Alteration of Dynein Function Affects α-Synuclein Degradation via the Autophagosome-Lysosome Pathway

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
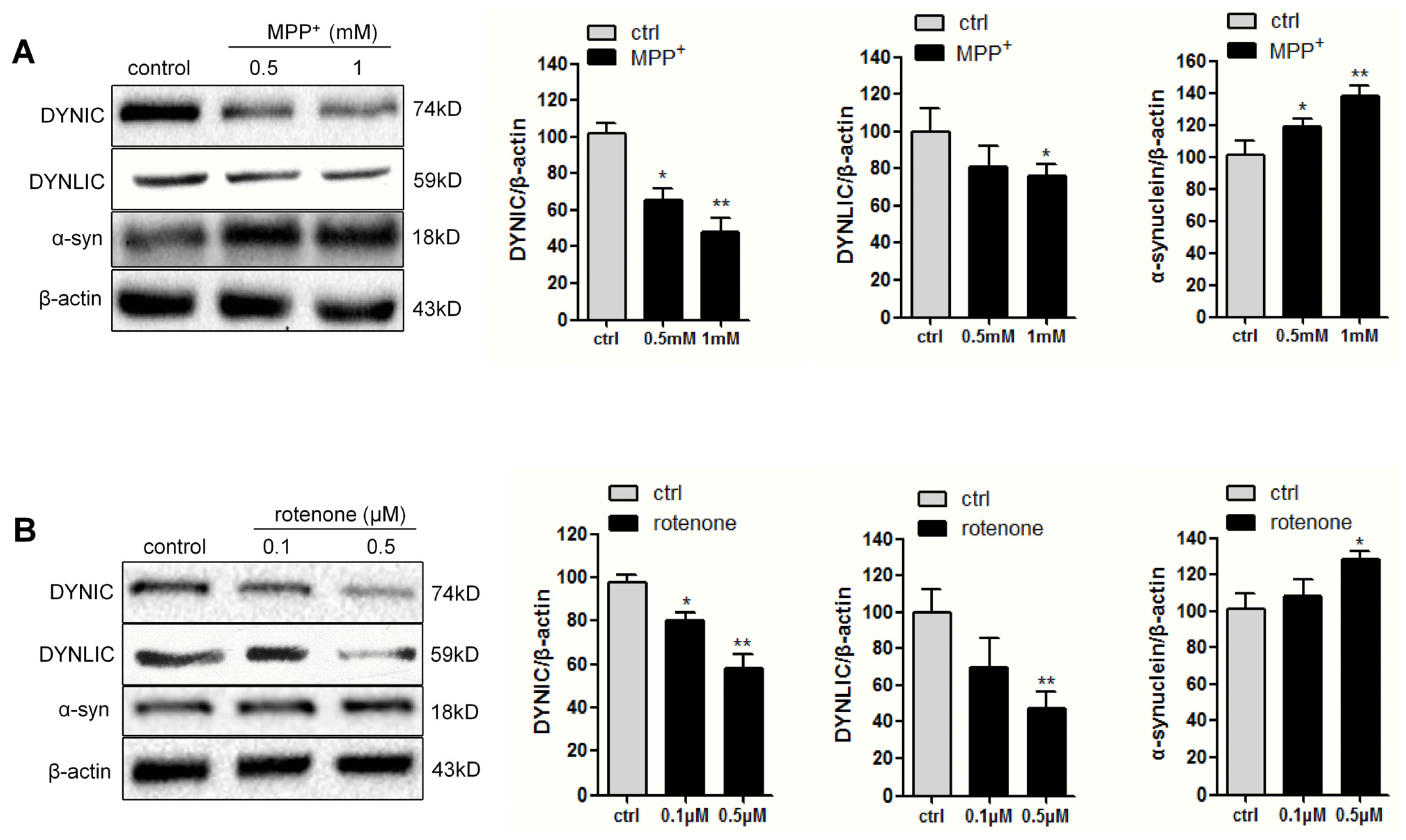
