 33–44. [Google Scholar]
- Tsang, M.; Friesel, R.; Kudoh, T.; Dawid, I.B. Identification of Sef, a novel modulator of FGF signalling. Nat. Cell. Biol 2002, 4, 165–169. [Google Scholar]
- Heinrich, R.; Neel, B.G.; Rapoport, T.A. Mathematical models of protein kinase signal transduction. Mol. Cell 2002, 9, 957–970. [Google Scholar]
- Locasale, J.W.; Shaw, A.S.; Chakraborty, A.K. Scaffold proteins confer diverse regulatory properties to protein kinase cascades. Proc. Natl. Acad. Sci. USA 2007, 104, 13307–13312. [Google Scholar]
- Yu, W.; Fantl, W.J.; Harrowe, G.; Williams, L.T. Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr. Biol 1998, 8, 56–64. [Google Scholar]
- Ferrell, J.E., Jr. What do scaffold proteins really do? Sci. STKE 2000, 2000, pe1. [Google Scholar]
- Levchenko, A.; Bruck, J.; Sternberg, P.W. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. USA 2000, 97, 5818–5823. [Google Scholar]
- Dard, N.; Peter, M. Scaffold proteins in MAP kinase signaling: More than simple passive activating platforms. Bioessays 2006, 28, 146–156. [Google Scholar]
- Elion, E.A. The Ste5p scaffold. J. Cell Sci 2001, 114, 3967–3978. [Google Scholar]
- Engstrom, W.; Ward, A.; Moorwood, K. The role of scaffold proteins in JNK signalling. Cell Prolif 2010, 43, 56–66. [Google Scholar]
- Saito, H. Regulation of cross-talk in yeast MAPK signaling pathways. Curr. Opin. Microbiol 2010, 13, 677–683. [Google Scholar]
- Dhanasekaran, D.N.; Kashef, K.; Lee, C.M.; Xu, H.; Reddy, E.P. Scaffold proteins of MAP-kinase modules. Oncogene 2007, 26, 3185–3202. [Google Scholar]
- Sbroggio, M.; Carnevale, D.; Bertero, A.; Cifelli, G.; De Blasio, E.; Mascio, G.; Hirsch, E.; Bahou, W.F.; Turco, E.; Silengo, L.; et al. IQGAP1 regulates ERK1/2 and AKT signalling in the heart and sustains functional remodelling upon pressure overload. Cardiovasc. Res 2011, 91, 456–464. [Google Scholar]
- Ren, J.G.; Li, Z.; Sacks, D.B. IQGAP1 modulates activation of B-Raf. Proc. Natl. Acad. Sci. USA 2007, 104, 10465–10469. [Google Scholar]
- Roy, M.; Li, Z.; Sacks, D.B. IQGAP1 binds ERK2 and modulates its activity. J. Biol. Chem 2004, 279, 17329–17337. [Google Scholar]
- Roy, M.; Li, Z.; Sacks, D.B. IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol. Cell. Biol 2005, 25, 7940–7952. [Google Scholar]
- Kornfeld, K.; Hom, D.B.; Horvitz, H.R. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 1995, 83, 903–913. [Google Scholar]
- Sundaram, M.; Han, M. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 1995, 83, 889–901. [Google Scholar]
- Therrien, M.; Chang, H.C.; Solomon, N.M.; Karim, F.D.; Wassarman, D.A.; Rubin, G.M. KSR, a novel protein kinase required for RAS signal transduction. Cell 1995, 83, 879–888. [Google Scholar]
- Therrien, M.; Michaud, N.R.; Rubin, G.M.; Morrison, D.K. KSR modulates signal propagation within the MAPK cascade. Genes Dev 1996, 10, 2684–2695. [Google Scholar]
- Channavajhala, P.L.; Wu, L.; Cuozzo, J.W.; Hall, J.P.; Liu, W.; Lin, L.L.; Zhang, Y. Identification of a novel human kinase supporter of Ras (hKSR-2) that functions as a negative regulator of Cot (Tpl2) signaling. J. Biol. Chem 2003, 278, 47089–47097. [Google Scholar]
