Regulation of Phosphatidylethanolamine Homeostasis — The Critical Role of CTP:Phosphoethanolamine Cytidylyltransferase (Pcyt2)


Abstract
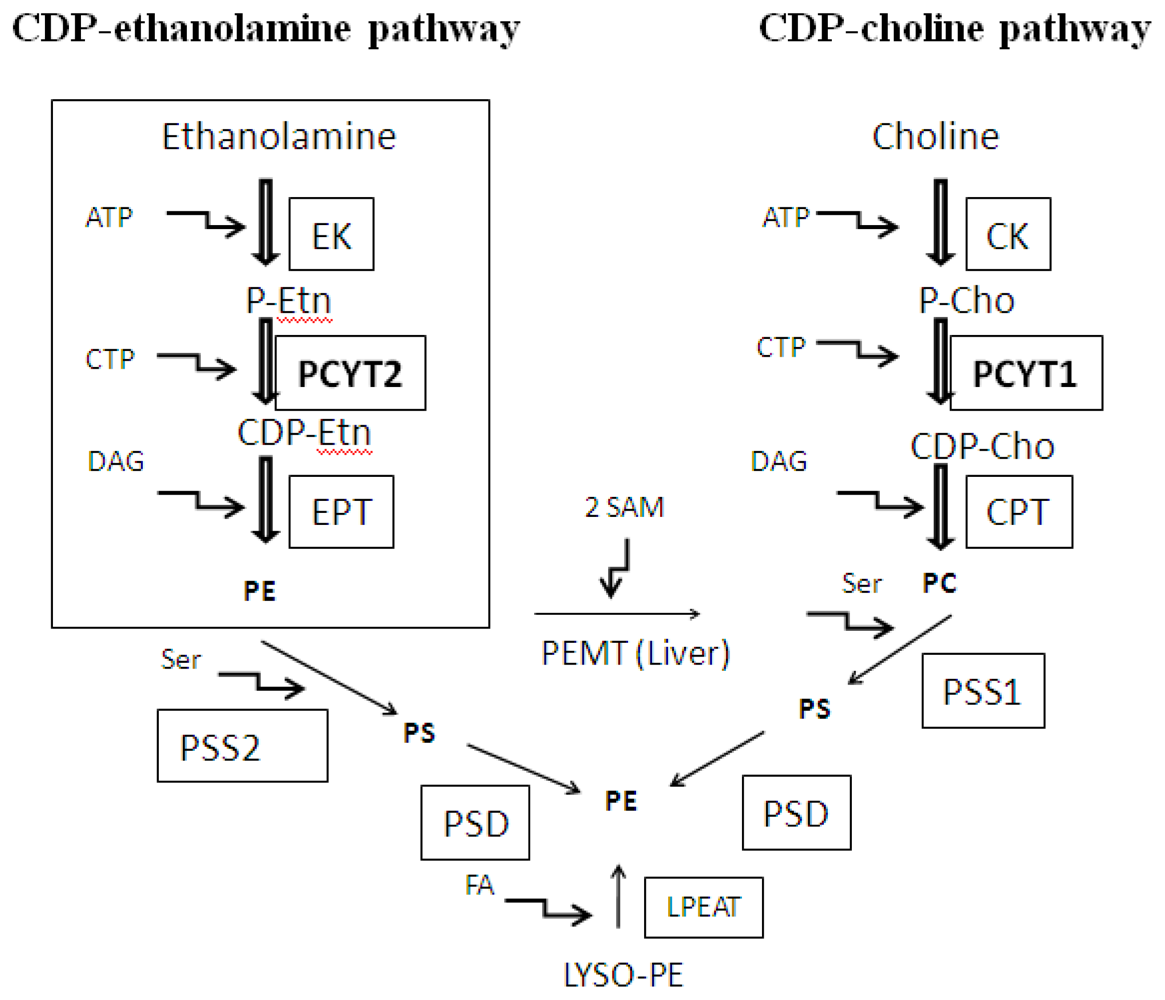
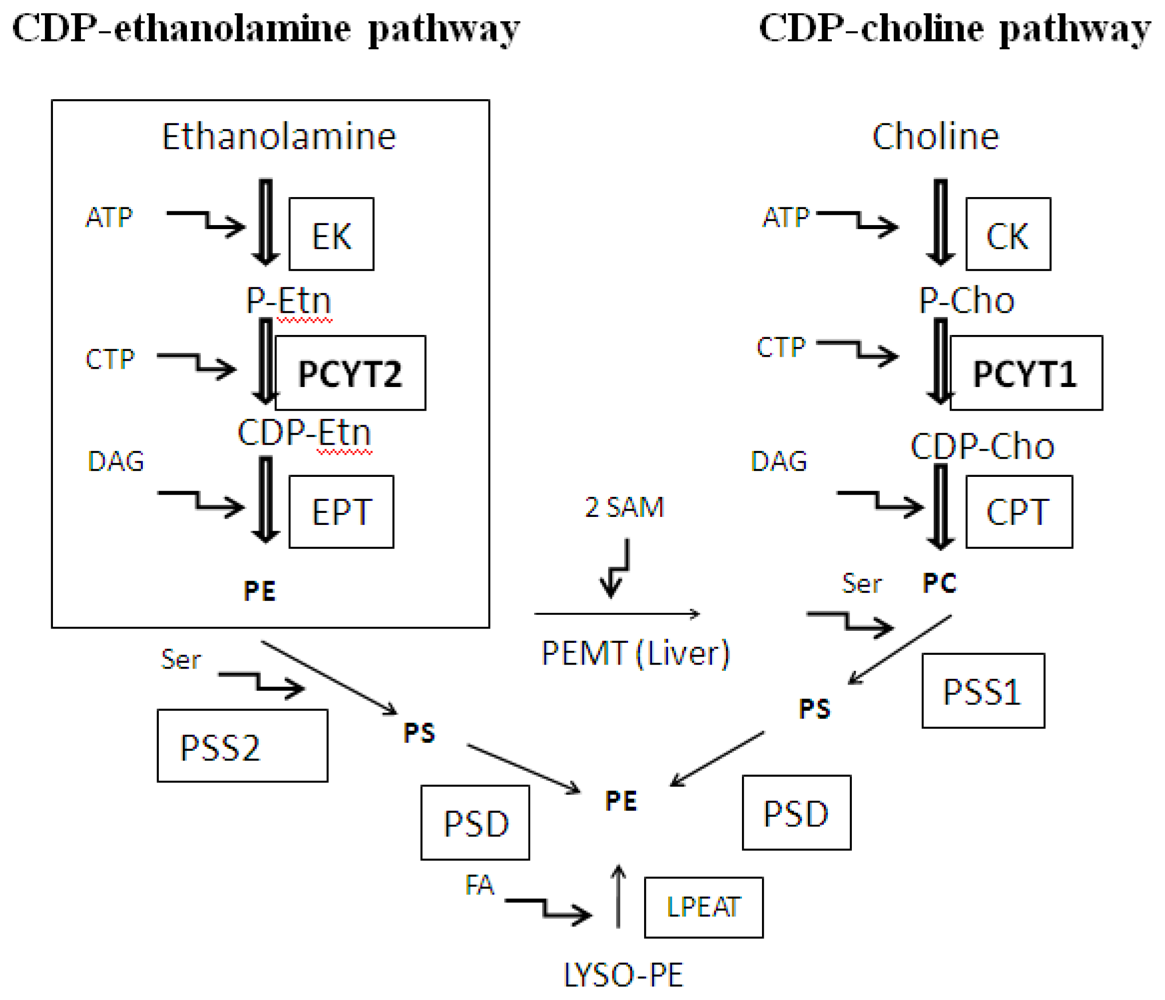
:1. Introduction
2. Substrate Utilization and Activity of Pcyt2
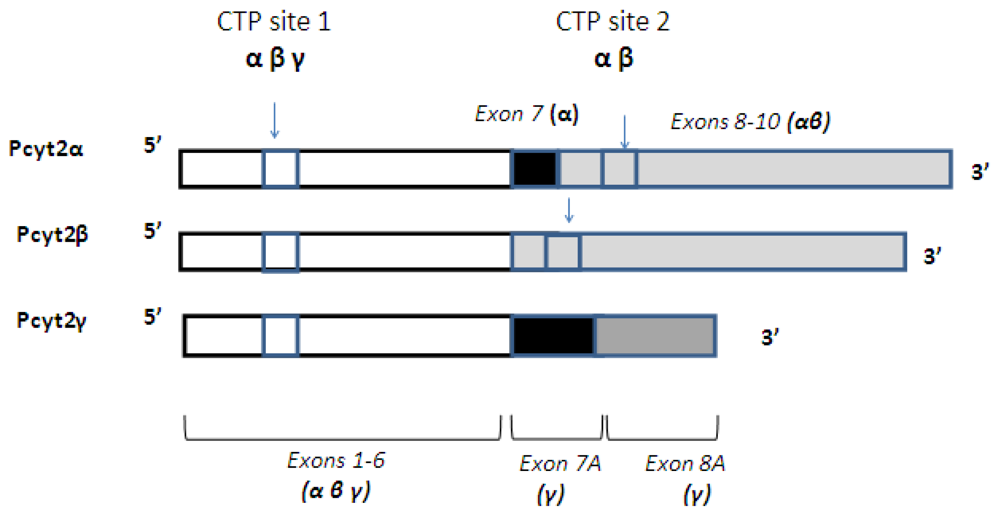
3. Alternative Splicing of Pcyt2
4. Transcriptional Regulation of Pcyt2
5. The Function of Pcyt2 in Cancer Cell Growth
6. The Role of Pcyt2 and PE in Post-Translational Modifications of Proteins
7. The Essentiality and Anti Obesity Function of Pcyt2
8. Pcyt2 Expression in the Metabolic Syndrome and Related Disorders
9. Conclusions
Acknowledgments
Conflict of Interest
References
- Deeba, F.; Tahseen, H.N.; Sharad, K.S.; Ahmad, N.; Akhtar, S.; Saleemuddin, M.; Mohammad, O. Phospholipid diversity: Correlation with membrane-membrane fusion events. Biochim. Biophys. Acta 2005, 1669, 170–181. [Google Scholar]
- Emoto, K.; Kobayashi, T.; Yamaji, A.; Aizawa, H.; Yahara, I.; Inoue, K.; Umeda, M. Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proc. Natl. Acad. Sci. USA 1996, 93, 12867–12872. [Google Scholar]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar]
- Emoto, K.; Toyama-Sorimachi, N.; Karasuyama, H.; Inoue, K.; Umeda, M. Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp. Cell Res 1997, 232, 430–434. [Google Scholar]
- Vance, J.E.; Tasseva, G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta 2012, in press. [Google Scholar]
- Gibellini, F.; Smith, T.K. The Kennedy pathway—de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar]
- Sundler, R. Ethanolaminephosphate cytidylyltransferase. Purification and characterization of the enzyme from rat liver. J. Biol. Chem 1975, 250, 8585–8590. [Google Scholar]
- Bladergroen, B.A.; van Golde, L.M. CTP:Phosphoethanolamine cytidylyltransferase. Biochim. Biophys. Acta 1997, 1348, 91–99. [Google Scholar]
- Ross, B.M.; Moszczynska, A.; Blusztajn, J.K.; Sherwin, A.; Lozano, A.; Kish, S.J. Phospholipid biosynthetic enzymes in human brain. Lipids 1997, 32, 351–358. [Google Scholar]
