Annexin A2: The Importance of Being Redox Sensitive



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Introduction to Reactive Oxygen Species
3. The Annexin Superfamily
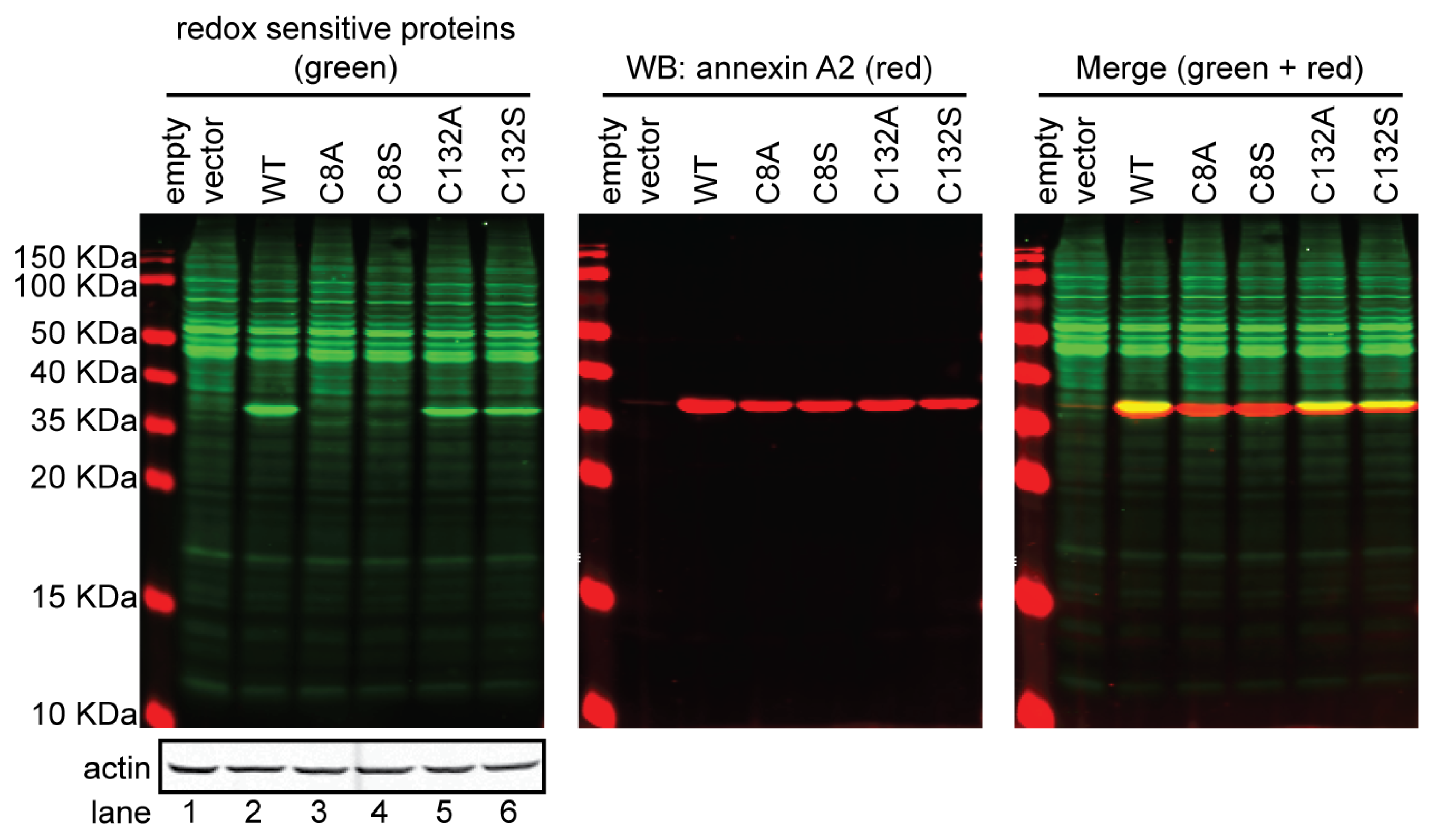
4. Annexin A2 Cysteine Reactivity and Functional Implications
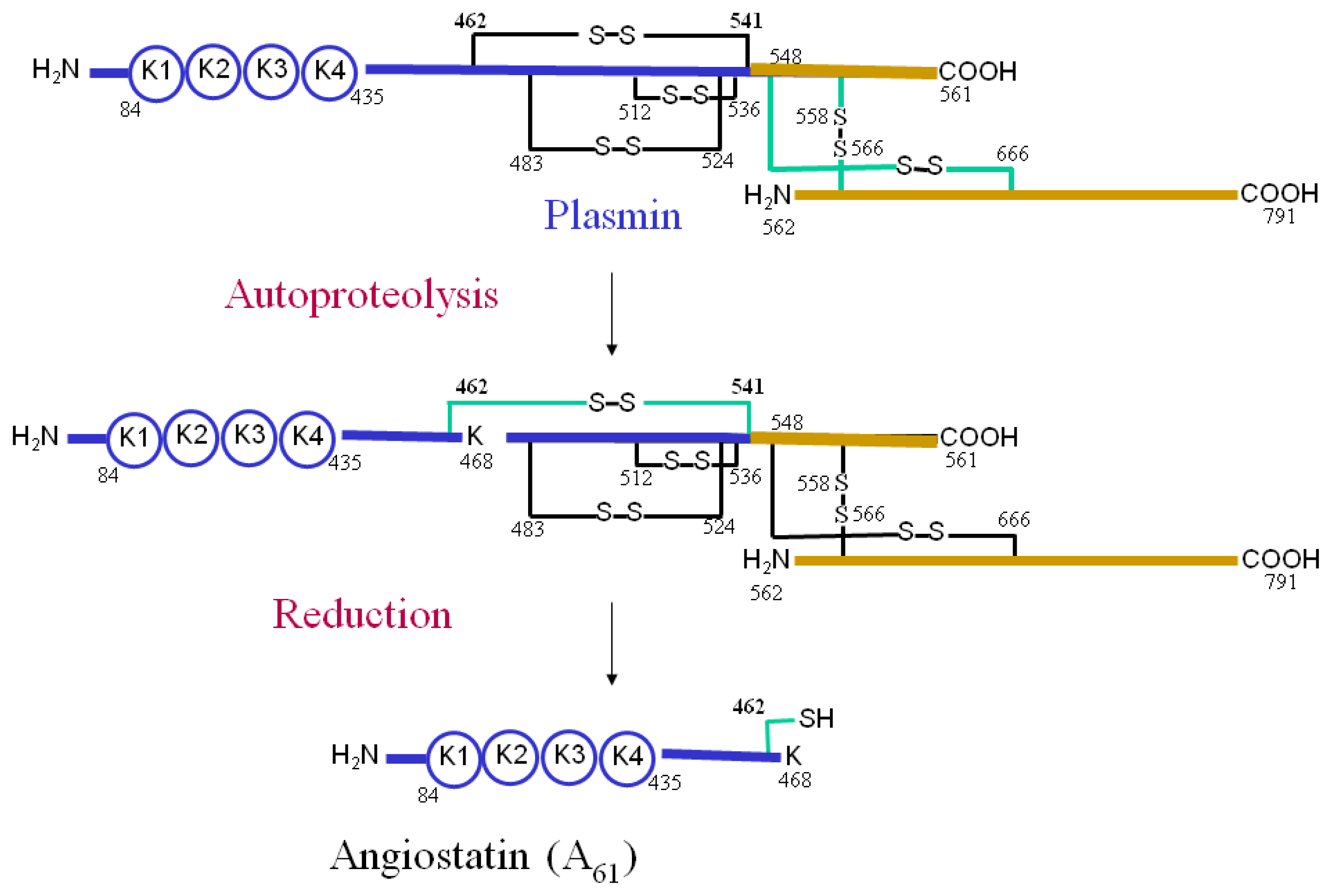
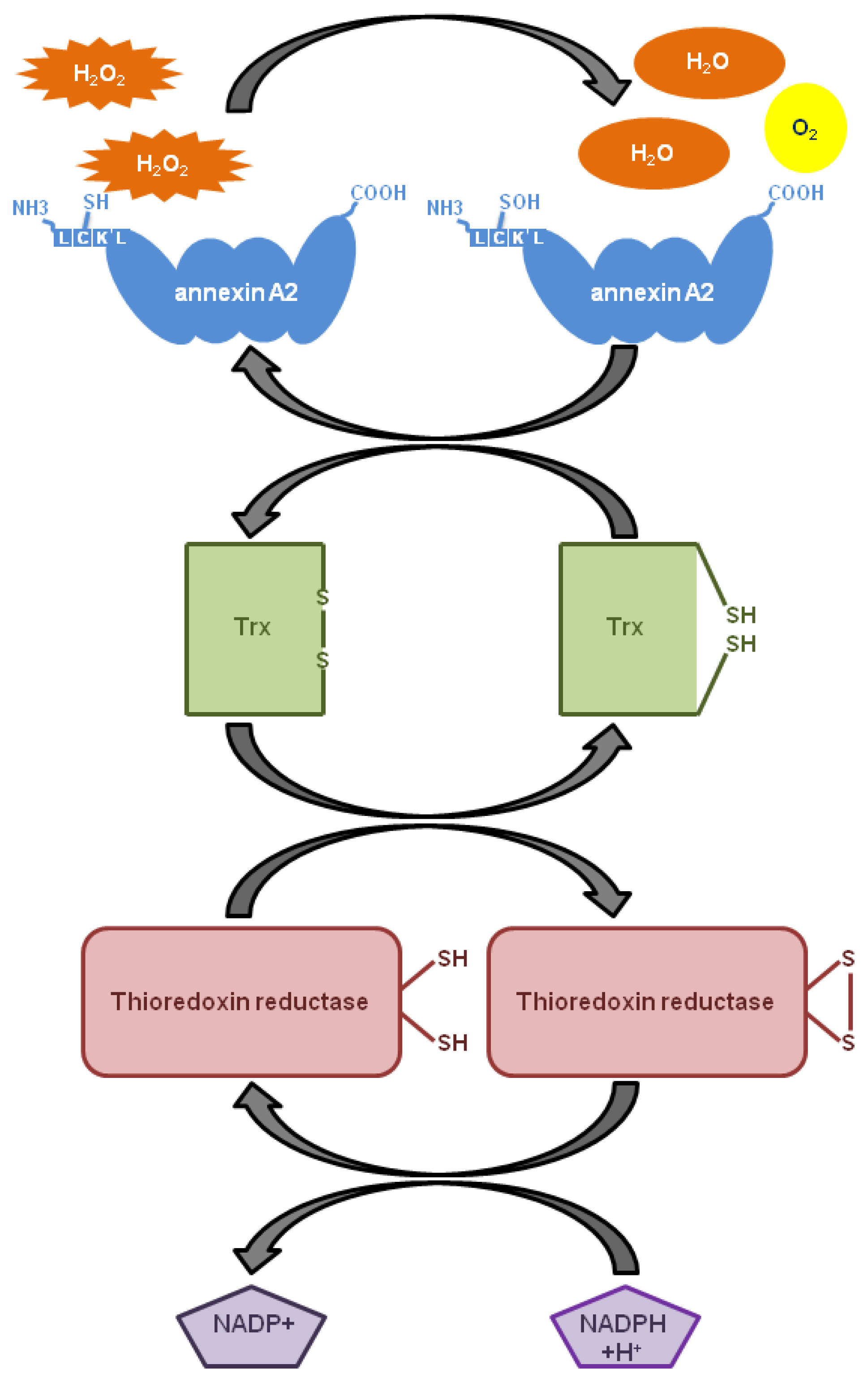
5. The Role of Annexin A2 As a Plasmin Reductase
6. The Role of Annexin A2 As a Cellular Redox Regulatory Protein
7. Annexin A2 Redox Status and Function in Disease
8. Annexin A2 Cellular Redox Regulatory Function Supports Tumour Growth
9. Annexin A2 Nuclear Translocation and Protection of DNA from Oxidative Damage
10. Conclusions and Future Directions
Acknowledgments
Conflict of Interest
References
- Rhee, S.G. Cell signalling. H2O2, a necessary evil for cell signalling. Science 2006, 312, 1882–1883. [Google Scholar]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signalling. Mol. Cell 2007, 26, 1–14. [Google Scholar]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox. Signal 2008, 10, 1343–1374. [Google Scholar]
- Jackson, A.L.; Loeb, L.A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat. Res 2001, 477, 7–21. [Google Scholar]
- Rubin, R.; Farber, J.L. Mechanisms of the killing of cultured hepatocytes by hydrogen peroxide. Arch. Biochem. Biophys 1984, 228, 450–459. [Google Scholar]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 2008, 68, 1777–1785. [Google Scholar]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumour cell metastasis. Science 2008, 320, 661–664. [Google Scholar]
- Madureira, P.A.; Hill, R.; Miller, V.A.; Giacomantonio, C.; Lee, P.W.; Waisman, D.M. Annexin A2 is a novel cellular redox regulatory protein involved in tumorigenesis. Oncotarget 2011, 2, 1075–1093. [Google Scholar]
- Pryor, W.A. Oxy-radicals and related species: Their formation, lifetimes, and reactions. Annu. Rev. Physiol 1986, 48, 657–667. [Google Scholar]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signalling messenger. Antioxid. Redox. Signal 2006, 8, 243–270. [Google Scholar]
- Kim, J.R.; Yoon, H.W.; Kwon, K.S.; Lee, S.R.; Rhee, S.G. Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal. Biochem 2000, 283, 214–221. [Google Scholar]
- Lindley, H. A study of the kinetics of the reaction between thiol compounds and choloracetamide. Biochem. J 1960, 74, 577–584. [Google Scholar]
- Rhee, S.G.; Jeong, W.; Chang, T.S.; Woo, H.A. Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: Its discovery, mechanism of action, and biological significance. Kidney Int. Suppl 2007, 106, S3–S8. [Google Scholar]
- Berndt, C.; Lillig, C.H.; Holmgren, A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: Implications for diseases in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol 2007, 292, H1227–H1236. [Google Scholar]
- Shelton, M.D.; Chock, P.B.; Mieyal, J.J. Glutaredoxin: Role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox. Signal 2005, 7, 348–366. [Google Scholar]
- Storz, P. Reactive oxygen species in tumour progression. Front. Biosci 2005, 10, 1881–1896. [Google Scholar]