- Ohmachi, M.; Rocheleau, C.E.; Church, D.; Lambie, E.; Schedl, T.; Sundaram, M.V. C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr. Biol 2002, 12, 427–433. [Google Scholar]
- Klutho, P.J.; Costanzo-Garvey, D.L.; Lewis, R.E. Regulation of glucose homeostasis by KSR1 and MARK2. PLoS One 2011, 6, e29304. [Google Scholar]
- Lozano, J.; Xing, R.; Cai, Z.; Jensen, H.L.; Trempus, C.; Mark, W.; Cannon, R.; Kolesnick, R. Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res 2003, 63, 4232–4238. [Google Scholar]
- Nguyen, A.; Burack, W.R.; Stock, J.L.; Kortum, R.; Chaika, O.V.; Afkarian, M.; Muller, W.J.; Murphy, K.M.; Morrison, D.K.; Lewis, R.E.; et al. Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol. Cell. Biol 2002, 22, 3035–3045. [Google Scholar]
- Shalin, S.C.; Hernandez, C.M.; Dougherty, M.K.; Morrison, D.K.; Sweatt, J.D. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron 2006, 50, 765–779. [Google Scholar]
- Fusello, A.M.; Mandik-Nayak, L.; Shih, F.; Lewis, R.E.; Allen, P.M.; Shaw, A.S. The MAPK scaffold kinase suppressor of Ras is involved in ERK activation by stress and proinflammatory cytokines and induction of arthritis. J. Immunol 2006, 177, 6152–6158. [Google Scholar]
- Kortum, R.L.; Johnson, H.J.; Costanzo, D.L.; Volle, D.J.; Razidlo, G.L.; Fusello, A.M.; Shaw, A.S.; Lewis, R.E. The molecular scaffold kinase suppressor of Ras 1 is a modifier of RasV12-induced and replicative senescence. Mol. Cell. Biol 2006, 26, 2202–2214. [Google Scholar]
- Morrison, D.K. KSR: A MAPK scaffold of the Ras pathway? J. Cell Sci 2001, 114, 1609–1612. [Google Scholar]
- Muller, J.; Ory, S.; Copeland, T.; Piwnica-Worms, H.; Morrison, D.K. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol. Cell 2001, 8, 983–993. [Google Scholar]
- Roy, F.; Therrien, M. MAP kinase module: The Ksr connection. Curr. Biol 2002, 12, R325–R327. [Google Scholar]
- Michaud, N.R.; Therrien, M.; Cacace, A.; Edsall, L.C.; Spiegel, S.; Rubin, G.M.; Morrison, D.K. KSR stimulates Raf-1 activity in a kinase-independent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 12792–12796. [Google Scholar]
- Channavajhala, P.L.; Rao, V.R.; Spaulding, V.; Lin, L.L.; Zhang, Y.G. hKSR-2 inhibits MEKK3-activated MAP kinase and NF-kappaB pathways in inflammation. Biochem. Biophys. Res. Commun 2005, 334, 1214–1218. [Google Scholar]
- Denouel-Galy, A.; Douville, E.M.; Warne, P.H.; Papin, C.; Laugier, D.; Calothy, G.; Downward, J.; Eychene, A. Murine Ksr interacts with MEK and inhibits Ras-induced transformation. Curr. Biol 1998, 8, 46–55. [Google Scholar]
- Roy, S.; Wyse, B.; Hancock, J.F. H-Ras signaling and K-Ras signaling are differentially dependent on endocytosis. Mol. Cell. Biol 2002, 22, 5128–5140. [Google Scholar]
- Boudeau, J.; Miranda-Saavedra, D.; Barton, G.J.; Alessi, D.R. Emerging roles of pseudokinases. Trends Cell Biol 2006, 16, 443–452. [Google Scholar]
- Goettel, J.A.; Liang, D.; Hilliard, V.C.; Edelblum, K.L.; Broadus, M.R.; Gould, K.L.; Hanks, S.K.; Polk, D.B. KSR1 is a functional protein kinase capable of serine autophosphorylation and direct phosphorylation of MEK1. Exp. Cell. Res 2011, 317, 452–463. [Google Scholar]
- Sugimoto, T.; Stewart, S.; Han, M.; Guan, K.L. The kinase suppressor of Ras (KSR) modulates growth factor and Ras signaling by uncoupling Elk-1 phosphorylation from MAP kinase activation. EMBO J 1998, 17, 1717–1727. [Google Scholar]