- Vermeulen, P.S.; Geelen, M.J.; van Golde, L.M. Substrate specificity of CTP: phosphoethanolamine cytidylyltransferase purified from rat liver. Biochim. Biophys. Acta 1994, 1211, 343–349. [Google Scholar]
- Vermeulen, P.S.; Tijburg, L.B.; Geelen, M.J.; van Golde, L.M. Immunological characterization, lipid dependence, and subcellular localization of CTP:phosphoethanolamine cytidylyltransferase purified from rat liver. comparison with CTP:phosphocholine cytidylyltransferase. J. Biol. Chem 1993, 268, 7458–7464. [Google Scholar]
- Houweling, M.; Tijburg, L.B.; Vaartjes, W.J.; van Golde, L.M. Phosphatidylethanolamine metabolism in rat liver after partial hepatectomy. control of biosynthesis of phosphatidylethanolamine by the availability of ethanolamine. Biochem. J 1992, 283, 55–61. [Google Scholar]
- Tijburg, L.B.; Vermeulen, P.S.; Schmitz, M.G.; van Golde, L.M. Okadaic acid inhibits phosphatidylethanolamine biosynthesis in rat hepatocytes. Biochem. Biophys. Res. Commun 1992, 182, 1226–1231. [Google Scholar]
- Tijburg, L.B.; Houweling, M.; Geelen, J.H.; van Golde, L.M. Stimulation of phosphatidylethanolamine synthesis in isolated rat hepatocytes by phorbol 12-myristate 13-acetate. Biochim. Biophys. Acta 1987, 922, 184–190. [Google Scholar]
- Bleijerveld, O.B.; Klein, W.; Vaandrager, A.B.; Helms, J.B.; Houweling, M. Control of the CDPethanolamine pathway in mammalian cells: Effect of ctp:phosphoethanolamine cytidylyltransferase overexpression and the amount of intracellular diacylglycerol. Biochem. J 2004, 379, 711–719. [Google Scholar]
- Tijburg, L.B.; Schuurmans, E.A.; Geelen, M.J.; van Golde, L.M. Effects of vasopressin on the synthesis of phosphatidylethanolamines and phosphatidylcholines by isolated rat hepatocytes. Biochim. Biophys. Acta 1987, 919, 49–57. [Google Scholar]
- Van Hellemond, J.J.; Slot, J.W.; Geelen, M.J.; van Golde, L.M.; Vermeulen, P.S. Ultrastructural localization of CTP:phosphoethanolamine cytidylyltransferase in rat liver. J. Biol. Chem 1994, 269, 15415–15418. [Google Scholar]
