Mitochondrial and Nuclear DNA Damage and Repair in Age-Related Macular Degeneration




{kind=link}
{kind=link}
{kind=link}
Abstract
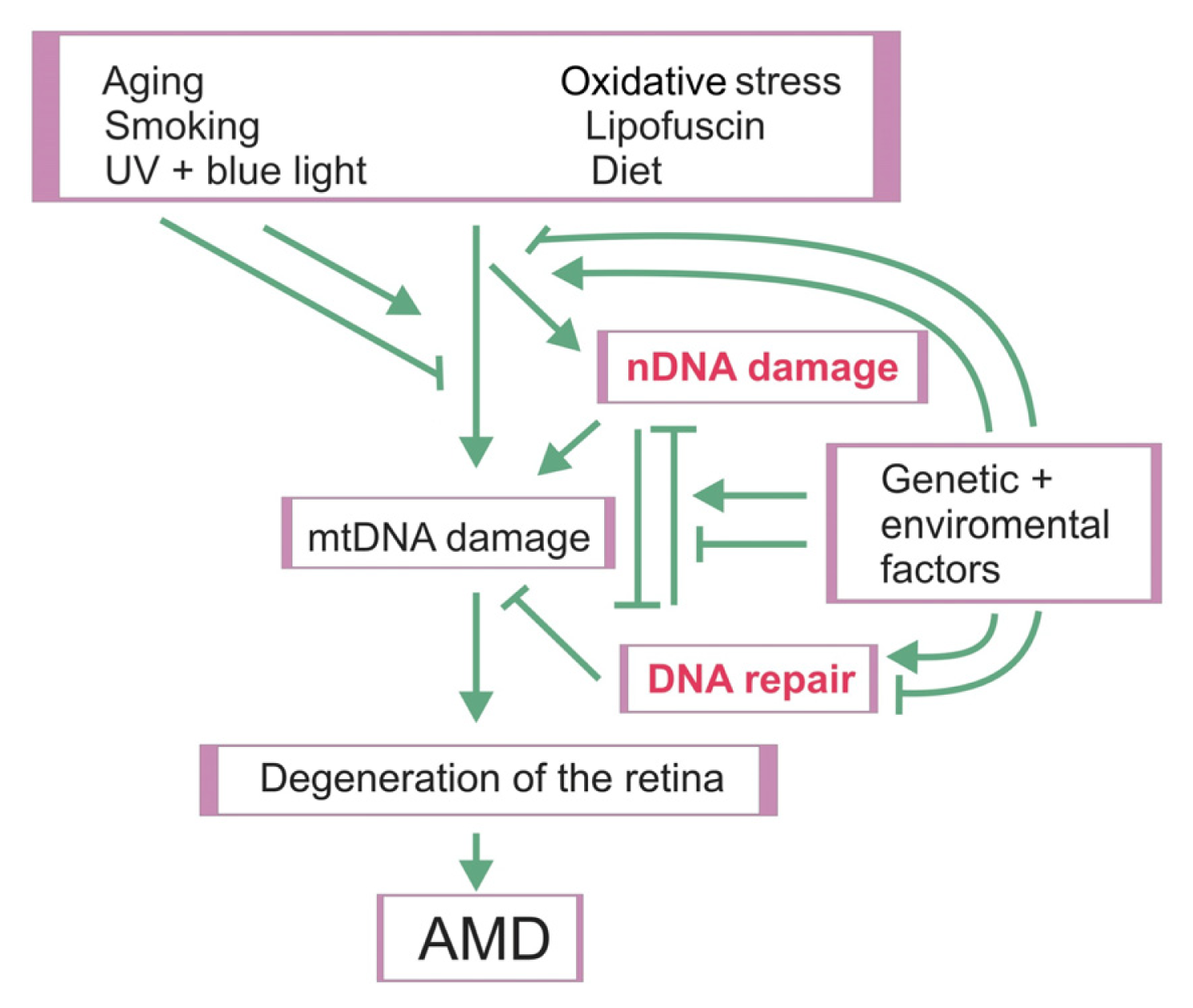
:1. Introduction
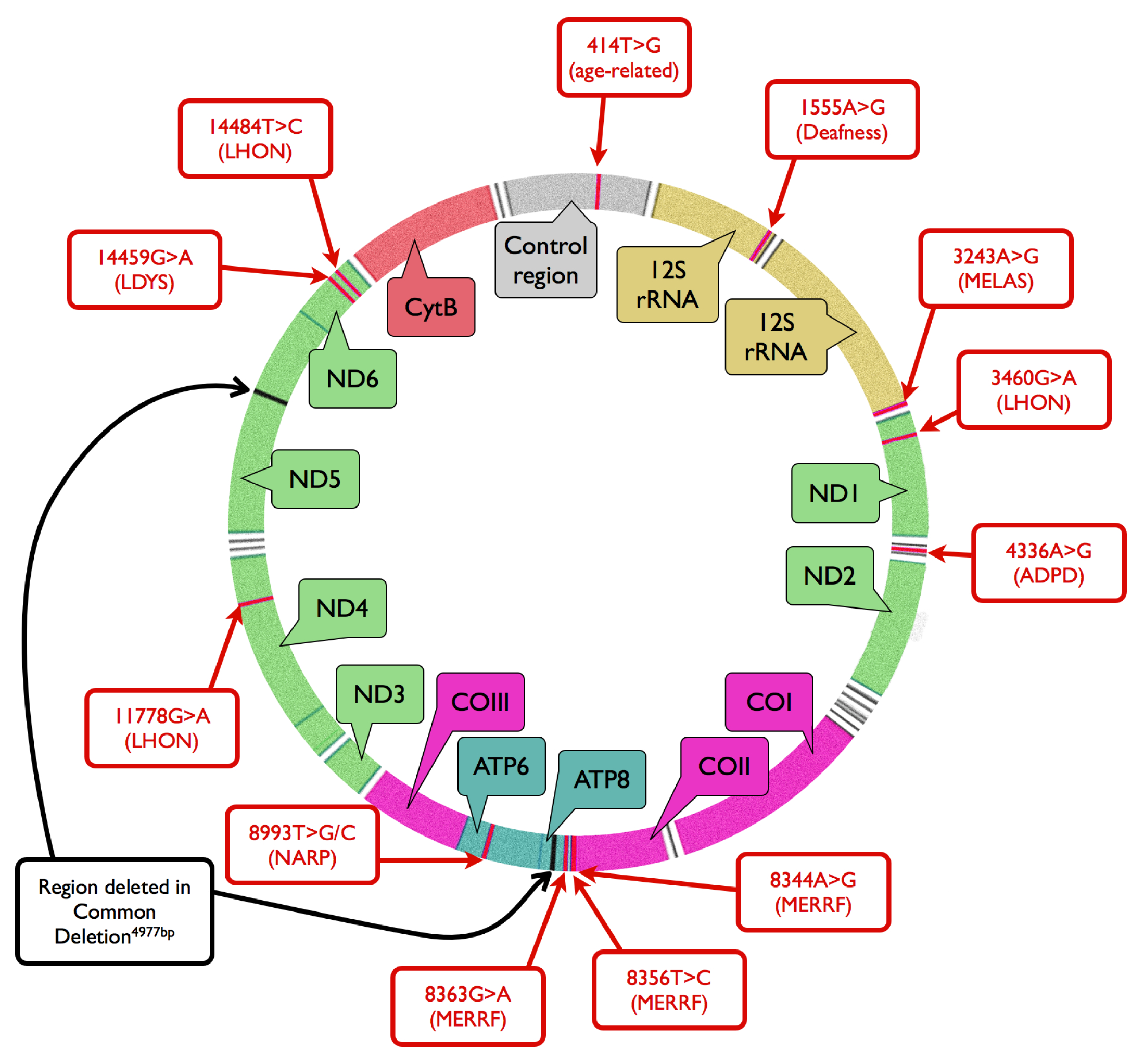
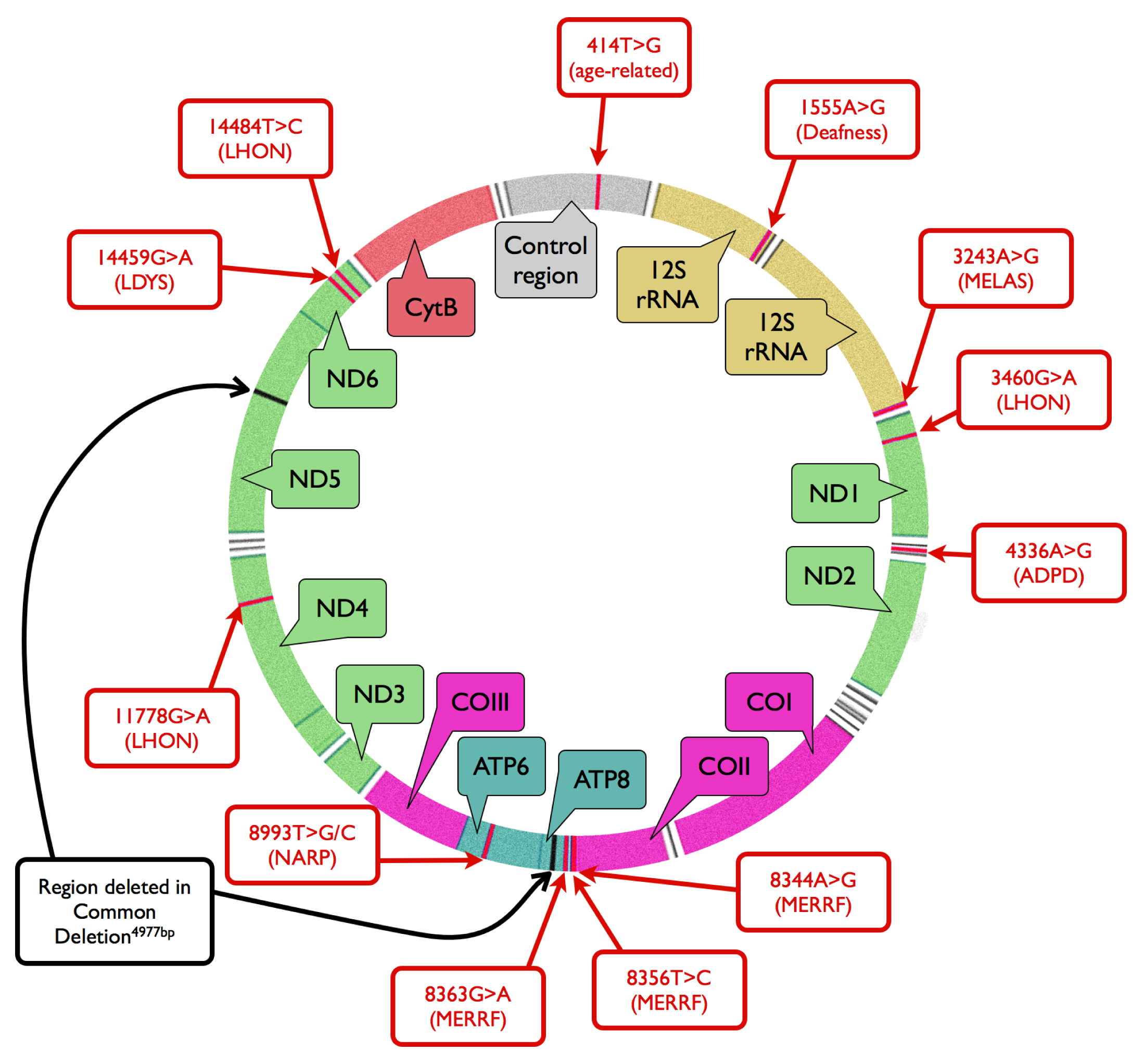
2. Mitochondrial Mutagenesis
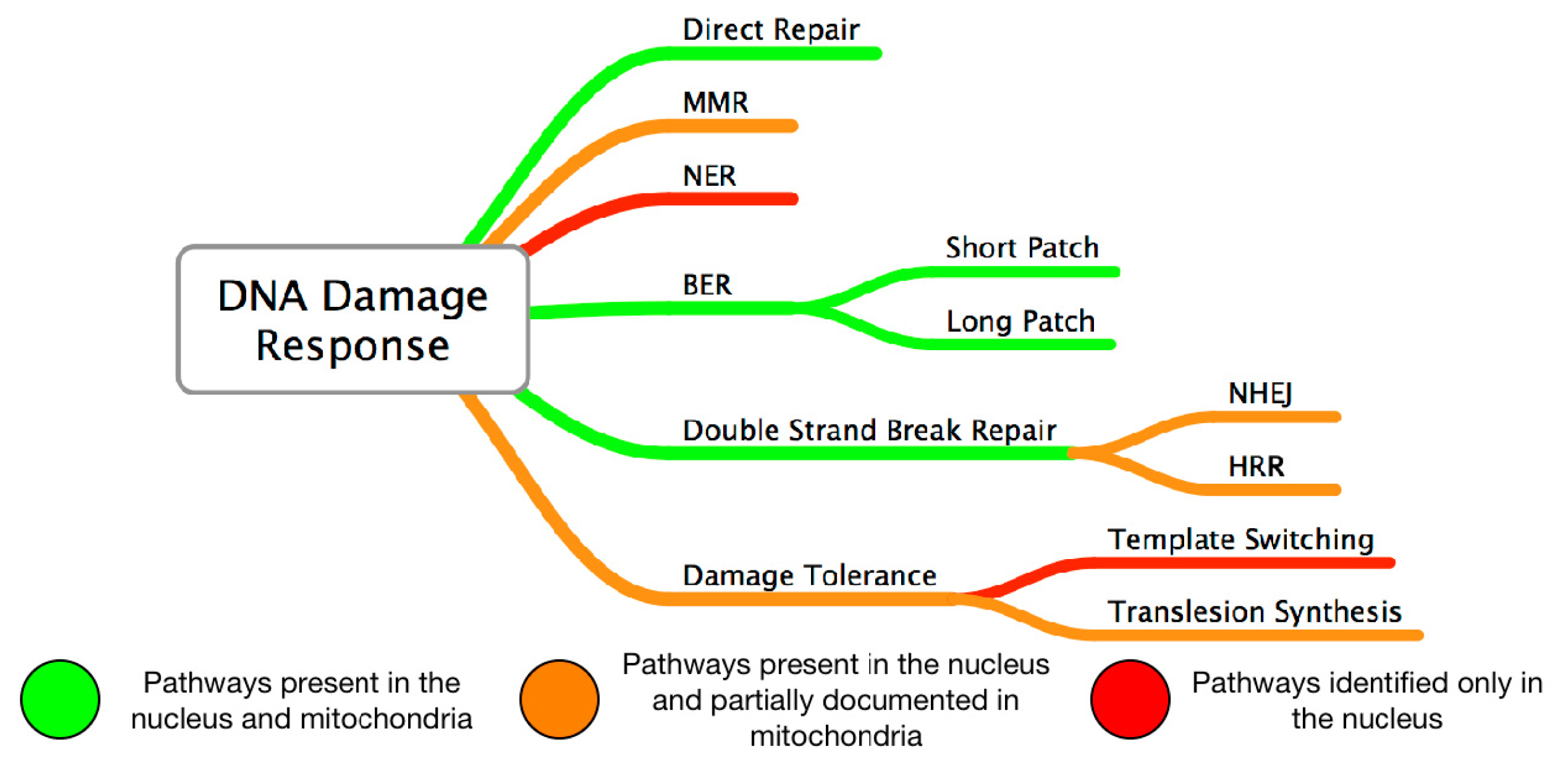
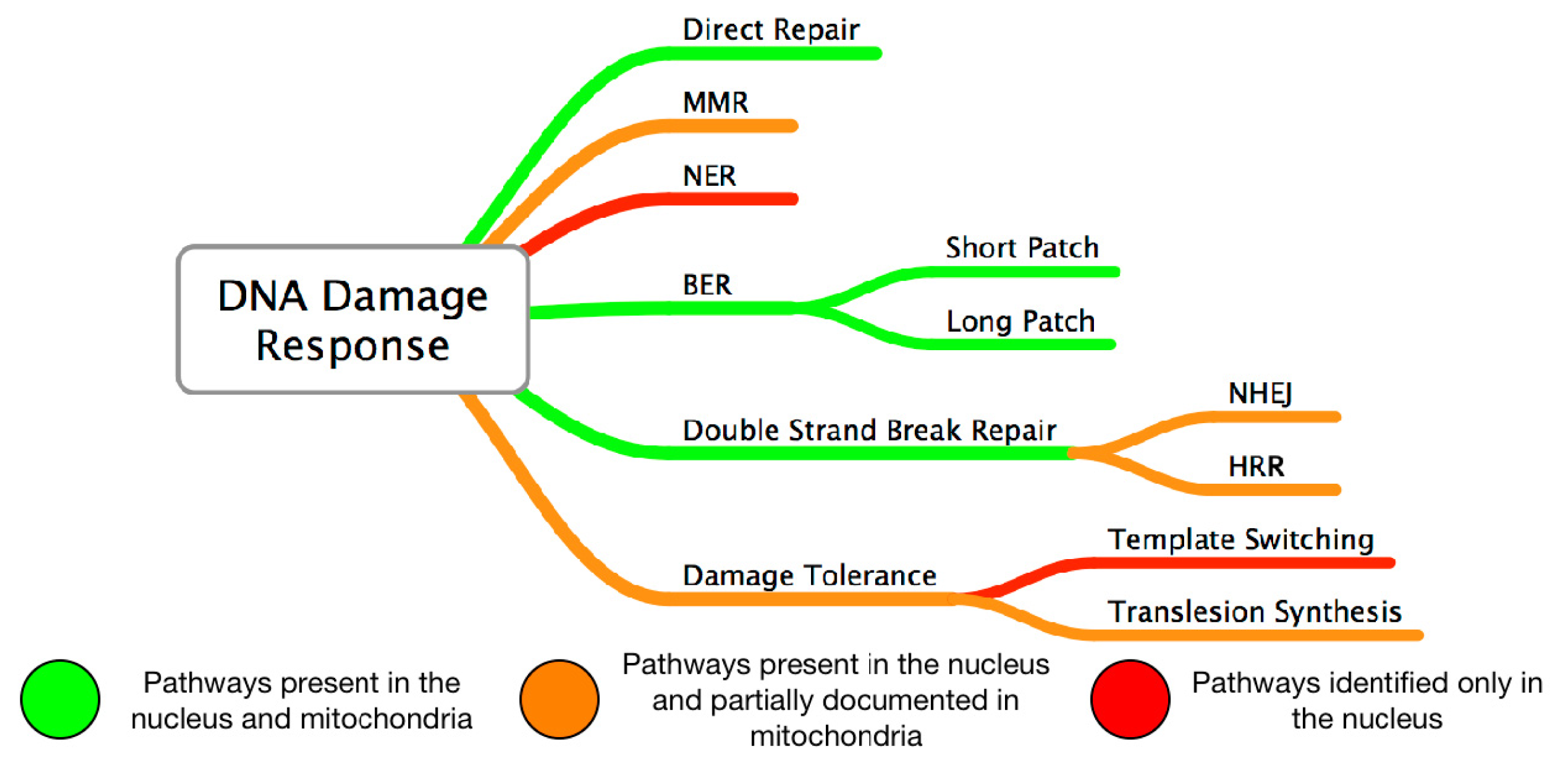
3. Mitochondrial DNA Damage and Repair in AMD
4. Nuclear DNA Damage and Repair in AMD
5. Discussion
6. Conclusions
Acknowledgments
References
- McCall, M.R.; Frei, B. Can antioxidant vitamins materially reduce oxidative damage in humans? Free Radic. Biol. Med 1999, 26, 1034–1053. [Google Scholar]
- Naviaux, R.K. Oxidative Shielding or Oxidative Stress? J. Pharmacol. Exp. Ther 2012, 342, 608–618. [Google Scholar]
- Ohia, S.E.; Opere, C.A.; Leday, A.M. Pharmacological consequences of oxidative stress in ocular tissues. Mutat. Res 2005, 579, 22–36. [Google Scholar]
- Vegh, M.J.; de Waard, M.C.; van der Pluijm, I.; Ridwan, Y.; Sassen, M.J.; van Nierop, P.; van der Schors, R.C.; Li, K.W.; Hoeijmakers, J.H.; Smit, A.B.; et al. Synaptic proteome changes in a DNA repair deficient ercc1 mouse model of accelerated aging. J. Proteome Res 2012, 11, 1855–1867. [Google Scholar]
- Cai, X.; McGinnis, J.F. Oxidative stress: The achilles’ heel of neurodegenerative diseases of the retina. Front Biosci 2012, 17, 1976–1995. [Google Scholar]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med 2008, 358, 2606–2617. [Google Scholar]
- Durik, M.; Kavousi, M.; van der Pluijm, I.; Isaacs, A.; Cheng, C.; Verdonk, K.; Loot, A.E.; Oeseburg, H.; Bhaggoe, U.M.; Leijten, F.; et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 2012, 126, 468–478. [Google Scholar]
- Khandhadia, S.; Lotery, A. Oxidation and age-related macular degeneration: Insights from molecular biology. Expert Rev. Mol. Med 2010, 12, e34. [Google Scholar]