- Yan, F.; Polk, D.B. Kinase suppressor of ras is necessary for tumor necrosis factor alpha activation of extracellular signal-regulated kinase/mitogen-activated protein kinase in intestinal epithelial cells. Cancer Res 2001, 61, 963–969. [Google Scholar]
- Robinson, M.J.; Harkins, P.C.; Zhang, J.; Baer, R.; Haycock, J.W.; Cobb, M.H.; Goldsmith, E.J. Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5′-triphosphate. Biochemistry 1996, 35, 5641–5646. [Google Scholar]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar]
- Brennan, D.F.; Dar, A.C.; Hertz, N.T.; Chao, W.C.; Burlingame, A.L.; Shokat, K.M.; Barford, D. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 2011, 472, 366–369. [Google Scholar]
- Hu, J.; Yu, H.; Kornev, A.P.; Zhao, J.; Filbert, E.L.; Taylor, S.S.; Shaw, A.S. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. USA 2011, 108, 6067–6072. [Google Scholar]
- Ory, S.; Zhou, M.; Conrads, T.P.; Veenstra, T.D.; Morrison, D.K. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr. Biol 2003, 13, 1356–1364. [Google Scholar]
- Matheny, S.A.; White, M.A. Signaling threshold regulation by the Ras effector IMP. J. Biol. Chem 2009, 284, 11007–11011. [Google Scholar]
- Chen, C.; Lewis, R.E.; White, M.A. IMP modulates KSR1-dependent multivalent complex formation to specify ERK1/2 pathway activation and response thresholds. J. Biol. Chem 2008, 283, 12789–12796. [Google Scholar]
- Howe, C.L.; Valletta, J.S.; Rusnak, A.S.; Mobley, W.C. NGF signaling from clathrin-coated vesicles: Evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron 2001, 32, 801–814. [Google Scholar]
- Jiang, X.; Sorkin, A. Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol. Biol. Cell 2002, 13, 1522–1535. [Google Scholar]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar]
- Wu, C.; Lai, C.F.; Mobley, W.C. Nerve growth factor activates persistent Rap1 signaling in endosomes. J. Neurosci 2001, 21, 5406–5416. [Google Scholar]
- Teis, D.; Wunderlich, W.; Huber, L.A. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev. Cell 2002, 3, 803–814. [Google Scholar]
- Lunin, V.V.; Munger, C.; Wagner, J.; Ye, Z.; Cygler, M.; Sacher, M. The structure of the MAPK scaffold, MP1, bound to its partner, p14. A complex with a critical role in endosomal map kinase signaling. J. Biol. Chem 2004, 279, 23422–23430. [Google Scholar]
- Bohn, G.; Allroth, A.; Brandes, G.; Thiel, J.; Glocker, E.; Schaffer, A.A.; Rathinam, C.; Taub, N.; Teis, D.; Zeidler, C.; et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat. Med 2007, 13, 38–45. [Google Scholar]
- Teis, D.; Taub, N.; Kurzbauer, R.; Hilber, D.; de Araujo, M.E.; Erlacher, M.; Offterdinger, M.; Villunger, A.; Geley, S.; Bohn, G.; et al. p14-MP1-MEK1 signaling regulates endosomal traffic and cellular proliferation during tissue homeostasis. J. Cell Biol 2006, 175, 861–868. [Google Scholar]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar]
- Ferguson, S.S.; Downey, W.E., 3rd; Colapietro, A.M.; Barak, L.S.; Menard, L.; Caron, M.G. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 1996, 271, 363–366. [Google Scholar]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu. Rev. Biochem 1998, 67, 653–692. [Google Scholar]