- Dechamps, S.; Wengelnik, K.; Berry-Sterkers, L.; Cerdan, R.; Vial, H.J.; Gannoun-Zaki, L. The Kennedy phospholipid biosynthesis pathways are refractory to genetic disruption in plasmodium berghei and therefore appear essential in blood stages. Mol. Biochem. Parasitol 2010, 173, 69–80. [Google Scholar]
- Bladergroen, B.A.; Geelen, M.J.; Reddy, A.C.; Declercq, P.E.; van Golde, L.M. Channelling of intermediates in the biosynthesis of phosphatidylcholine and phosphatidylethanolamine in mammalian cells. Biochem. J 1998, 334, 511–517. [Google Scholar]
- Min-Seok, R.; Kawamata, Y.; Nakamura, H.; Ohta, A.; Takagi, M. Isolation and characterization of ECT1 gene encoding CTP: phosphoethanolamine cytidylyltransferase of Saccharomyces cerevisiae. J. Biochem 1996, 120, 1040–1047. [Google Scholar]
- Nakashima, A.; Hosaka, K.; Nikawa, J. Cloning of a human cDNA for CTP-phosphoethanolamine cytidylyltransferase by complementation in vivo of a yeast mutant. J. Biol. Chem 1997, 272, 9567–9572. [Google Scholar]
- Bladergroen, B.A.; Houweling, M.; Geelen, M.J.; van Golde, L.M. Cloning and expression of CTP:phosphoethanolamine cytidylyltransferase cDNA from rat liver. Biochem. J 1999, 343, 107–114. [Google Scholar]
- Poloumienko, A.; Cote, A.; Quee, A.T.; Zhu, L.; Bakovic, M. Genomic organization and differential splicing of the mouse and human Pcyt2 genes. Gene 2004, 325, 145–155. [Google Scholar]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–229. [Google Scholar]
- Tie, A.; Bakovic, M. Alternative splicing of CTP:phosphoethanolamine cytidylyltransferase produces two isoforms that differ in catalytic properties. J. Lipid Res 2007, 48, 2172–2181. [Google Scholar]
- Zhu, L.; Bakovic, M. Breast cancer cells adapt to metabolic stress by increasing ethanolamine phospholipid synthesis and CTP:ethanolaminephosphate cytidylyltransferase-Pcyt2 activity. Biochem. Cell Biol 2012, 90, 188–199. [Google Scholar]
- Expasy-Swiss Institute of Biotechnology Bioinformatics Resource Portal. Available online: http://www.expacy.org/ accessed on 1 October 2012.