2.1. Dynein Expression Is Decreased in Neurotoxins-Treated PC12 Cells
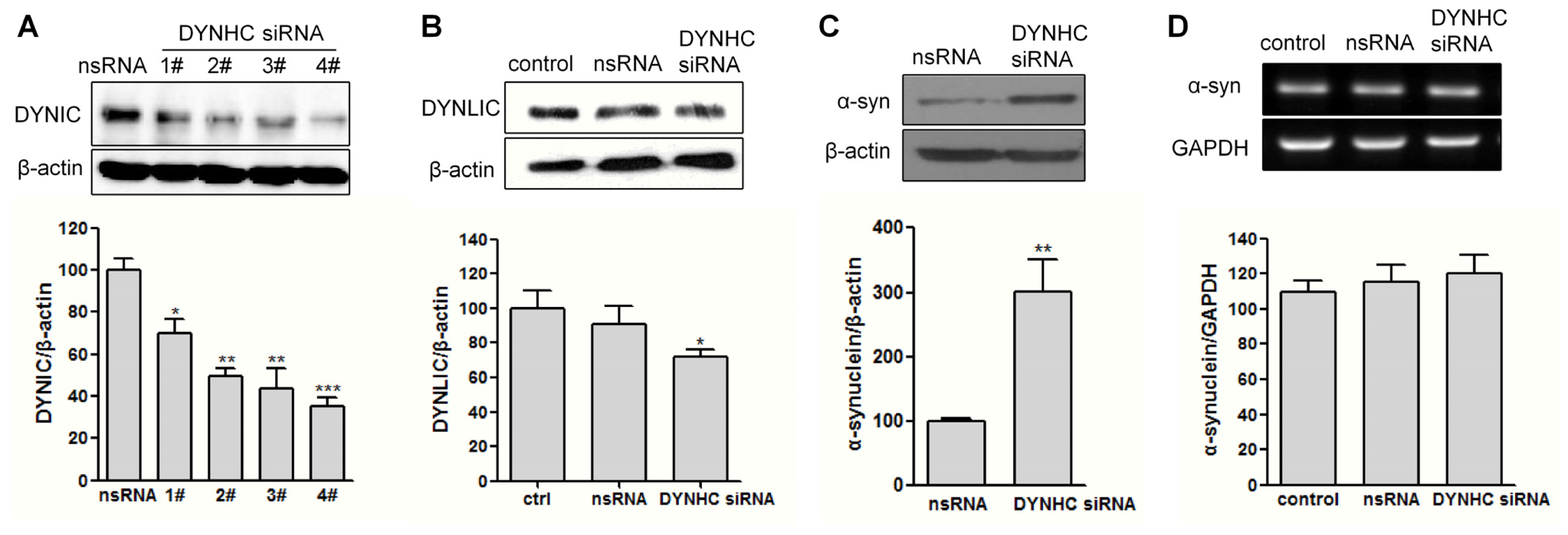