- Kovacs, J.J.; Hara, M.R.; Davenport, C.L.; Kim, J.; Lefkowitz, R.J. Arrestin development: Emerging roles for beta-arrestins in developmental signaling pathways. Dev. Cell 2009, 17, 443–458. [Google Scholar]
- Gurevich, E.V.; Gurevich, V.V. Arrestins: Ubiquitous regulators of cellular signaling pathways. Genome Biol 2006, 7, 236. [Google Scholar]
- Martin, N.P.; Lefkowitz, R.J.; Shenoy, S.K. Regulation of V2 vasopressin receptor degradation by agonist-promoted ubiquitination. J. Biol. Chem 2003, 278, 45954–45959. [Google Scholar]
- Shenoy, S.K.; Lefkowitz, R.J. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta-arrestin deubiquitination. J. Biol. Chem 2003, 278, 14498–14506. [Google Scholar]
- Shenoy, S.K.; McDonald, P.H.; Kohout, T.A.; Lefkowitz, R.J. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 2001, 294, 1307–1313. [Google Scholar]
- Shenoy, S.K.; Xiao, K.; Venkataramanan, V.; Snyder, P.M.; Freedman, N.J.; Weissman, A.M. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the beta2-adrenergic receptor. J. Biol. Chem 2008, 283, 22166–22176. [Google Scholar]
- Shenoy, S.K.; Barak, L.S.; Xiao, K.; Ahn, S.; Berthouze, M.; Shukla, A.K.; Luttrell, L.M.; Lefkowitz, R.J. Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J. Biol. Chem 2007, 282, 29549–29562. [Google Scholar]
- Ma, L.; Pei, G. Beta-arrestin signaling and regulation of transcription. J. Cell Sci 2007, 120, 213–218. [Google Scholar]
- Shenoy, S.K.; Drake, M.T.; Nelson, C.D.; Houtz, D.A.; Xiao, K.; Madabushi, S.; Reiter, E.; Premont, R.T.; Lichtarge, O.; Lefkowitz, R.J. Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem 2006, 281, 1261–1273. [Google Scholar]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol 2007, 69, 483–510. [Google Scholar]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Dery, O.; Mullins, R.D.; Bunnett, N.W. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol 2000, 148, 1267–1281. [Google Scholar]
- Lin, F.T.; Miller, W.E.; Luttrell, L.M.; Lefkowitz, R.J. Feedback regulation of beta-arrestin1 function by extracellular signal-regulated kinases. J. Biol. Chem 1999, 274, 15971–15974. [Google Scholar]
- Lin, F.T.; Krueger, K.M.; Kendall, H.E.; Daaka, Y.; Fredericks, Z.L.; Pitcher, J.A.; Lefkowitz, R.J. Clathrin-mediated endocytosis of the beta-adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-arrestin1. J. Biol. Chem 1997, 272, 31051–31057. [Google Scholar]
- McDonald, P.H.; Chow, C.W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. Beta-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar]
- Conner, D.A.; Mathier, M.A.; Mortensen, R.M.; Christe, M.; Vatner, S.F.; Seidman, C.E.; Seidman, J.G. Beta-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to beta-adrenergic stimulation. Circ Res 1997, 81, 1021–1026. [Google Scholar]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar]
- Kohout, T.A.; Lin, F.S.; Perry, S.J.; Conner, D.A.; Lefkowitz, R.J. beta-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. USA 2001, 98, 1601–1606. [Google Scholar]
- Kouhara, H.; Hadari, Y.R.; Spivak-Kroizman, T.; Schilling, J.; Bar-Sagi, D.; Lax, I.; Schlessinger, J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 1997, 89, 693–702. [Google Scholar]