- Johnson, C.M.; Yuan, Z.; Bakovic, M. Characterization of transcription factors and cis-acting elements that regulate human CTP: phosphoethanolamine cytidylyltransferase (Pcyt2). Biochim. Biophys. Acta 2005, 1735, 230–235. [Google Scholar]
- Zhu, L.; Johnson, C.; Bakovic, M. Stimulation of the human CTP:phosphoethanolamine cytidylyltransferase gene by early growth response protein 1. J. Lipid Res 2008, 49, 2197–2211. [Google Scholar]
- Zhu, L.; Michel, V.; Bakovic, M. Regulation of the mouse CTP: Phosphoethanolamine cytidylyltransferase gene Pcyt2 during myogenesis. Gene 2009, 447, 51–59. [Google Scholar]
- Zhu, L.; Bakovic, M. Liver X receptor agonists inhibit the phospholipid regulatory gene CTP: Phosphoethanolamine cytidylyltransferase-Pcyt2. Res. Lett. Biochem. 2008, 2008, 801849:1–801849:5. [Google Scholar]
- Ando, H.; Horibata, Y.; Yamashita, S.; Oyama, T.; Sugimoto, H. Low-density lipoprotein and oxysterols suppress the transcription of CTP: Phosphoethanolamine cytidylyltransferase in vitro. Biochim. Biophys. Acta 2010, 1801, 487–495. [Google Scholar]
- Shan, L.; Chen, Y.A.; Davis, L.; Han, G.; Zhu, W.; Molina, A.D.; Arango, H.; Lapolla, J.P.; Hoffman, M.S.; Sellers, T.; et al. Measurement of phospholipids may improve diagnostic accuracy in ovarian cancer. PLoS One 2012, 7, e46846. [Google Scholar]
- Bakovic, M.; Fullerton, M.D.; Michel, V. Metabolic and molecular aspects of ethanolamine phospholipid biosynthesis: The role of CTP:Phosphoethanolamine cytidylyltransferase (Pcyt2). Biochem. Cell Biol 2007, 85, 283–300. [Google Scholar]
- Vink, S.R.; van Blitterswijk, W.J.; Schellens, J.H.; Verheij, M. Rationale and clinical application of alkylphospholipid analogues in combination with radiotherapy. Cancer Treat. Rev 2007, 33, 191–202. [Google Scholar]
- Reddy, J.K.; Azarnoff, D.L.; Hignite, C.E. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature 1980, 283, 397–398. [Google Scholar]
- Mizuguchi, H.; Kudo, N.; Ohya, T.; Kawashima, Y. Effects of tiadenol and di-(2-ethylhexyl)phthalate on the metabolism of phosphatidylcholine and phosphatidylethanolamine in the liver of rats: Comparison with clofibric acid. Biochem. Pharmacol 1999, 57, 869–876. [Google Scholar]
- Kudo, N.; Mizuguchi, H.; Yamamoto, A.; Kawashima, Y. Alterations by perfluorooctanoic acid of glycerolipid metabolism in rat liver. Chem. Biol. Interact 1999, 118, 69–83. [Google Scholar]
- Boggs, K.P.; Rock, C.O.; Jackowski, S. Lysophosphatidylcholine and 1-O-octadecyl-2-O-methylrac-glycero-3-phosphocholine inhibit the CDP-choline pathway of phosphatidylcholine synthesis at the ctp:phosphocholine cytidylyltransferase step. J. Biol. Chem 1995, 270, 7757–7764. [Google Scholar]
- Jimenez-Lopez, J.M.; Carrasco, M.P.; Segovia, J.L.; Marco, C. Hexadecylphosphocholine inhibits phosphatidylcholine synthesis via both the methylation of phosphatidylethanolamine and CDP-choline pathways in HepG2 cells. Int. J. Biochem. Cell Biol 2004, 36, 153–161. [Google Scholar]
- Zhou, X.; Arthur, G. Effect of 1-O-octadecyl-2-O-methyl-glycerophosphocholine on phosphatidylcholine and phosphatidylethanolamine synthesis in MCF-7 and A549 cells and its relationship to inhibition of cell proliferation. Eur. J. Biochem 1995, 232, 881–888. [Google Scholar]
- Bhoumik, A.; Fichtman, B.; Derossi, C.; Breitwieser, W.; Kluger, H.M.; Davis, S.; Subtil, A.; Meltzer, P.; Krajewski, S.; Jones, N.; et al. Suppressor role of activating transcription factor 2 (ATF2) in skin cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 1674–1679. [Google Scholar]
- Provenzani, A.; Fronza, R.; Loreni, F.; Pascale, A.; Amadio, M.; Quattrone, A. Global alterations in mRNA polysomal recruitment in a cell model of colorectal cancer progression to metastasis. Carcinogenesis 2006, 27, 1323–1333. [Google Scholar]
- Kim, B.C.; Shin, A.; Hong, C.W.; Sohn, D.K.; Han, K.S.; Ryu, K.H.; Park, B.J.; Nam, J.H.; Park, J.W.; Chang, H.J.; et al. Association of colorectal adenoma with components of metabolic syndrome. Cancer Causes Control 2012, 23, 727–735. [Google Scholar]
- Selga, E.; Morales, C.; Noe, V.; Peinado, M.A.; Ciudad, C.J. Role of Caveolin 1, E-Cadherin, Enolase 2 and PKCalpha on resistance to methotrexate in human HT29 colon cancer cells. BMC Med. Genomics 2008, 1, 35. [Google Scholar]