- Shabalina, I.G.; Nedergaard, J. Mitochondrial (“mild”) uncoupling and ROS production: Physiologically relevant or not? Biochem. Soc. Trans 2011, 39, 1305–1309. [Google Scholar]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol 2012, 942, 157–183. [Google Scholar]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Gabrieli, C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar]
- Jarrett, S.G.; Lewin, A.S.; Boulton, M.E. The importance of mitochondria in age-related and inherited eye disorders. Ophthalmic Res 2010, 44, 179–190. [Google Scholar]
- Harman, D. Free radical theory of aging: An update: Increasing the functional life span. Ann. N. Y. Acad. Sci 2006, 1067, 10–21. [Google Scholar]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Lightowlers, R.N.; Dietrich, A. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2011, 1813, 186–200. [Google Scholar]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res 2004, 567, 1–61. [Google Scholar]
- Nair, J.; Strand, S.; Frank, N.; Knauft, J.; Wesch, H.; Galle, P.R.; Bartsch, H. Apoptosis and age-dependant induction of nuclear and mitochondrial etheno-DNA adducts in Long-Evans Cinnamon (LEC) rats: Enhanced DNA damage by dietary curcumin upon copper accumulation. Carcinogenesis 2005, 26, 1307–1315. [Google Scholar]
- Chalam, K.V.; Khetpal, V.; Rusovici, R.; Balaiya, S. A review: Role of ultraviolet radiation in age-related macular degeneration. Eye Contact Lens 2011, 37, 225–232. [Google Scholar]
- Zhang, H.; Maguire, D.J.; Zhang, M.; Zhang, L.; Okunieff, P. Elevated mitochondrial DNA copy number and POL-γ expression but decreased expression of TFAM in murine intestine following therapeutic dose irradiation. Adv. Exp. Med. Biol 2011, 701, 201–206. [Google Scholar]
- Bacman, S.R.; Williams, S.L.; Moraes, C.T. Intra- and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks. Nucleic Acids Res 2009, 37, 4218–4226. [Google Scholar]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol 2012, 13, 659–671. [Google Scholar]