- Delahaye, L.; Rocchi, S.; Van Obberghen, E. Potential involvement of FRS2 in insulin signaling. Endocrinology 2000, 141, 621–628. [Google Scholar]
- Hadari, Y.R.; Gotoh, N.; Kouhara, H.; Lax, I.; Schlessinger, J. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 8578–8583. [Google Scholar]
- Kurokawa, K.; Iwashita, T.; Murakami, H.; Hayashi, H.; Kawai, K.; Takahashi, M. Identification of SNT/FRS2 docking site on RET receptor tyrosine kinase and its role for signal transduction. Oncogene 2001, 20, 1929–1938. [Google Scholar]
- Meakin, S.O.; MacDonald, J.I.; Gryz, E.A.; Kubu, C.J.; Verdi, J.M. The signaling adapter FRS-2 competes with Shc for binding to the nerve growth factor receptor TrkA. A model for discriminating proliferation and differentiation. J. Biol. Chem 1999, 274, 9861–9870. [Google Scholar]
- Melillo, R.M.; Santoro, M.; Ong, S.H.; Billaud, M.; Fusco, A.; Hadari, Y.R.; Schlessinger, J.; Lax, I. Docking protein FRS2 links the protein tyrosine kinase RET and its oncogenic forms with the mitogen-activated protein kinase signaling cascade. Mol. Cell. Biol 2001, 21, 4177–4187. [Google Scholar]
- Yamada, S.; Taketomi, T.; Yoshimura, A. Model analysis of difference between EGF pathway and FGF pathway. Biochem. Biophys. Res. Commun 2004, 314, 1113–1120. [Google Scholar]
- Ong, S.H.; Guy, G.R.; Hadari, Y.R.; Laks, S.; Gotoh, N.; Schlessinger, J.; Lax, I. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol. Cell. Biol 2000, 20, 979–989. [Google Scholar]
- Dhalluin, C.; Yan, K.S.; Plotnikova, O.; Lee, K.W.; Zeng, L.; Kuti, M.; Mujtaba, S.; Goldfarb, M.P.; Zhou, M.M. Structural basis of SNT PTB domain interactions with distinct neurotrophic receptors. Mol. Cell 2000, 6, 921–929. [Google Scholar]
- Easton, J.B.; Royer, A.R.; Middlemas, D.S. The protein tyrosine phosphatase, Shp2, is required for the complete activation of the RAS/MAPK pathway by brain-derived neurotrophic factor. J. Neurochem 2006, 97, 834–845. [Google Scholar]
- Xu, H.; Lee, K.W.; Goldfarb, M. Novel recognition motif on fibroblast growth factor receptor mediates direct association and activation of SNT adapter proteins. J. Biol. Chem 1998, 273, 17987–17990. [Google Scholar]
- Hadari, Y.R.; Kouhara, H.; Lax, I.; Schlessinger, J. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol. Cell. Biol 1998, 18, 3966–3973. [Google Scholar]
- Ong, S.H.; Hadari, Y.R.; Gotoh, N.; Guy, G.R.; Schlessinger, J.; Lax, I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 6074–6079. [Google Scholar]
- Wong, A.; Lamothe, B.; Lee, A.; Schlessinger, J.; Lax, I. FRS2 alpha attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc. Natl. Acad. Sci. USA 2002, 99, 6684–6689. [Google Scholar]
- McDougall, K.; Kubu, C.; Verdi, J.M.; Meakin, S.O. Developmental expression patterns of the signaling adapters FRS-2 and FRS-3 during early embryogenesis. Mech. Dev 2001, 103, 145–148. [Google Scholar]
- Gotoh, N.; Laks, S.; Nakashima, M.; Lax, I.; Schlessinger, J. FRS2 family docking proteins with overlapping roles in activation of MAP kinase have distinct spatial-temporal patterns of expression of their transcripts. FEBS Lett 2004, 564, 14–18. [Google Scholar]