- Karimpour, S.; Lou, J.; Lin, L.L.; Rene, L.M.; Lagunas, L.; Ma, X.; Karra, S.; Bradbury, C.M.; Markovina, S.; Goswami, P.C.; et al. Thioredoxin reductase regulates AP-1 activity as well as thioredoxin nuclear localization via active cysteines in response to ionizing radiation. Oncogene 2002, 21, 6317–6327. [Google Scholar]
- Hansen, J.M.; Go, Y.M.; Jones, D.P. Nuclear and mitochondrial compartmentation of oxidative stress and redox signalling. Annu. Rev. Pharmacol. Toxicol 2006, 46, 215–234. [Google Scholar]
- Go, Y.M.; Ziegler, T.R.; Johnson, J.M.; Gu, L.; Hansen, J.M.; Jones, D.P. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic. Biol. Med 2007, 42, 363–370. [Google Scholar]
- Halvey, P.J.; Hansen, J.M.; Johnson, J.M.; Go, Y.M.; Samali, A.; Jones, D.P. Selective oxidative stress in cell nuclei by nuclear-targeted d-amino acid oxidase. Antioxid. Redox. Signal 2007, 9, 807–816. [Google Scholar]
- Chiarugi, P.; Cirri, P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem. Sci 2003, 28, 509–514. [Google Scholar]
- Rhee, S.G.; Kang, S.W.; Jeong, W.; Chang, T.S.; Yang, K.S.; Woo, H.A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol 2005, 17, 183–189. [Google Scholar]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signalling. Cell 2005, 121, 667–670. [Google Scholar]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001, 2, 222–228. [Google Scholar]
- Dolado, I.; Swat, A.; Ajenjo, N.; de Vita, G.; Cuadrado, A.; Nebreda, A.R. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar]
- Hansen, J.M.; Watson, W.H.; Jones, D.P. Compartmentation of Nrf-2 redox control: Regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol. Sci 2004, 82, 308–317. [Google Scholar]
- Liu, H.; Colavitti, R.; Rovira, I.I.; Finkel, T. Redox-dependent transcriptional regulation. Circ. Res 2005, 97, 967–974. [Google Scholar]
- Wei, S.J.; Botero, A.; Hirota, K.; Bradbury, C.M.; Markovina, S.; Laszlo, A.; Spitz, D.R.; Goswami, P.C.; Yodoi, J.; Gius, D. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res 2000, 60, 6688–6695. [Google Scholar]
- Go, Y.M.; Pohl, J.; Jones, D.P. Quantification of redox conditions in the nucleus. Methods Mol. Biol 2009, 464, 303–317. [Google Scholar]
- Waisman, D.M. Annexin II tetramer: Structure and function. Mol. Cell. Biochem. 1995, 149–150, 301–322. [Google Scholar]
- Filipenko, N.R.; Kang, H.M.; Waisman, D.M. Characterization of the Ca2+-binding sites of annexin II tetramer. J. Biol. Chem 2000, 275, 38877–38884. [Google Scholar]
- Moss, S.E.; Morgan, R.O. The annexins. Genome Biol 2004, 5, 219. [Google Scholar]
- Gorecka, K.M.; Konopka-Postupolska, D.; Hennig, J.; Buchet, R.; Pikula, S. Peroxidase activity of annexin 1 from Arabidopsis thaliana. Biochem. Biophys. Res. Commun 2005, 336, 868–875. [Google Scholar]
- Laohavisit, A.; Mortimer, J.C.; Demidchik, V.; Coxon, K.M.; Stancombe, M.A.; Macpherson, N.; Brownlee, C.; Hofmann, A.; Webb, A.A.; Miedema, H.; et al. Zea mays annexins modulate cytosolic free Ca2+ and generate a Ca2+-permeable conductance. Plant Cell 2009, 21, 479–493. [Google Scholar]
- Gidrol, X.; Sabelli, P.A.; Fern, Y.S.; Kush, A.K. Annexin-like protein from Arabidopsis thaliana rescues delta oxyR mutant of Escherichia coli from H2O2 stress. Proc. Natl. Acad. Sci. USA 1996, 93, 11268–11273. [Google Scholar]