- Rasmussen, A.K.; Rasmussen, L.J. Targeting of O6-MeG DNA methyltransferase (MGMT) to mitochondria protects against alkylation induced cell death. Mitochondrion 2005, 5, 411–417. [Google Scholar]
- Waters, R.; Moustacchi, E. Dose dependence of the excision of ultraviolet-induced pyrimidine dimers from nuclear deoxyribonucleic acids of haploid and diploid Saccharomyces cerevisiae. J. Bacteriol 1975, 121, 901–906. [Google Scholar]
- LeDoux, S.P.; Wilson, G.L. Base excision repair of mitochondrial DNA damage in mammalian cells. Prog. Nucleic Acid Res. Mol. Biol 2001, 68, 273–284. [Google Scholar]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar]
- Bharati, S.; Krokan, H.E.; Kristiansen, L.; Otterlei, M.; Slupphaug, G. Human mitochondrial uracil-DNA glycosylase preform (UNG1) is processed to two forms one of which is resistant to inhibition by AP sites. Nucleic Acids Res 1998, 26, 4953–4959. [Google Scholar]
- Ruchko, M.V.; Gorodnya, O.M.; Zuleta, A.; Pastukh, V.M.; Gillespie, M.N. The DNA glycosylase Ogg1 defends against oxidant-induced mtDNA damage and apoptosis in pulmonary artery endothelial cells. Free Radic. Biol. Med 2011, 50, 1107–1113. [Google Scholar]
- Hu, J.; de Souza-Pinto, N.C.; Haraguchi, K.; Hogue, B.A.; Jaruga, P.; Greenberg, M.M.; Dizdaroglu, M.; Bohr, V.A. Repair of formamidopyrimidines in DNA involves different glycosylases: Role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem 2005, 280, 40544–40551. [Google Scholar]
- Stierum, R.H.; Dianov, G.L.; Bohr, V.A. Single-nucleotide patch base excision repair of uracil in DNA by mitochondrial protein extracts. Nucleic Acids Res 1999, 27, 3712–3719. [Google Scholar]
- Akbari, M.; Visnes, T.; Krokan, H.E.; Otterlei, M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair 2008, 7, 605–616. [Google Scholar]
- Liu, P.; Qian, L.; Sung, J.-S.; de Souza-Pinto, N.C.; Zheng, L.; Bogenhagen, D.F.; Bohr, V.A.; Wilson, D.M.; Shen, B.; Demple, B. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell. Biol 2008, 28, 4975–4987. [Google Scholar]