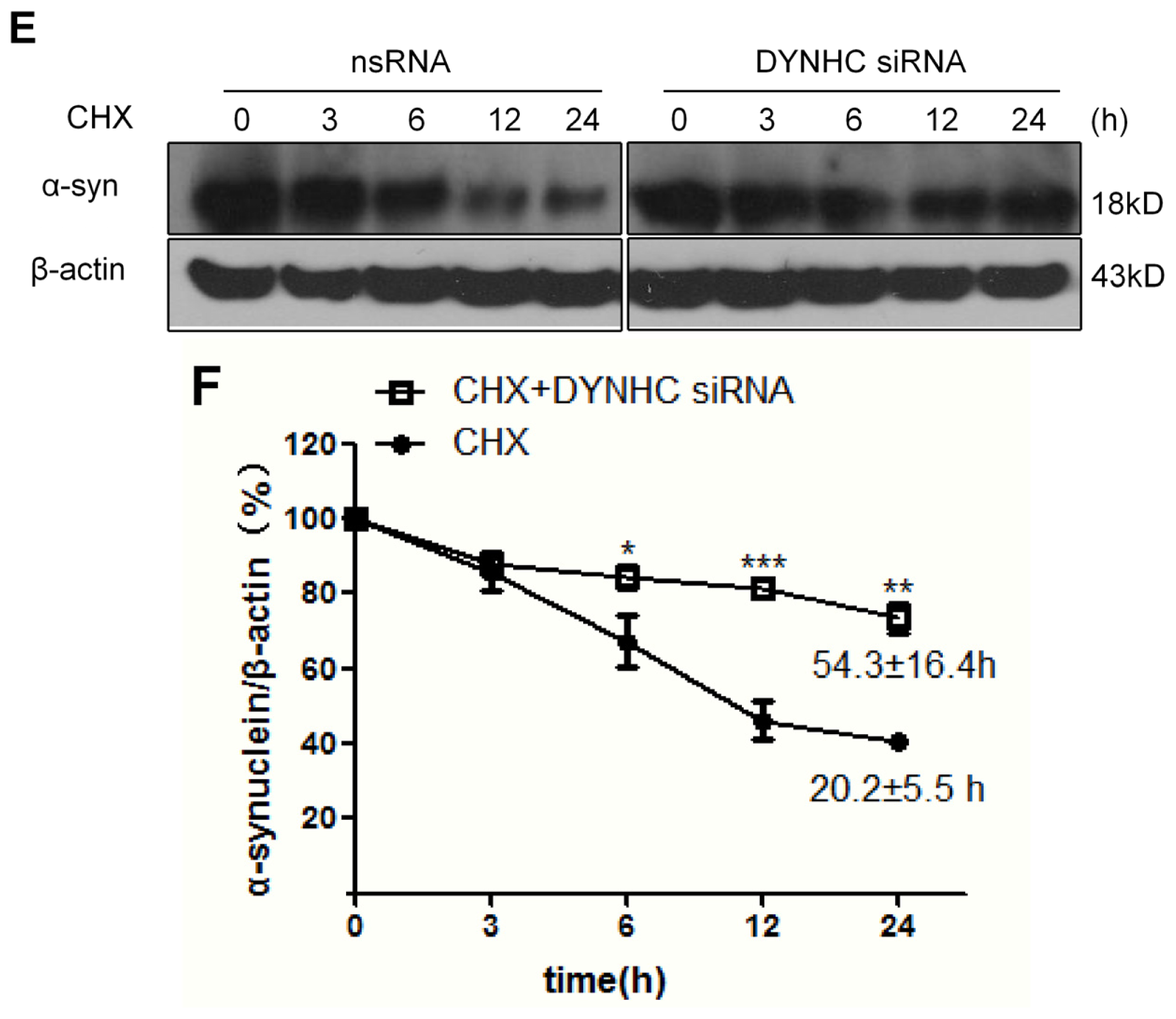
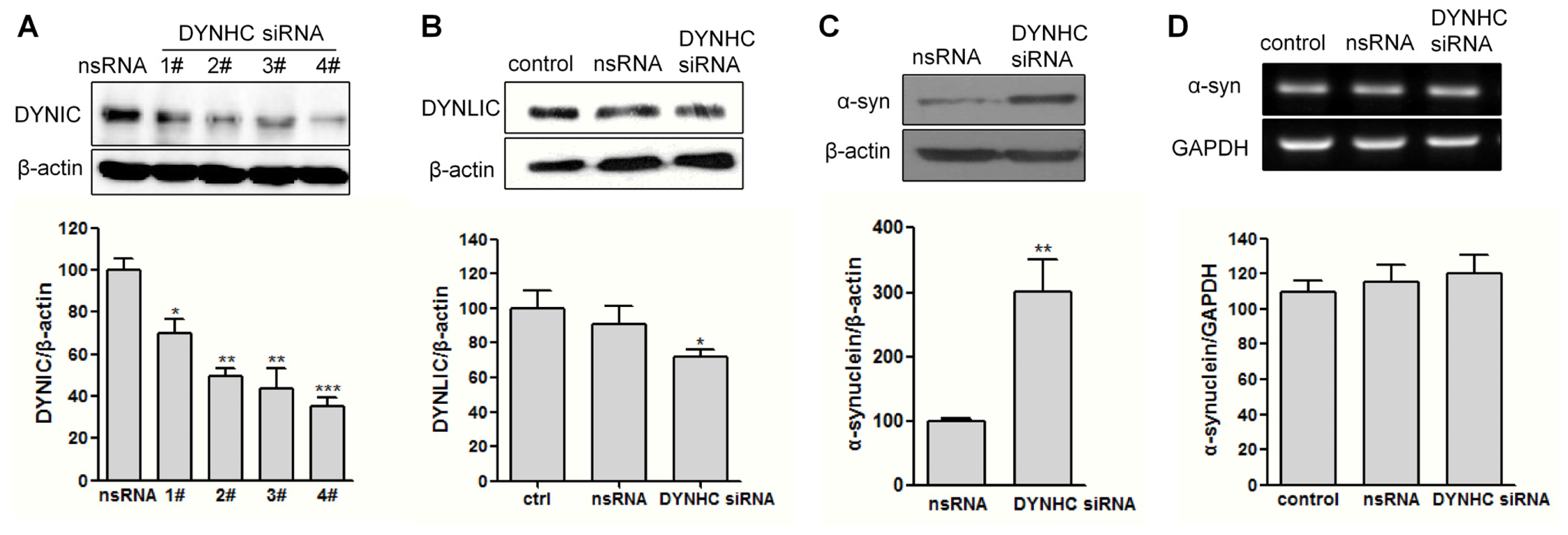