- Konopka-Postupolska, D.; Clark, G.; Goch, G.; Debski, J.; Floras, K.; Cantero, A.; Fijolek, B.; Roux, S.; Hennig, J. The role of annexin 1 in drought stress in Arabidopsis. Plant Physiol 2009, 150, 1394–1410. [Google Scholar]
- Jami, S.K.; Clark, G.B.; Turlapati, S.A.; Handley, C.; Roux, S.J.; Kirti, P.B. Ectopic expression of an annexin from Brassica juncea confers tolerance to abiotic and biotic stress treatments in transgenic tobacco. Plant Physiol. Biochem 2008, 46, 1019–1030. [Google Scholar]
- Divya, K.; Jami, S.K.; Kirti, P.B. Constitutive expression of mustard annexin, AnnBj1 enhances abiotic stress tolerance and fiber quality in cotton under stress. Plant Mol. Biol 2010, 73, 293–308. [Google Scholar]
- Zhou, L.; Duan, J.; Wang, X.M.; Zhang, H.M.; Duan, M.X.; Liu, J.Y. Characterization of a novel annexin gene from cotton (Gossypium hirsutum cv. CRI 35) and antioxidative role of its recombinant protein. J. Integr. Plant Biol 2011, 53, 347–357. [Google Scholar]
- Zhang, Y.W.Q.; Zhang, X.; Liu, X.; Wang, P.; Hou, Y. Cloning and characterization of an annexin gene from Cynanchum komarovii that enhances tolerance to drought and fusarium oxysporum in transgenic cotton. J. Plant Biol 2011, 54, 303–313. [Google Scholar]
- Clark, G.; Konopka-Postupolska, D.; Hennig, J.; Roux, S. Is annexin 1 a multifunctional protein during stress responses? Plant Signal. Behav 2010, 5, 303–307. [Google Scholar]
- Khalaj, V.; Azarian, B.; Enayati, S.; Vaziri, B. Annexin C4 in A. fumigatus: A proteomics approach to understand the function. J. Proteomics 2011, 74, 1950–1958. [Google Scholar]
- Gerke, V.; Weber, K. Calcium-dependent conformational changes in the 36-kDa subunit of intestinal protein I related to the cellular 36-kDa target of Rous sarcoma virus tyrosine kinase. J. Biol. Chem 1985, 260, 1688–1695. [Google Scholar]
- Zokas, L.; Glenney, J.R. The calpactin light chain is tightly linked to the cytoskeletal form of calpactin I: Studies using monoclonal antibodies to calpactin subunits. J. Cell Biol 1987, 105, 2111–2121. [Google Scholar]
- Madureira, P.A.; O’Connell, P.A.; Surette, A.P.; Miller, V.A.; Waisman, D.M. The biochemistry and regulation of S100A10: A multifunctional plasminogen receptor involved in oncogenesis. J. Biomed. Biotechnol 2012, 2012, 353687. [Google Scholar]
- Madureira, P.A.; Surette, A.P.; Phipps, K.D.; Taboski, M.A.; Miller, V.A.; Waisman, D.M. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood 2011, 118, 4789–4797. [Google Scholar]
- Johnsson, N.; Marriott, G.; Weber, K. p36, the major cytoplasmic substrate of src tyrosine protein kinase, binds to its p11 regulatory subunit via a short amino-terminal amphiphatic helix. EMBO J 1988, 7, 2435–2442. [Google Scholar]
- Sullivan, D.M.; Wehr, N.B.; Fergusson, M.M.; Levine, R.L.; Finkel, T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry 2000, 39, 11121–11128. [Google Scholar]
- Caplan, J.F.; Filipenko, N.R.; Fitzpatrick, S.L.; Waisman, D.M. Regulation of annexin A2 by reversible glutathionylation. J. Biol. Chem 2004, 279, 7740–7750. [Google Scholar]
- Gould, K.L.; Woodgett, J.R.; Isacke, C.M.; Hunter, T. The protein-tyrosine kinase substrate p36 is also a substrate for protein kinase C in vitro and in vivo. Mol. Cell. Biol 1986, 6, 2738–2744. [Google Scholar]