- Boesch, P.; Ibrahim, N.; Dietrich, A.; Lightowlers, R.N. Membrane association of mitochondrial DNA facilitates base excision repair in mammalian mitochondria. Nucleic Acids Res 2010, 38, 1478–1488. [Google Scholar]
- Kalifa, L.; Beutner, G.; Phadnis, N.; Sheu, S.-S.; Sia, E.A. Evidence for a role of FEN1 in maintaining mitochondrial DNA integrity. DNA Repair 2009, 8, 1242–1249. [Google Scholar]
- Copeland, W.C.; Longley, M.J. DNA2 resolves expanding flap in mitochondrial base excision repair. Mol. Cell 2008, 32, 457–458. [Google Scholar]
- Ralph, S.J.; Rodriguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sanchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation-why mitochondria are targets for cancer therapy. Mol. Asp. Med 2010, 31, 145–170. [Google Scholar]
- Mason, P.A.; Matheson, E.C.; Hall, A.G.; Lightowlers, R.N. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res 2003, 31, 1052–1058. [Google Scholar]
- Kraytsberg, Y.; Schwartz, M.; Brown, T.A.; Ebralidse, K.; Kunz, W.S.; Clayton, D.A.; Vissing, J.; Khrapko, K. Recombination of human mitochondrial DNA. Science 2004, 304, 981. [Google Scholar]
- Sage, J.M.; Gildemeister, O.S.; Knight, K.L. Discovery of a novel function for human Rad51: Maintenance of the mitochondrial genome. J. Biol. Chem 2010, 285, 18984–18990. [Google Scholar]
- Lakshmipathy, U.; Campbell, C. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res 1999, 27, 1198–1204. [Google Scholar]
- Liu, P.; Demple, B. DNA repair in mammalian mitochondria: Much more than we thought? Environ. Mol. Mutagen 2010, 51, 417–426. [Google Scholar]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012. [Google Scholar] [CrossRef]
- Poe, B.G.; Navratil, M.; Arriaga, E.A. Absolute quantitation of a heteroplasmic mitochondrial DNA deletion using a multiplex three-primer real-time PCR assay. Anal. Biochem 2007, 362, 193–200. [Google Scholar]