- Selga, E.; Oleaga, C.; Ramirez, S.; de Almagro, M.C.; Noe, V.; Ciudad, C.J. Networking of differentially expressed genes in human cancer cells resistant to methotrexate. Genome Med. 2009, 1, 83.1–83.16. [Google Scholar]
- Crunkhorn, S.; Dearie, F.; Mantzoros, C.; Gami, H.; da Silva, W.S.; Espinoza, D.; Faucette, R.; Barry, K.; Bianco, A.C.; Patti, M.E. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: Potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J. Biol. Chem 2007, 282, 15439–15450. [Google Scholar]
- Yamawaki, H.; Iwai, N. Cytotoxicity of water-soluble fullerene in vascular endothelial cells. Am. J. Physiol. Cell. Physiol 2006, 290, C1495–C1502. [Google Scholar]
- Yang, X.; Pratley, R.E.; Tokraks, S.; Bogardus, C.; Permana, P.A. Microarray profiling of skeletal muscle tissues from equally obese, non-diabetic insulin-sensitive and insulin-resistant pima indians. Diabetologia 2002, 45, 1584–1593. [Google Scholar]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 2009, 9, 327–338. [Google Scholar]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab 2007, 6, 38–54. [Google Scholar]
- Granneman, J.G.; Li, P.; Zhu, Z.; Lu, Y. Metabolic and cellular plasticity in white adipose tissue I: Effects of β3-adrenergic receptor activation. Am. J. Physiol. Endocrinol. Metab 2005, 289, E608–E616. [Google Scholar]
- Koza, R.A.; Nikonova, L.; Hogan, J.; Rim, J.S.; Mendoza, T.; Faulk, C.; Skaf, J.; Kozak, L.P. Changes in gene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet 2006, 2, e81. [Google Scholar]
- De Wit, N.J.; Bosch-Vermeulen, H.; de Groot, P.J.; Hooiveld, G.J.; Bromhaar, M.M.; Jansen, J.; Muller, M.; van der Meer, R. The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC Med. Genomics 2008, 1, 1–14. [Google Scholar]
- Li, H.; Xie, Z.; Lin, J.; Song, H.; Wang, Q.; Wang, K.; Su, M.; Qiu, Y.; Zhao, T.; Song, K.; et al. Transcriptomic and metabonomic profiling of obesity-prone and obesity-resistant rats under high fat diet. J. Proteome Res. 2008, 7, 4775–4783. [Google Scholar]
- Bektas, M.; Allende, M.L.; Lee, B.G.; Chen, W.; Amar, M.J.; Remaley, A.T.; Saba, J.D.; Proia, R.L. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem 2010, 285, 10880–10889. [Google Scholar]
- Huster, D.; Purnat, T.D.; Burkhead, J.L.; Ralle, M.; Fiehn, O.; Stuckert, F.; Olson, N.E.; Teupser, D.; Lutsenko, S. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of wilson disease. J. Biol. Chem 2007, 282, 8343–8355. [Google Scholar]
- Tanaka, T.; Kono, T.; Terasaki, F.; Yasui, K.; Soyama, A.; Otsuka, K.; Fujita, S.; Yamane, K.; Manabe, M.; Usui, K.; et al. Thiamine prevents obesity and obesity-associated metabolic disorders in OLETF rats. J. Nutr. Sci. Vitaminol. (Tokyo) 2010, 56, 335–346. [Google Scholar]
- Signorell, A.; Jelk, J.; Rauch, M.; Butikofer, P. Phosphatidylethanolamine is the precursor of the ethanolamine phosphoglycerol moiety bound to eukaryotic elongation Factor 1A. J. Biol. Chem 2008, 283, 20320–20329. [Google Scholar]
- Gibellini, F.; Hunter, W.N.; Smith, T.K. The ethanolamine branch of the kennedy pathway is essential in the bloodstream form of Trypanosoma brucei. Mol. Microbiol 2009, 73, 826–843. [Google Scholar]
- Signorell, A.; Gluenz, E.; Rettig, J.; Schneider, A.; Shaw, M.K.; Gull, K.; Butikofer, P. Perturbation of phosphatidylethanolamine synthesis affects mitochondrial morphology and cell-cycle progression in procyclic-form Trypanosoma brucei. Mol. Microbiol 2009, 72, 1068–1079. [Google Scholar]
- Girardi, J.P.; Pereira, L.; Bakovic, M. De novo synthesis of phospholipids is coupled with autophagosome formation. Med. Hypotheses 2011, 77, 1083–1087. [Google Scholar]
- Pereira, L.; Girardi, J.P.; Bakovic, M. Forms, crosstalks, and the role of phospholipid biosynthesis in autophagy. Int. J. Cell. Biol. 2012, 2012, 931956:1–931956:10. [Google Scholar]
- Bakovic, M. University of Guelph: Guelph, ON, Canada; Unpublished work; 2012. [Google Scholar]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar]
- Li, H.; Liu, Y.; Shin, S.; Sun, Y.; Loring, J.F.; Mattson, M.P.; Rao, M.S.; Zhan, M. Transcriptome coexpression map of human embryonic stem cells. BMC Genomics 2006, 7, 103.1–103.15. [Google Scholar]