- Luo, W.; Yan, G.; Li, L.; Wang, Z.; Liu, H.; Zhou, S.; Liu, S.; Tang, M.; Yi, W.; Dong, Z.; et al. Epstein-Barr virus latent membrane protein 1 mediates serine 25 phosphorylation and nuclear entry of annexin A2 via PI-PLC-PKCalpha/PKCbeta pathway. Mol. Carcinog. 2008, 47, 934–946. [Google Scholar]
- Eberhard, D.A.; Karns, L.R.; VandenBerg, S.R.; Creutz, C.E. Control of the nuclear-cytoplasmic partitioning of annexin II by a nuclear export signal and by p11 binding. J. Cell. Sci 2001, 114, 3155–3166. [Google Scholar]
- Kwon, M.; Caplan, J.F.; Filipenko, N.R.; Choi, K.S.; Fitzpatrick, S.L.; Zhang, L.; Waisman, D.M. Identification of annexin II heterotetramer as a plasmin reductase. J. Biol. Chem 2002, 277, 10903–10911. [Google Scholar]
- Filipenko, N.R.; Waisman, D.M. The C terminus of annexin II mediates binding to F-actin. J. Biol. Chem 2001, 276, 5310–5315. [Google Scholar]
- Jost, M.; Thiel, C.; Weber, K.; Gerke, V. Mapping of three unique Ca2+-binding sites in human annexin II. Eur. J. Biochem 1992, 207, 923–930. [Google Scholar]
- Jost, M.; Weber, K.; Gerke, V. Annexin II contains two types of Ca2+-binding sites. Biochem. J 1994, 298, 553–559. [Google Scholar]
- Choi, K.S.; Fitzpatrick, S.L.; Filipenko, N.R.; Fogg, D.K.; Kassam, G.; Magliocco, A.M.; Waisman, D.M. Regulation of plasmin-dependent fibrin clot lysis by annexin II heterotetramer. J. Biol. Chem 2001, 276, 25212–25221. [Google Scholar]
- Aukrust, I.; Hollas, H.; Strand, E.; Evensen, L.; Trave, G.; Flatmark, T.; Vedeler, A. The mRNA-binding site of annexin A2 resides in helices C–D of its domain IV. J. Mol. Biol 2007, 368, 1367–1378. [Google Scholar]
- Kassam, G.; Manro, A.; Braat, C.E.; Louie, P.; Fitzpatrick, S.L.; Waisman, D.M. Characterization of the heparin binding properties of annexin II tetramer. J. Biol. Chem 1997, 272, 15093–15100. [Google Scholar]
- Turnay, J.; Lecona, E.; Fernández-Lizarbe, S.; Guzmán-Aránguez, A.; Fernández, M.P.; Olmo, N.; Lizarbe, M.A. Structure-function relationship in annexin A13, the founder member of the vertebrate family of annexins. Biochem. J 2005, 389, 899–911. [Google Scholar]
- Courtneidge, S.; Ralston, R.; Alitalo, K.; Bishop, J.M. Subcellular location of an abundant substrate (p36) for tyrosine-specific protein kinases. Mol. Cell. Biol 1983, 3, 340–350. [Google Scholar]
- Arrigo, A.P.; Darlix, J.L.; Spahr, P.F. A cellular protein phosphorylated by the avian sarcoma virus transforming gene product is associated with ribonucleoprotein particles. EMBO J 1983, 2, 309–315. [Google Scholar]
- Vishwanatha, J.K.; Jindal, H.K.; Davis, R.G. The role of primer recognition proteins in DNA replication: Association with nuclear matrix in HeLa cells. J. Cell Sci 1992, 101, 25–34. [Google Scholar]
- Chung, C.Y.; Erickson, H.P. Cell surface annexin II is a high affinity receptor for the alternatively spliced segment of tenascin-C. J. Cell Biol 1994, 126, 539–548. [Google Scholar]
- Surette, A.P.; Madureira, P.A.; Phipps, K.D.; Miller, V.A.; Svenningsson, P.; Waisman, D.M. Regulation of fibrinolysis by S100A10 in vivo. Blood 2011, 118, 3172–3181. [Google Scholar]