- Tatuch, Y.; Christodoulou, J.; Feigenbaum, A.; Clarke, J.T.; Wherret, J.; Smith, C.; Rudd, N.; Petrova-Benedict, R.; Robinson, B.H. Heteroplasmic mtDNA mutation (T-G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am. J. Hum. Genet 1992, 50, 852–858. [Google Scholar]
- Chinnery, P.F.; Zwijnenburg, P.J.; Walker, M.; Howell, N.; Taylor, R.W.; Lightowlers, R.N.; Bindoff, L.; Turnbull, D.M. Nonrandom tissue distribution of mutant mtDNA. Am. J. Med. Genet 1999, 85, 498–501. [Google Scholar]
- Jarrett, S.G.; Lin, H.; Godley, B.F.; Boulton, M.E. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res 2008, 27, 596–607. [Google Scholar]
- Pitceathly, R.D.; Rahman, S.; Hanna, M.G. Single deletions in mitochondrial DNA—Molecular mechanisms and disease phenotypes in clinical practice. Neuromuscul. Disord 2012, 22, 577–586. [Google Scholar]
- Barreau, E.; Brossas, J.Y.; Courtois, Y.; Treton, J.A. Accumulation of mitochondrial DNA deletions in human retina during aging. Invest. Ophthalmol. Vis. Sci 1996, 37, 384–391. [Google Scholar]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Neufeld, A.H. Increased mitochondrial DNA damage and down-regulation of DNA repair enzymes in aged rodent retinal pigment epithelium and choroid. Mol. Vis 2008, 14, 644–651. [Google Scholar]
- Ballinger, S.W.; van Houten, B.; Jin, G.F.; Conklin, C.A.; Godley, B.F. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp. Eye Res 1999, 68, 765–772. [Google Scholar]
- Jarrett, S.G.; Boulton, M.E. Antioxidant up-regulation and increased nuclear DNA protection play key roles in adaptation to oxidative stress in epithelial cells. Free Radic. Biol. Med 2005, 38, 1382–1391. [Google Scholar]
- Jarrett, S.G.; Boulton, M.E. Poly(ADP-ribose) polymerase offers protection against oxidative and alkylation damage to the nuclear and mitochondrial genomes of the retinal pigment epithelium. Ophthalmic Res 2007, 39, 213–223. [Google Scholar]