2.2. The α-Synuclein Clearance Is Impaired in Dynein-Silenced PC12 Cells
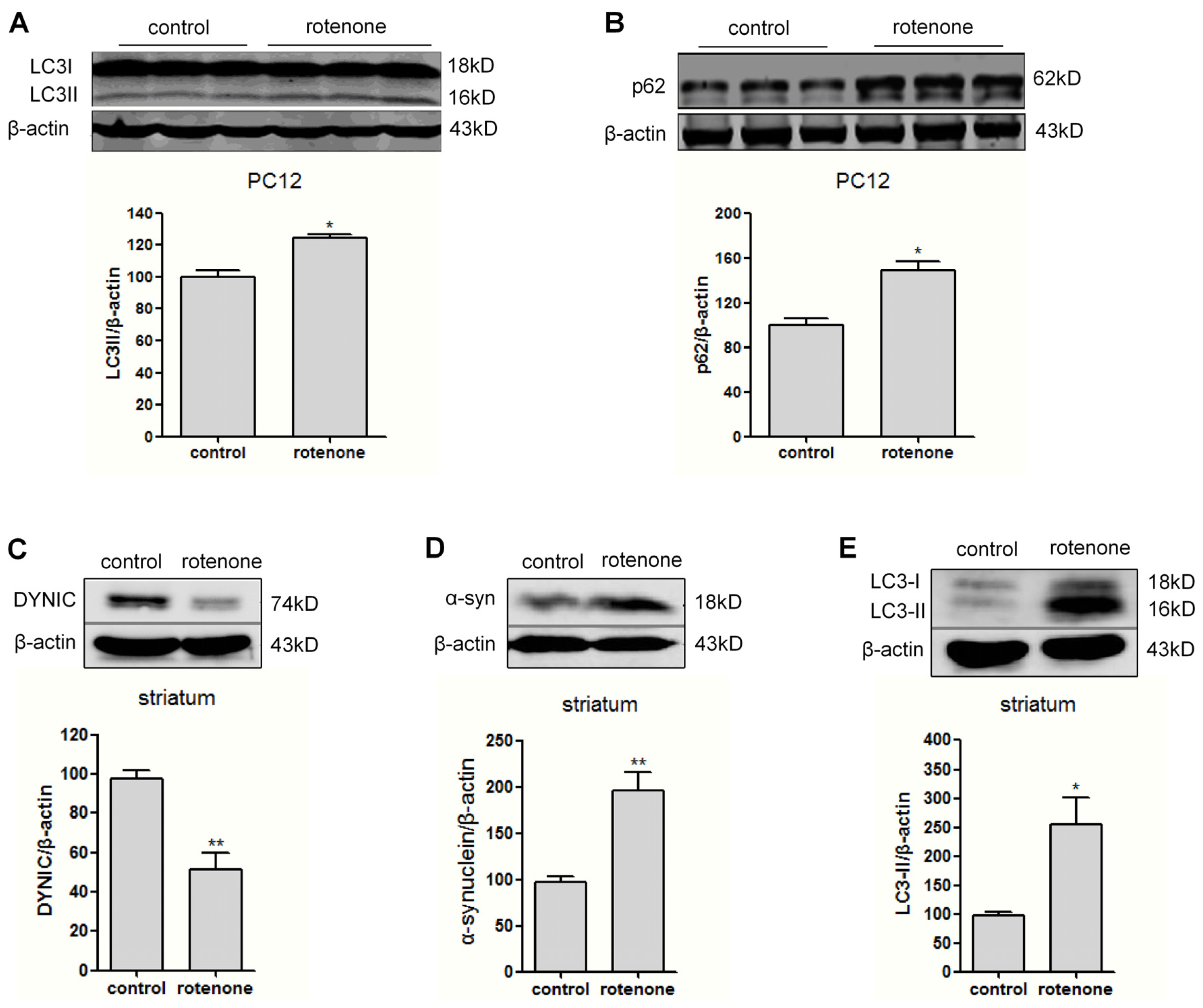