- Giritharan, G.; Li, M.W.; de Sebastiano, F.; Esteban, F.J.; Horcajadas, J.A.; Lloyd, K.C.; Donjacour, A.; Maltepe, E.; Rinaudo, P.F. Effect of ICSI on gene expression and development of mouse preimplantation embryos. Hum. Reprod 2010, 25, 3012–3024. [Google Scholar]
- Swiss, V.A.; Nguyen, T.; Dugas, J.; Ibrahim, A.; Barres, B.; Androulakis, I.P.; Casaccia, P. Identification of a gene regulatory network necessary for the initiation of oligodendrocyte differentiation. PLoS One 2011, 6, e18088. [Google Scholar]
- Mizoi, J.; Nakamura, M.; Nishida, I. Defects in CTP:phosphorylethanolamine cytidylyltransferase affect embryonic and postembryonic development in arabidopsis. Plant Cell 2006, 18, 3370–3385. [Google Scholar]
- Fullerton, M.D.; Hakimuddin, F.; Bakovic, M. Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2). Mol. Cell. Biol 2007, 27, 3327–3336. [Google Scholar]
- Dobrosotskaya, I.Y.; Seegmiller, A.C.; Brown, M.S.; Goldstein, J.L.; Rawson, R.B. Regulation of SREBP processing and membrane lipid production by phospholipids in drosophila. Science 2002, 296, 879–883. [Google Scholar]
- Lim, H.Y.; Wang, W.; Wessells, R.J.; Ocorr, K.; Bodmer, R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in drosophila. Genes Dev 2011, 25, 189–200. [Google Scholar]
- Leonardi, R.; Frank, M.W.; Jackson, P.D.; Rock, C.O.; Jackowski, S. Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J. Biol. Chem 2009, 284, 27077–27089. [Google Scholar]
- Fullerton, M.D.; Hakimuddin, F.; Bonen, A.; Bakovic, M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J. Biol. Chem 2009, 284, 25704–25713. [Google Scholar]
- Steenbergen, R.; Nanowski, T.S.; Beigneux, A.; Kulinski, A.; Young, S.G.; Vance, J.E. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem 2005, 280, 40032–40040. [Google Scholar]
- Fullerton, M.D.; Bakovic, M. Complementation of the metabolic defect in CTP:phosphoethanolamine cytidylyltransferase (Pcyt2)-deficient primary hepatocytes. Metabolism 2010, 59, 1691–1700. [Google Scholar]
- Singh, R.K.; Fullerton, M.D.; Vine, D.; Bakovic, M. Mechanism of hypertriglyceridemia in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J. Lipid Res 2012, 53, 1811–1822. [Google Scholar]
- Hussain, M.M.; Rava, P.; Walsh, M.; Rana, M.; Iqbal, J. Multiple functions of microsomal triglyceride transfer protein. Nutr. Metab. (Lond) 2012, 9, 1–16. [Google Scholar]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol 2011, 12, 408–415. [Google Scholar]
- Jotwani, A.; Richerson, D.; Motta, I.; Julca-Zevallos, O.; Melia, T. Approaches to the study of Atg8-mediated membrane dynamics in vitro. Methods Cell Biol 2012, 108, 93–116. [Google Scholar]
- Li, L.; Chen, Y.; Gibson, S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell. Signal 2012, 25, 50–65. [Google Scholar]
- Houweling, M.; Klein, W.; Geelen, M.J. Regulation of phosphatidylcholine and phosphatidylethanolamine synthesis in rat hepatocytes by 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR). Biochem. J 2002, 362, 97–104. [Google Scholar]
- Leick, L.; Fentz, J.; Bienso, R.S.; Knudsen, J.G.; Jeppesen, J.; Kiens, B.; Wojtaszewski, J.F.; Pilegaard, H. PGC-1{α} is required for AICAR-induced expression of GLUT4 and mitochondrial proteins in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab 2010, 299, E456–E465. [Google Scholar]
- Erickson, K.A.; Smith, M.E.; Anthonymuthu, T.S.; Evanson, M.J.; Brassfield, E.S.; Hodson, A.E.; Bressler, M.A.; Tucker, B.J.; Thatcher, M.O.; Prince, J.T.; et al. AICAR inhibits ceramide biosynthesis in skeletal muscle. Diabetol. Metab. Syndr. 2012, 4, 1–7. [Google Scholar]
- Wei, P.; Pan, D.; Mao, C.; Wang, Y.X. RNF34 is a cold-regulated E3 ubiquitin ligase for PGC-1α and modulates brown fat cell metabolism. Mol. Cell. Biol 2012, 32, 266–275. [Google Scholar]
- Li, Z.; Agellon, L.B.; Vance, D.E. Phosphatidylcholine homeostasis and liver failure. J. Biol. Chem 2005, 280, 37798–37802. [Google Scholar]
- Page, G.L.; Laight, D.; Cummings, M.H. Thiamine deficiency in diabetes mellitus and the impact of thiamine replacement on glucose metabolism and vascular disease. Int. J. Clin. Pract 2011, 65, 684–690. [Google Scholar]