- O’Connell, P.A.; Madureira, P.A.; Berman, J.N.; Liwski, R.S.; Waisman, D.M. Regulation of S100A10 by the PML-RAR-alpha oncoprotein. Blood 2011, 117, 4095–4105. [Google Scholar]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol 2005, 6, 449–461. [Google Scholar]
- Filipenko, N.R.; MacLeod, T.J.; Yoon, C.S.; Waisman, D.M. Annexin A2 is a novel RNA-binding protein. J. Biol. Chem 2004, 279, 8723–8731. [Google Scholar]
- Jindal, H.K.; Chaney, W.G.; Anderson, C.W.; Davis, R.G.; Vishwanatha, J.K. The protein-tyrosine kinase substrate, calpactin I heavy chain (p36), is part of the primer recognition protein complex that interacts with DNA polymerase alpha. J. Biol. Chem 1991, 266, 5169–5176. [Google Scholar]
- Madureira, P.A.; Hill, R.; Lee, P.W.; Waisman, D.M. Genotoxic agents promote the nuclear accumulation of annexin A2: Role of annexin A2 in mitigating DNA damage. PLoS One 2012, 7, e50591. [Google Scholar]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol 2010, 27, 221–224. [Google Scholar]
- Hajjar, K.A.; Mauri, L.; Jacovina, A.T.; Zhong, F.; Mirza, U.A.; Padovan, J.C.; Chait, B.T. Tissue plasminogen activator binding to the annexin II tail domain. Direct modulation by homocysteine. J. Biol. Chem 1998, 273, 9987–9993. [Google Scholar]
- Sundaramoorthy, E.; Maiti, S.; Brahmachari, S.K.; Sengupta, S. Predicting protein homocysteinylation targets based on dihedral strain energy and pKa of cysteines. Proteins 2008, 71, 1475–1483. [Google Scholar]
- Felez, J.; Chanquia, C.J.; Fabregas, P.; Plow, E.F.; Miles, L.A. Competition between plasminogen and tissue plasminogen activator for cellular binding sites. Blood 1993, 82, 2433–2441. [Google Scholar]
- Felez, J.; Chanquia, C.J.; Levin, E.G.; Miles, L.A.; Plow, E.F. Binding of tissue plasminogen activator to human monocytes and monocytoid cells. Blood 1991, 78, 2318–2327. [Google Scholar]
- Felez, J.; Miles, L.A.; Fabregas, P.; Jardi, M.; Plow, E.F.; Lijnen, R.H. Characterization of cellular binding sites and interactive regions within reactants required for enhancement of plasminogen activation by tPA on the surface of leukocytic cells. Thromb. Haemost 1996, 76, 577–584. [Google Scholar]
- Roda, O.; Valero, M.L.; Peiro, S.; Andreu, D.; Real, F.X.; Navarro, P. New insights into the tPA-annexin A2 interaction. Is annexin A2 CYS8 the sole requirement for this association? J. Biol. Chem 2003, 278, 5702–5709. [Google Scholar]
- Singh, T.K.; Liu, L. Modification of cysteine residues by N-ethylmaleimide inhibits annexin II tetramer mediated liposome aggregation. Arch. Biochem. Biophys 2000, 381, 235–240. [Google Scholar]
- Condino-Neto, A.; Newburger, P.E. NADPH oxidase activity and cytochrome b558 content of human Epstein-Barr-virus-transformed B lymphocytes correlate with expression of genes encoding components of the oxidase system. Arch. Biochem. Biophys 1998, 360, 158–164. [Google Scholar]
- Newburger, P.E.; Dai, Q.; Whitney, C. In vitro regulation of human phagocyte cytochrome b heavy and light chain gene expression by bacterial lipopolysaccharide and recombinant human cytokines. J. Biol. Chem 1991, 266, 16171–16177. [Google Scholar]