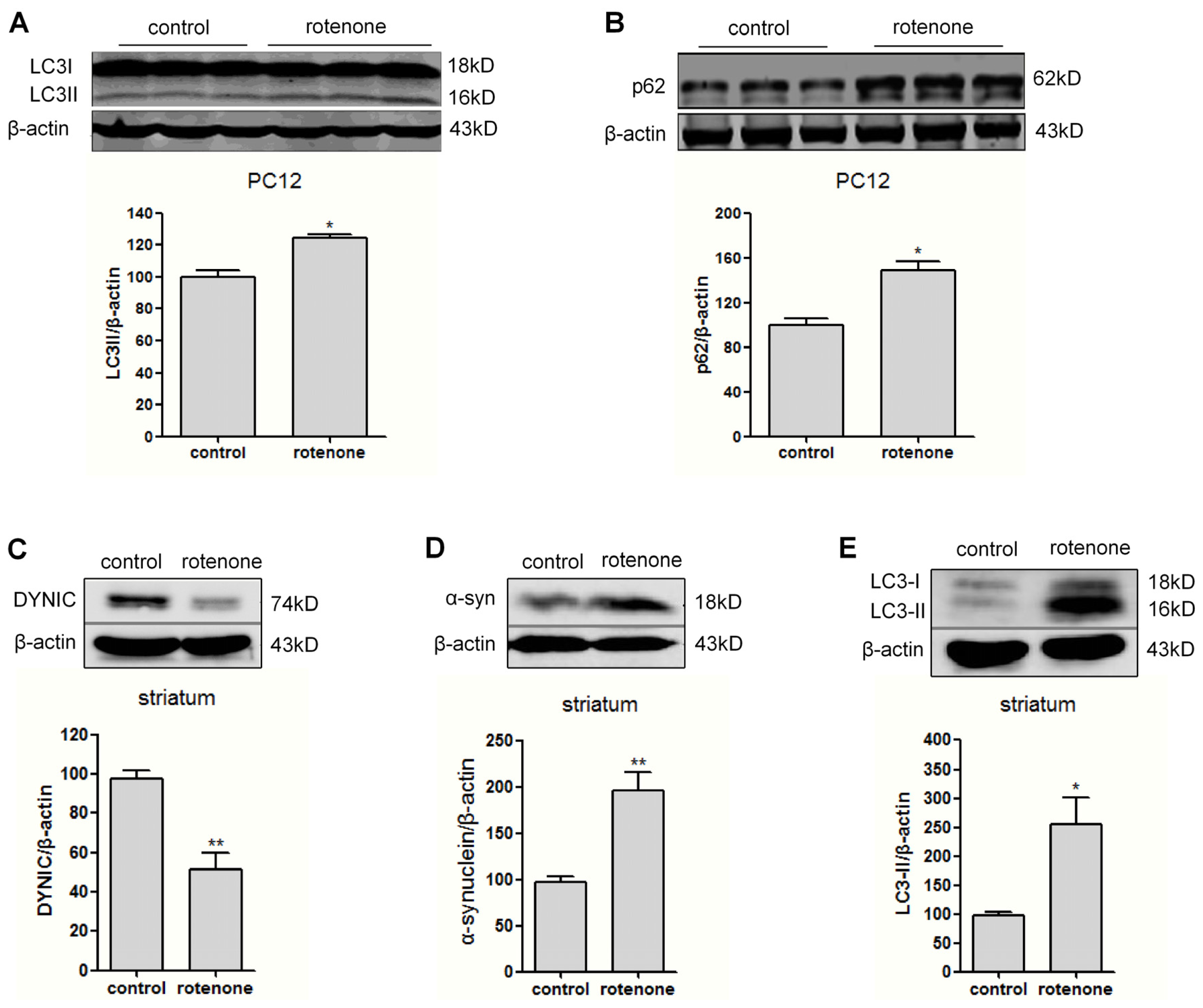
2.3. Autophagy Is Activated in Rotenone-Intoxicated PC12 Cells