- Godley, B.F.; Shamsi, F.A.; Liang, F.Q.; Jarrett, S.G.; Davies, S.; Boulton, M.J. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. Biol. Chem 2005, 280, 21061–21066. [Google Scholar]
- Liang, F.Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res 2003, 76, 397–403. [Google Scholar]
- Kenney, M.C.; Atilano, S.R.; Boyer, D.; Chwa, M.; Chak, G.; Chinichian, S.; Coskun, P.; Wallace, D.C.; Nesburn, A.B.; Udar, N.S. Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Invest. Ophthalmol. Vis. Sci 2010, 51, 4289–4297. [Google Scholar]
- Udar, N.; Atilano, S.R.; Memarzadeh, M.; Boyer, D.S.; Chwa, M.; Lu, S.; Maguen, B.; Langberg, J.; Coskun, P.; Wallace, D.C.; et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 2009, 50, 2966–2974. [Google Scholar]
- Karunadharma, P.P.; Nordgaard, C.L; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar]
- Lin, H.; Xu, H.; Liang, F.Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 2011, 52, 3521–3529. [Google Scholar]
- Park, M.J.; Park, J.H.; Hahm, S.H.; Ko, S.I.; Lee, Y.R.; Chung, J.H.; Sohn, S.Y.; Cho, Y.; Kang, L.W.; Han, Y.S. Repair activities of human 8-oxoguanine DNA glycosylase are stimulated by the interaction with human checkpoint sensor Rad9-Rad1-Hus1 complex. DNA Repair 2009, 8, 1190–1200. [Google Scholar]
- Garinis, G.A.; van der Horst, G.T.; Vijg, J.; Hoeijmakers, J.H. DNA damage and ageing: New-age ideas for an age-old problem. Nat. Cell Biol 2008, 10, 1241–1247. [Google Scholar]
- Dunn, K.C.; Aotaki-Keen, A.E.; Putkey, F.R.; Hjelmeland, L.M. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res 1996, 62, 155–169. [Google Scholar]
- Kalariya, N.M.; Ramana, K.V.; Srivastava, S.K.; van Kuijk, F.J. Genotoxic effects of carotenoid breakdown products in human retinal pigment epithelial cells. Curr. Eye Res 2009, 3, 737–747. [Google Scholar]
- Delaney, S.; Jarem, D.A.; Volle, C.B.; Yennie, C.J. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic. Res 2012, 46, 420–441. [Google Scholar]