- De Keulenaer, G.W.; Alexander, R.W.; Ushio-Fukai, M.; Ishizaka, N.; Griendling, K.K. Tumour necrosis factor alpha activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem. J 1998, 329, 653–657. [Google Scholar]
- Gauss, K.A.; Nelson-Overton, L.K.; Siemsen, D.W.; Gao, Y.; DeLeo, F.R.; Quinn, M.T. Role of NF-kappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumour necrosis factor-alpha. J. Leukoc. Biol 2007, 82, 729–741. [Google Scholar]
- Kamizato, M.; Nishida, K.; Masuda, K.; Takeo, K.; Yamamoto, Y.; Kawai, T.; Teshima-Kondo, S.; Tanahashi, T.; Rokutan, K. Interleukin 10 inhibits interferon gamma- and tumour necrosis factor alpha-stimulated activation of NADPH oxidase 1 in human colonic epithelial cells and the mouse colon. J. Gastroenterol 2009, 44, 1172–1184. [Google Scholar]
- Li, X.J.; Marchal, C.C.; Stull, N.D.; Stahelin, R.V.; Dinauer, M.C. p47phox Phox homology domain regulates plasma membrane but not phagosome neutrophil NADPH oxidase activation. J. Biol. Chem 2010, 285, 35169–35179. [Google Scholar]
- Moe, K.T.; Aulia, S.; Jiang, F.; Chua, Y.L.; Koh, T.H.; Wong, M.C.; Dusting, G.J. Differential upregulation of Nox homologues of NADPH oxidase by tumour necrosis factor-alpha in human aortic smooth muscle and embryonic kidney cells. J. Cell. Mol. Med 2006, 10, 231–239. [Google Scholar]
- Yoshida, L.S.; Tsunawaki, S. Expression of NADPH oxidases and enhanced H2O2-generating activity in human coronary artery endothelial cells upon induction with tumour necrosis factor-alpha. Int. Immunopharmacol 2008, 8, 1377–1385. [Google Scholar]
- Frey, R.S.; Rahman, A.; Kefer, J.C.; Minshall, R.D.; Malik, A.B. PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ. Res 2002, 90, 1012–1019. [Google Scholar]
- Akimaru, K.; Utsumi, T.; Sato, E.F.; Klostergaard, J.; Inoue, M.; Utsumi, K. Role of tyrosyl phosphorylation in neutrophil priming by tumour necrosis factor-alpha and granulocyte colony stimulating factor. Arch. Biochem. Biophys 1992, 298, 703–709. [Google Scholar]
- Dewas, C.; Dang, P.M.; Gougerot-Pocidalo, M.A.; El-Benna, J. TNF-alpha induces phosphorylation of p47(phox) in human neutrophils: Partial phosphorylation of p47phox is a common event of priming of human neutrophils by TNF-alpha and granulocyte-macrophage colony-stimulating factor. J. Immunol 2003, 171, 4392–4398. [Google Scholar]
- Utsumi, T.; Klostergaard, J.; Akimaru, K.; Edashige, K.; Sato, E.F.; Utsumi, K. Modulation of TNF-alpha-priming and stimulation-dependent superoxide generation in human neutrophils by protein kinase inhibitors. Arch. Biochem. Biophys 1992, 294, 271–278. [Google Scholar]
- Dang, P.M.; Stensballe, A.; Boussetta, T.; Raad, H.; Dewas, C.; Kroviarski, Y.; Hayem, G.; Jensen, O.N.; Gougerot-Pocidalo, M.A.; El-Benna, J. A specific p47phox -serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Invest 2006, 116, 2033–2043. [Google Scholar]
- Chenevier-Gobeaux, C.; Simonneau, C.; Therond, P.; Bonnefont-Rousselot, D.; Poiraudeau, S.; Ekindjian, O.G.; Borderie, D. Implication of cytosolic phospholipase A2 (cPLA2) in the regulation of human synoviocyte NADPH oxidase (Nox2) activity. Life Sci 2007, 81, 1050–1058. [Google Scholar]
- Kim, Y.S.; Morgan, M.J.; Choksi, S.; Liu, Z.G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 2007, 26, 675–687. [Google Scholar]