- Vomastek, T.; Schaeffer, H.J.; Tarcsafalvi, A.; Smolkin, M.E.; Bissonette, E.A.; Weber, M.J. Modular construction of a signaling scaffold: MORG1 interacts with components of the ERK cascade and links ERK signaling to specific agonists. Proc. Natl. Acad. Sci. USA 2004, 101, 6981–6986. [Google Scholar]
- Hopfer, U.; Hopfer, H.; Jablonski, K.; Stahl, R.A.; Wolf, G. The novel WD-repeat protein Morg1 acts as a molecular scaffold for hypoxia-inducible factor prolyl hydroxylase 3 (PHD3). J. Biol. Chem 2006, 281, 8645–8655. [Google Scholar]
- Haase, D.; Keiner, S.; Mawrin, C.; Wolf, G. Reduced Morg1 expression in ischemic human brain. Neurosci. Lett 2009, 455, 46–50. [Google Scholar]
- Hammerschmidt, E.; Loeffler, I.; Wolf, G. Morg1 heterozygous mice are protected from acute renal ischemia-reperfusion injury. Am. J. Physiol. Renal. Physiol 2009, 297, F1273–F1287. [Google Scholar]
- Stahr, A.; Frahm, C.; Kretz, A.; Bondeva, T.; Witte, O.W.; Wolf, G. Morg1(+/−) heterozygous mice are protected from experimentally induced focal cerebral ischemia. Brain Res 2012, 1482, 22–31. [Google Scholar]
- Simons, K.; Gerl, M.J. Revitalizing membrane rafts: New tools and insights. Nat. Rev 2010, 11, 688–699. [Google Scholar]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harbor Perspect. Biol 2011, 3, a004697. [Google Scholar]
- Bickel, P.E.; Scherer, P.E.; Schnitzer, J.E.; Oh, P.; Lisanti, M.P.; Lodish, H.F. Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J. Biol. Chem 1997, 272, 13793–13802. [Google Scholar]
- Schulte, T.; Paschke, K.A.; Laessing, U.; Lottspeich, F.; Stuermer, C.A. Reggie-1 and reggie-2, two cell surface proteins expressed by retinal ganglion cells during axon regeneration. Development 1997, 124, 577–587. [Google Scholar]
- Banning, A.; Tomasovic, A.; Tikkanen, R. Functional aspects of membrane association of reggie/flotillin proteins. Curr. Protein Peptide Sci 2011, 12, 725–735. [Google Scholar]
- Babuke, T.; Ruonala, M.; Meister, M.; Amaddii, M.; Genzler, C.; Esposito, A.; Tikkanen, R. Hetero-oligomerization of reggie-1/flotillin-2 and reggie-2/flotillin-1 is required for their endocytosis. Cell. Signal 2009, 21, 1287–1297. [Google Scholar]
- Babuke, T.; Tikkanen, R. Dissecting the molecular function of reggie/flotillin proteins. Eur. J. Cell Biol 2007, 86, 525–532. [Google Scholar]
- Frick, M.; Bright, N.A.; Riento, K.; Bray, A.; Merrified, C.; Nichols, B.J. Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr. Biol 2007, 17, 1151–1156. [Google Scholar]
- Morrow, I.C.; Rea, S.; Martin, S.; Prior, I.A.; Prohaska, R.; Hancock, J.F.; James, D.E.; Parton, R.G. Flotillin-1/reggie-2 traffics to surface raft domains via a novel golgi-independent pathway. Identification of a novel membrane targeting domain and a role for palmitoylation. J. Biol. Chem 2002, 277, 48834–48841. [Google Scholar]
- Neumann-Giesen, C.; Falkenbach, B.; Beicht, P.; Claasen, S.; Luers, G.; Stuermer, C.A.; Herzog, V.; Tikkanen, R. Membrane and raft association of reggie-1/flotillin-2: Role of myristoylation, palmitoylation and oligomerization and induction of filopodia by overexpression. Biochem. J 2004, 378, 509–518. [Google Scholar]
- Neumann-Giesen, C.; Fernow, I.; Amaddii, M.; Tikkanen, R. Role of EGF-induced tyrosine phosphorylation of reggie-1/flotillin-2 in cell spreading and signaling to the actin cytoskeleton. J. Cell Sci 2007, 120, 395–406. [Google Scholar]