- Beard, W.A.; Batra, V.K.; Wilson, S.H. DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine. Mutat. Res 2010, 703, 18–23. [Google Scholar]
- Synowiec, E.; Blasiak, J.; Zaras, M.; Szaflik, J.; Szaflik, J.P. Association between polymorphisms of the DNA base excision repair genes MUTYH and hOGG1 and age–related macular degeneration. Exp. Eye Res 2012, 98, 58–66. [Google Scholar]
- Totan, Y.; Yagci, R.; Bardak, Y.; Ozyurt, H.; Kendir, F.; Yilmaz, G.; Sahin, S.; Sahin, T.U. Oxidative macromolecular damage in age-related macular degeneration. Curr. Eye Res 2009, 34, 1089–1093. [Google Scholar]
- Strunnikova, N.; Hilmer, S.; Flippin, J.; Robinson, M.; Hoffman, E.; Csaky, K.G. Differences in gene expression profiles in dermal fibroblasts from control and patients with age-related macular degeneration elicited by oxidative injury. Free Radic. Biol. Med 2005, 39, 781–796. [Google Scholar]
- Kaarniranta, K.; Salminen, A.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J. Alzheimers Dis 2011, 24, 615–631. [Google Scholar]
- Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Yates, J.R.; Bradley, M.; Moore, A.T.; Bird, A.C. Genetic Factors in AMD Study. Smoking and age related macular degeneration: The number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br. J. Ophthalmol 2006, 90, 75–80. [Google Scholar]
- Hecht, S.S. Progress and challenges in selected areas of tobacco carcinogenesis. Chem. Res. Toxicol 2008, 21, 160–171. [Google Scholar]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar]
- Yang, Z.; Harrison, C.M.; Chuang, G.C.; Ballinger, S.W. The role of tobacco smoke induced mitochondrial damage in vascular dysfunction and atherosclerosis. Mutat. Res 2007, 621, 61–74. [Google Scholar]
- Fischer, F.; Hamann, A.; Osiewacz, H.D. Mitochondrial quality control: An integrated network of pathways. Trends Biochem. Sci 2012, 37, 284–292. [Google Scholar]