- Liu, L.; Enright, E.; Sun, P.; Tsai, S.Y.; Mehta, P.; Beckman, D.L.; Terrian, D.M. Inactivation of annexin II tetramer by S-nitrosoglutathione. Eur. J. Biochem 2002, 269, 4277–4286. [Google Scholar]
- Kwon, M.; Yoon, C.S.; Jeong, W.; Rhee, S.G.; Waisman, D.M. Annexin A2-S100A10 heterotetramer, a novel substrate of thioredoxin. J. Biol. Chem 2005, 280, 23584–23592. [Google Scholar]
- Nogueira, V.; Park, Y.; Chen, C.C.; Xu, P.Z.; Chen, M.L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem 2004, 279, 32804–32812. [Google Scholar]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell Mol. Biol 2009, 41, 348–357. [Google Scholar]
- Jamaluddin, M.; Wiktorowicz, J.E.; Soman, K.V.; Boldogh, I.; Forbus, J.D.; Spratt, H.; Garofalo, R.P.; Brasier, A.R. Role of peroxiredoxin 1 and peroxiredoxin 4 in protection of respiratory syncytial virus-induced cysteinyl oxidation of nuclear cytoskeletal proteins. J. Virol 2010, 84, 9533–9545. [Google Scholar]
- De Marco, F.; Bucaj, E.; Foppoli, C.; Fiorini, A.; Blarzino, C.; Filipi, K.; Giorgi, A.; Schinina, M.E.; di Domenico, F.; Coccia, R.; et al. Oxidative stress in HPV-driven viral carcinogenesis: Redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One 2012, 7, e34366. [Google Scholar]
- Tanaka, T.; Akatsuka, S.; Ozeki, M.; Shirase, T.; Hiai, H.; Toyokuni, S. Redox regulation of annexin 2 and its implications for oxidative stress-induced renal carcinogenesis and metastasis. Oncogene 2004, 23, 3980–3989. [Google Scholar]
- Kim, J.S.; Kim, E.J.; Kim, H.J.; Yang, J.Y.; Hwang, G.S.; Kim, C.W. Proteomic and metabolomic analysis of H2O2-induced premature senescent human mesenchymal stem cells. Exp. Gerontol 2011, 46, 500–510. [Google Scholar]
- Wang, Z.C.; E, D.; Batu, D.L.; Saixi, Y.L.; Zhang, B.; Ren, L.Q. 2D-DIGE proteomic analysis of changes in estrogen/progesterone-induced rat breast hyperplasia upon treatment with the Mongolian remedy RuXian-I. Molecules 2011, 16, 3048–3065. [Google Scholar]
- Alexandre, J.; Batteux, F.; Nicco, C.; Chereau, C.; Laurent, A.; Guillevin, L.; Weill, B.; Goldwasser, F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int. J. Cancer 2006, 119, 41–48. [Google Scholar]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [Google Scholar]
- O’Connell, P.A.; Waisman, D.M. Regulation of plasmin generation by the annexin A2 heterotetramer: A shift in perspective. Future Oncol 2012, 8, 763–765. [Google Scholar]
- Bugge, T.H.; Kombrinck, K.W.; Xiao, Q.; Holmback, K.; Daugherty, C.C.; Witte, D.P.; Degen, J.L. Growth and dissemination of Lewis lung carcinoma in plasminogen-deficient mice. Blood 1997, 90, 4522–4531. [Google Scholar]
- Bugge, T.H.; Lund, L.R.; Kombrinck, K.K.; Nielsen, B.S.; Holmback, K.; Drew, A.F.; Flick, M.J.; Witte, D.P.; Dano, K.; Degen, J.L. Reduced metastasis of Polyoma virus middle T antigen-induced mammary cancer in plasminogen-deficient mice. Oncogene 1998, 16, 3097–3104. [Google Scholar]
- Miles, L.A.; Hawley, S.B.; Baik, N.; Andronicos, N.M.; Castellino, F.J.; Parmer, R.J. Plasminogen receptors: The sine qua non of cell surface plasminogen activation. Front. Biosci 2005, 10, 1754–1762. [Google Scholar]