- Solis, G.P.; Hoegg, M.; Munderloh, C.; Schrock, Y.; Malaga-Trillo, E.; Rivera-Milla, E.; Stuermer, C.A. Reggie/flotillin proteins are organized into stable tetramers in membrane microdomains. Biochem. J 2007, 403, 313–322. [Google Scholar]
- Dermine, J.F.; Duclos, S.; Garin, J.; St-Louis, F.; Rea, S.; Parton, R.G.; Desjardins, M. Flotillin-1-enriched lipid raft domains accumulate on maturing phagosomes. J. Biol. Chem 2001, 276, 18507–18512. [Google Scholar]
- Glebov, O.O.; Bright, N.A.; Nichols, B.J. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat. Cell. Biol 2006, 8, 46–54. [Google Scholar]
- Santamaria, A.; Castellanos, E.; Gomez, V.; Benedit, P.; Renau-Piqueras, J.; Morote, J.; Reventos, J.; Thomson, T.M.; Paciucci, R. PTOV1 enables the nuclear translocation and mitogenic activity of flotillin-1, a major protein of lipid rafts. Mol. Cell. Biol 2005, 25, 1900–1911. [Google Scholar]
- Stuermer, C.A.; Lang, D.M.; Kirsch, F.; Wiechers, M.; Deininger, S.O.; Plattner, H. Glycosylphosphatidyl inositol-anchored proteins and fyn kinase assemble in noncaveolar plasma membrane microdomains defined by reggie-1 and −2. Mol. Biol. Cell 2001, 12, 3031–3045. [Google Scholar]
- Raemaekers, T.; Peric, A.; Baatsen, P.; Sannerud, R.; Declerck, I.; Baert, V.; Michiels, C.; Annaert, W. ARF6-mediated endosomal transport of Telencephalin affects dendritic filopodia-to-spine maturation. EMBO J 2012, 31, 3252–3269. [Google Scholar]
- Banning, A.; Ockenga, W.; Finger, F.; Siebrasse, P.; Tikkanen, R. Transcriptional regulation of flotillins by the extracellularly regulated kinases and retinoid X receptor complexes. PLoS One 2012, 7, e45514. [Google Scholar]
- Amaddii, M.; Meister, M.; Banning, A.; Tomasovic, A.; Mooz, J.; Rajalingam, K.; Tikkanen, R. Flotillin-1/reggie-2 protein plays dual role in activation of receptor-tyrosine kinase/mitogen-activated protein kinase signaling. J. Biol. Chem 2012, 287, 7265–7278. [Google Scholar]
- Lin, C.; Wu, Z.; Lin, X.; Yu, C.; Shi, T.; Zeng, Y.; Wang, X.; Li, J.; Song, L. Knockdown of FLOT1 impairs cell proliferation and tumorigenicity in breast cancer through upregulation of FOXO3a. Clin. Cancer Res 2011, 17, 3089–3099. [Google Scholar]
- Cremona, M.L.; Matthies, H.J.; Pau, K.; Bowton, E.; Speed, N.; Lute, B.J.; Anderson, M.; Sen, N.; Robertson, S.D.; Vaughan, R.A.; et al. Flotillin-1 is essential for PKC-triggered endocytosis and membrane microdomain localization of DAT. Nat. Neurosci 2011, 14, 469–477. [Google Scholar]
- Langhorst, M.F.; Solis, G.P.; Hannbeck, S.; Plattner, H.; Stuermer, C.A. Linking membrane microdomains to the cytoskeleton: Regulation of the lateral mobility of reggie-1/flotillin-2 by interaction with actin. FEBS Lett 2007, 581, 4697–4703. [Google Scholar]
- Hazarika, P.; McCarty, M.F.; Prieto, V.G.; George, S.; Babu, D.; Koul, D.; Bar-Eli, M.; Duvic, M. Up-regulation of Flotillin-2 is associated with melanoma progression and modulates expression of the thrombin receptor protease activated receptor 1. Cancer Res 2004, 64, 7361–7369. [Google Scholar]