- Rothig, H.J.; Reinauer, H.; Hollmann, S. Synthesis rate of phospholipids, cytochrome b5 and P450 in thiamine deficient rats following treatment with thiamine. Hoppe Seylers Z. Physiol. Chem 1972, 353, 1562. [Google Scholar]
- Czesny, S.; Dettmers, J.M.; Rinchard, J.; Dabrowski, K. Linking egg thiamine and fatty acid concentrations of lake michigan lake trout with early life stage mortality. J. Aquat. Anim. Health 2009, 21, 262–271. [Google Scholar]

{kind=link}
{kind=link}
| GEO Reference | Pcyt2 Expression | Study summary | Reference |
|---|---|---|---|
| GDS2648/1420493_a_at/ Pcyt2/Mus musculus /The effect of palmitate on myoblast cell line | Up-regulated in the presence of palmitate | Analysis: C2C12 myotubes Function: Palmitate decreases the expression of PPAR co-activator 1 (PGC-1) which increases lipogenesis. Research goal: To determine a link between over-nutrition, obesity, and PGC-1 expression | [47] |
| GDS1553/209577_at/ PCYT2/Homo sapiens/Fullerene effect on vascular endothelial cells | Up-regulated in cells treated with fullerane | Analysis: Umbilical vein endothelial cells Function: Nanomaterials are known for its ability to potentially injure endothelial cells which may cause cell death. Research goal: To get an insight into the effects of a nanomaterial (fullerenes) on endothelial injury and toxicity. | [48] |
| GDS157/D84307_at/ PCYT2 /Homo sapiens/Type 2 diabetes and insulin resistance (HuGeneFL) | Down-regulated in insulin-resistant (IR) muscle | Analysis: Vastus lateralis muscle samples of insulin-sensitive and IR equally obese, non-diabetic Pima Indians. Research goal: Identification of differentially expressed skeletal muscle genes in insulin resistance. | [49] |
| GDS3666/A_51_P432504 /Pcyt2/Mus musculus/SIRT1 deficiency effect on the liver | Up-regulated in Sirtuin (SIRT1) null mice | Analysis: Liver specific SIRT1 knockout (SIRT1 LKO) C57BL/6 mice fed ad libitum Study goal: To examine the role of SIRT1 in the regulation of hepatic lipid homeostasis. Function: SIRT1, a NAD+-dependent protein deacetylase, is a significant regulator of energy metabolism in response to changes in the availability of nutrients. | [50] |
| GDS2813/1420493_a_at/ Pcyt2/Mus musculus/Brown adipose tissue | Up-regulated in brown adipose tissue in comparison to white adipose tissue | Analysis: Interscapular brown fat tissue and epididymal white fat from male C57Bl6 mice. Research goal: To identify differential gene expression profiles between brown and white adipose tissue. | [51] |
| GDS1225/103914_at/ Pcyt2/Mus musculus/White adipose tissue remodeling: response to β3-adrenergic receptor activation | Expression level decreases from day 1 to day 6 of treatments | Analysis: Epididymal white adipose tissue from 3 to 4 months old male Bl6 mice treated with an agonist of beta(3)-adrenergic receptors(CL 316243) for 0, 1, 3 or 6 days. Function: CL 316243 caused remodeling of white adipose tissue and expanded its catabolic activity. Research goal: Investigation of potential anti-diabetic and anti-obesity effect of CL 316243. | [52] |
| GDS1225/103914_at/ Pcyt2/Mus musculus/White adipose tissue remodeling: response to β3-adrenergic receptor activation | Expression level decreases from day 1 to day 6 of treatments | Analysis: Epididymal white adipose tissue from 3 to 4 months old male Bl6 mice treated with an agonist of beta(3)-adrenergic receptors(CL 316243) for 0, 1, 3 or 6 days. Function: CL 316243 caused remodeling of white adipose tissue and expanded its catabolic activity. Research goal: Investigation of potential anti-diabetic and anti-obesity effect of CL 316243. | [52] |
| GDS2319/357445/Pcyt2/ Mus musculus/High and low weight gainers: adipose tissue | Up-regulated in adipose tissue of high-weight gainers | Analysis: Inguinal adipose tissue of C57BL/6J males exhibiting high or low weight gain after 4 weeks on a high-fat diet. Function: Genes involved in vascularization and tissue remodeling control susceptibility to obesogenic phenotype. Research goal: To examine the role of epigenetic mechanisms in the susceptibility to obesity. | [53] |