- Ballinger, S.W.; Patterson, C.; Yan, C.N.; Doan, R.; Burow, D.L.; Young, C.G.; Yakes, F.M.; van Houten, B.; Ballinger, C.A.; Freeman, B.A.; et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ. Res 2000, 86, 960–966. [Google Scholar]
- Knight-Lozano, C.A.; Young, C.G.; Burow, D.L.; Hu, Z.Y.; Uyeminami, D.; Pinkerton, K.E.; Ischiropoulos, H.; Ballinger, S.W. Cigarette smoke exposure and hypercholesterolemia increase mitochondrial damage in cardiovascular tissues. Circulation 2002, 105, 849–854. [Google Scholar]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar]
- Yang, Z.; Knight, C.A.; Mamerow, M.M.; Vickers, K.; Penn, A.; Postlethwait, E.M.; Ballinger, S.W. Prenatal environmental tobacco smoke exposure promotes adult atherogenesis and mitochondrial damage in apolipoprotein E−/− mice fed a chow diet. Circulation 2004, 110, 3715–3720. [Google Scholar]
- Alexandrov, K.; Rojas, M.; Satarug, S. The critical DNA damage by benzo(a)pyrene in lung tissues of smokers and approaches to preventing its formation. Toxicol. Lett 2010, 198, 63–68. [Google Scholar]
- Blasiak, J.; Synowiec, E.; Salminen, A.; Kaarniranta, K. Genetic variability in dna repair proteins in age-related macular degeneration. Int. J. Mol. Sci 2012, 13, 13378–13397. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and Nuclear DNA Damage and Repair in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2013, 14, 2996-3010. https://doi.org/10.3390/ijms14022996
Blasiak J, Glowacki S, Kauppinen A, Kaarniranta K. Mitochondrial and Nuclear DNA Damage and Repair in Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2013; 14(2):2996-3010. https://doi.org/10.3390/ijms14022996
Chicago/Turabian StyleBlasiak, Janusz, Sylwester Glowacki, Anu Kauppinen, and Kai Kaarniranta. 2013. "Mitochondrial and Nuclear DNA Damage and Repair in Age-Related Macular Degeneration" International Journal of Molecular Sciences 14, no. 2: 2996-3010. https://doi.org/10.3390/ijms14022996