- Kato, N.; Nakanishi, M.; Hirashima, N. Flotillin-1 regulates IgE receptor-mediated signaling in rat basophilic leukemia (RBL-2H3) cells. J. Immunol 2006, 177, 147–154. [Google Scholar]
- Limpert, A.S.; Karlo, J.C.; Landreth, G.E. Nerve growth factor stimulates the concentration of TrkA within lipid rafts and extracellular signal-regulated kinase activation through c-Cbl-associated protein. Mol. Cell. Biol 2007, 27, 5686–5698. [Google Scholar]
- Sugawara, Y.; Nishii, H.; Takahashi, T.; Yamauchi, J.; Mizuno, N.; Tago, K.; Itoh, H. The lipid raft proteins flotillins/reggies interact with Galphaq and are involved in Gq-mediated p38 mitogen-activated protein kinase activation through tyrosine kinase. Cell. Signal 2007, 19, 1301–1308. [Google Scholar]
- Riento, K.; Frick, M.; Schafer, I.; Nichols, B.J. Endocytosis of flotillin-1 and flotillin-2 is regulated by Fyn kinase. J. Cell Sci 2009, 122, 912–918. [Google Scholar]
- Baumann, C.A.; Ribon, V.; Kanzaki, M.; Thurmond, D.C.; Mora, S.; Shigematsu, S.; Bickel, P.E.; Pessin, J.E.; Saltiel, A.R. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature 2000, 407, 202–207. [Google Scholar]
- Tomasovic, A.; Traub, S.; Tikkanen, R. Molecular networks in FGF signaling: Flotillin-1 and cbl-associated protein compete for the binding to fibroblast growth factor receptor substrate 2. PLoS One 2012, 7, e29739. [Google Scholar]
- Lax, I.; Wong, A.; Lamothe, B.; Lee, A.; Frost, A.; Hawes, J.; Schlessinger, J. The docking protein FRS2alpha controls a MAP kinase-mediated negative feedback mechanism for signaling by FGF receptors. Mol. Cell 2002, 10, 709–719. [Google Scholar]
- Wu, Y.; Chen, Z.; Ullrich, A. EGFR and FGFR signaling through FRS2 is subject to negative feedback control by ERK1/2. Biol. Chem 2003, 384, 1215–1226. [Google Scholar]
- Pullikuth, A.; McKinnon, E.; Schaeffer, H.J.; Catling, A.D. The MEK1 scaffolding protein MP1 regulates cell spreading by integrating PAK1 and Rho signals. Mol. Cell. Biol 2005, 25, 5119–5133. [Google Scholar]
- Slack-Davis, J.K.; Eblen, S.T.; Zecevic, M.; Boerner, S.A.; Tarcsafalvi, A.; Diaz, H.B.; Marshall, M.S.; Weber, M.J.; Parsons, J.T.; Catling, A.D. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol 2003, 162, 281–291. [Google Scholar]
- Vomastek, T.; Iwanicki, M.P.; Schaeffer, H.J.; Tarcsafalvi, A.; Parsons, J.T.; Weber, M.J. RACK1 targets the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway to link integrin engagement with focal adhesion disassembly and cell motility. Mol. Cell. Biol 2007, 27, 8296–8305. [Google Scholar]



© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Meister, M.; Tomasovic, A.; Banning, A.; Tikkanen, R. Mitogen-Activated Protein (MAP) Kinase Scaffolding Proteins: A Recount. Int. J. Mol. Sci. 2013, 14, 4854-4884. https://doi.org/10.3390/ijms14034854
Meister M, Tomasovic A, Banning A, Tikkanen R. Mitogen-Activated Protein (MAP) Kinase Scaffolding Proteins: A Recount. International Journal of Molecular Sciences. 2013; 14(3):4854-4884. https://doi.org/10.3390/ijms14034854
Chicago/Turabian StyleMeister, Melanie, Ana Tomasovic, Antje Banning, and Ritva Tikkanen. 2013. "Mitogen-Activated Protein (MAP) Kinase Scaffolding Proteins: A Recount" International Journal of Molecular Sciences 14, no. 3: 4854-4884. https://doi.org/10.3390/ijms14034854