| GDS3357/1420493_a_at/ Pcyt2/Mus musculus/High dietary fat effect on small intestine: time course | Up-regulated in distal, proximal and middle part of small intestine in animals fed high fat diet | Analysis: Small intestines of male C57BL/6J rodents fed a powdered high-fat purified diet for up to 8 weeks. Research goal: To examine the array of genes involved into molecular mechanism of diet induced obesity and insulin resistance and the role of small intestine in these processes. | [54] |
| GDS3677/rn7664/Pcyt2/ Rattus norvegicus/Highfat-diet model: liver | Up-regulated in obesity-resistant rats | Analysis: Hepatic transcript profile using cDNA microarrays in Obesity-prone(OP) and Obesity resistant(OR) phenotypes in Wistar rats on HFD for 16 weeks. Research goal: mRNA and metabolomic profiling of OP vs OR. | [55] |
| GDS3654/1420493_a_at/ Pcyt2/Mus musculus Sphingosine 1-phosphate lyase deficiency effect on liver | Down-regulated in S1P null mice | Analysis: Liver of mice (C57BL6/129sv) lacking sphingosine 1-phosphate lyase (S1P). Littermate Sgpl1+/+(wild-type) and Sgpl1+/− mice were used as controls. Function: S1P lyase controls the final step in shingolipid degradation to produce P-Etn and a fatty aldehyde. Research goal: To establish the link between the level of shingolipids and metabolic diseases. | [56] |
| GDS2509 1420493_a_at/Pcyt2/ Mus musculus/Wilsone disease model | Down-regulated in ATP7B null mice | Analysis: Livers of copper-transporting ATPase ATP7B null animals. Function: Genetic inactivation of ATP7B causes Wilson’s Disease (WD), a severe metabolic disorder associated with intracellular copper overload. Research goal: To provide insight into the initial events of copper-dependent liver pathology in WD. | [57] |
| GDS3682/256066/Pcyt2/ Rattus norvegicus/Thiamine effect on liver in type-2 diabetic rats (OLETF rats) | Up-regulated by Thiamine supplementation | Analysis: Liver blood parameters and cardiac functions were monitored in OLETF male rats on thiamine treatment for 51 weeks. Function: Thiamine treatment influenced obesity through the reduction of visceral adipose tissue. Research goal: The impact of thiamine supplementation on obesity and metabolic disorders in rats. | [58] |
| GDS3330/209577_at/ PCYT2/Homo Sapiens/Methotrexate resistance in cancer | Up-regulated in resistant in comparison to sensitive HT29 cells | Analysis: Cancer cells sensitive or resistant to methotrexate (MTX). Function: MTX is used in the treatment of cancer, but long term treatment may lead to drug resistance. Research goal: Networking of the genes differentially expressed in cell lines resistant to MTX. | [45,46] |
| GDS3334/2630092/Pcyt2/ Mus musculus/Skin carcinogenesis model | Down regulated in ATF2 null in comparison to WT | Analysis of papillomas initiated by DMBA/TPA treatment of epidermal keratinocytes deficient for activating transcriptional factor 2 (ATF2). Function: ATF2 regulates transcription in response to stress and growth factor stimuli Resarch goal: To get an insight into the role of ATF2 in skin cancer. | [42] |
| GDS756/209577_at/ PCYT2/Homo sapiens | Down-regulated in metastatic colon tumor | Analysis: Differential gene expression between SW480, a primary tumor colon cancer cell line, and SW620, an isogenic metastatic colon cancer cell line. Research goal: To get insight into the progression of cancer from primary tumor growth to metastasis. | [43] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pavlovic, Z.; Bakovic, M. Regulation of Phosphatidylethanolamine Homeostasis — The Critical Role of CTP:Phosphoethanolamine Cytidylyltransferase (Pcyt2). Int. J. Mol. Sci. 2013, 14, 2529-2550. https://doi.org/10.3390/ijms14022529
Pavlovic Z, Bakovic M. Regulation of Phosphatidylethanolamine Homeostasis — The Critical Role of CTP:Phosphoethanolamine Cytidylyltransferase (Pcyt2). International Journal of Molecular Sciences. 2013; 14(2):2529-2550. https://doi.org/10.3390/ijms14022529
Chicago/Turabian StylePavlovic, Zvezdan, and Marica Bakovic. 2013. "Regulation of Phosphatidylethanolamine Homeostasis — The Critical Role of CTP:Phosphoethanolamine Cytidylyltransferase (Pcyt2)" International Journal of Molecular Sciences 14, no. 2: 2529-2550. https://doi.org/10.3390/ijms14022529