- Mai, J.; Waisman, D.M.; Sloane, B.F. Cell surface complex of cathepsin B/annexin II tetramer in malignant progression. Biochim. Biophys. Acta 2000, 1477, 215–230. [Google Scholar]
- Takano, S.; Togawa, A.; Yoshitomi, H.; Shida, T.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Ohtsuka, M.; Kato, A.; Tomonaga, T.; et al. Annexin II overexpression predicts rapid recurrence after surgery in pancreatic cancer patients undergoing gemcitabine-adjuvant chemotherapy. Ann. Surg. Oncol. 2008, 15, 3157–3168. [Google Scholar]
- Chuthapisith, S.; Bean, B.E.; Cowley, G.; Eremin, J.M.; Samphao, S.; Layfield, R.; Kerr, I.D.; Wiseman, J.; El-Sheemy, M.; Sreenivasan, T.; et al. Annexins in human breast cancer: Possible predictors of pathological response to neoadjuvant chemotherapy. Eur. J. Cancer 2009, 45, 1274–1281. [Google Scholar]
- Sacre, S.M.; Moss, S.E. Intracellular localization of endothelial cell annexins is differentially regulated by oxidative stress. Exp. Cell Res 2002, 274, 254–263. [Google Scholar]
- Kovacs, I.; Ayaydin, F.; Oberschall, A.; Ipacs, I.; Bottka, S.; Pongor, S.; Dudits, D.; Toth, E.C. Immunolocalization of a novel annexin-like protein encoded by a stress and abscisic acid responsive gene in alfalfa. Plant J 1998, 15, 185–197. [Google Scholar]
- Rhee, H.J.; Kim, G.Y.; Huh, J.W.; Kim, S.W.; Na, D.S. Annexin I is a stress protein induced by heat, oxidative stress and a sulfhydryl-reactive agent. Eur. J. Biochem 2000, 267, 3220–3225. [Google Scholar]
- Clark, G.B.; Dauwalder, M.; Roux, S.J. Immunological and biochemical evidence for nuclear localization of annexin in peas. Plant Physiol. Biochem 1998, 36, 621–627. [Google Scholar]
- Waters, K.M.; Stenoien, D.L.; Sowa, M.B.; von Neubeck, C.H.; Chrisler, W.B.; Tan, R.; Sontag, R.L.; Weber, T.J. Annexin A2 modulates radiation-sensitive transcriptional programming and cell fate. Radiat. Res 2013, 179, 53–61. [Google Scholar]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem 1998, 273, 5858–5868. [Google Scholar]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991, 51, 6304–6311. [Google Scholar]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA 1992, 89, 7491–7495. [Google Scholar]
- Hill, R.; Bodzak, E.; Blough, M.D.; Lee, P.W. p53 Binding to the p21 promoter is dependent on the nature of DNA damage. Cell Cycle 2008, 7, 2535–2543. [Google Scholar]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell. Biol 2000, 151, 1381–1390. [Google Scholar]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar]
- Van Loon, B.; Markkanen, E.; Hubscher, U. Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010, 9, 604–661. [Google Scholar]


© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Madureira, P.A.; Waisman, D.M. Annexin A2: The Importance of Being Redox Sensitive. Int. J. Mol. Sci. 2013, 14, 3568-3594. https://doi.org/10.3390/ijms14023568
Madureira PA, Waisman DM. Annexin A2: The Importance of Being Redox Sensitive. International Journal of Molecular Sciences. 2013; 14(2):3568-3594. https://doi.org/10.3390/ijms14023568
Chicago/Turabian StyleMadureira, Patrícia A., and David M. Waisman. 2013. "Annexin A2: The Importance of Being Redox Sensitive" International Journal of Molecular Sciences 14, no. 2: 3568-3594. https://doi.org/10.3390/ijms14023568