and Rat Striatum
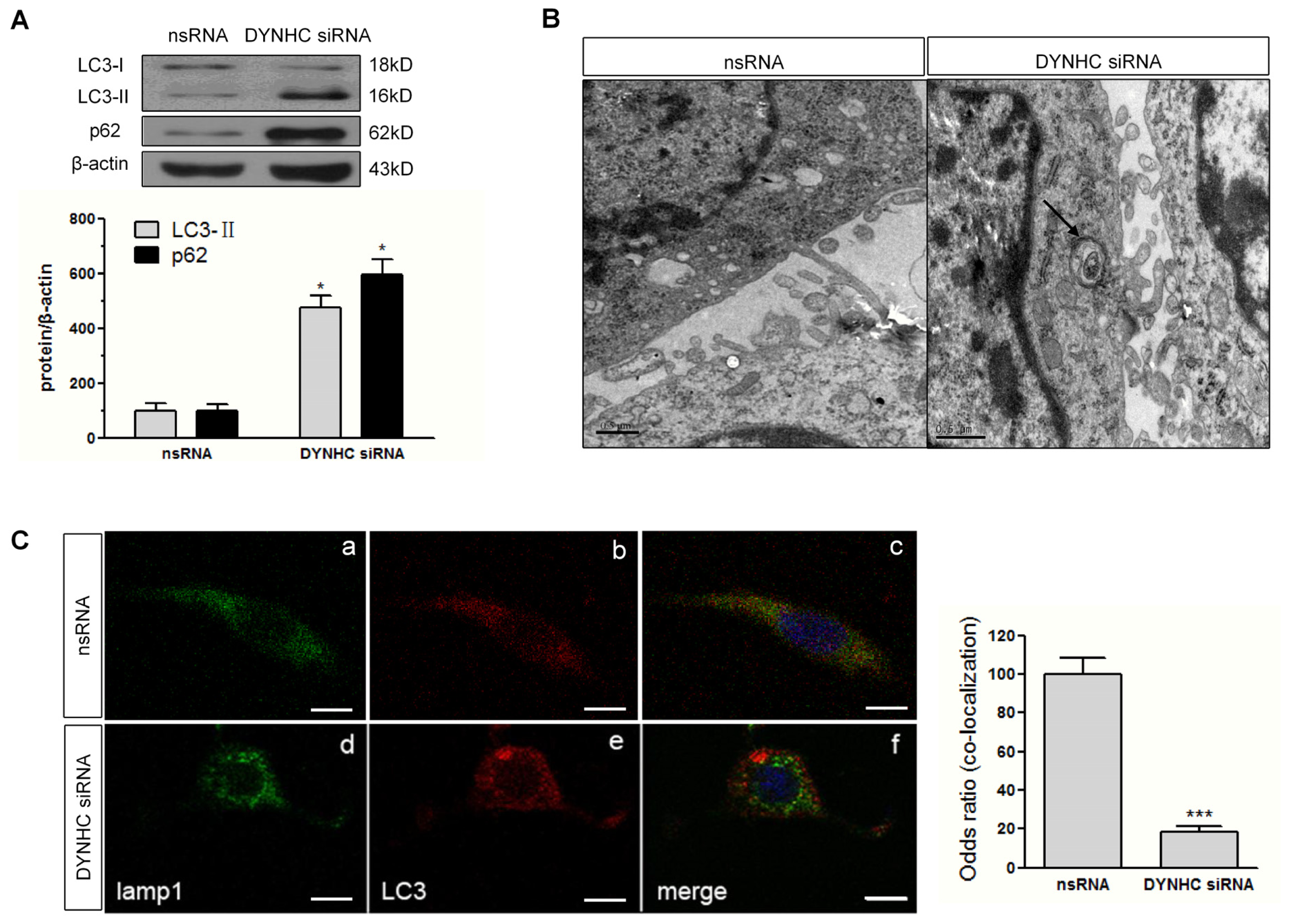
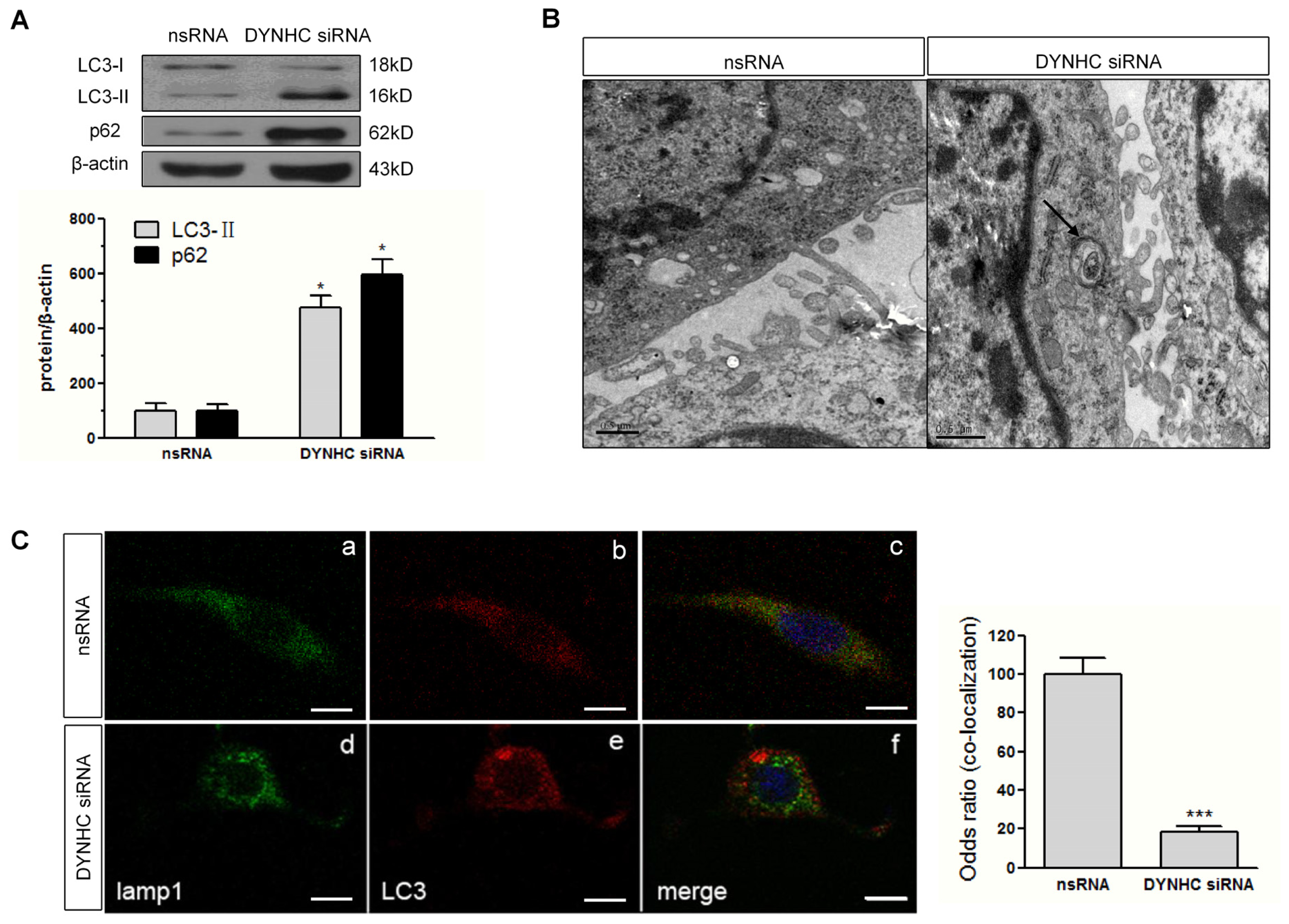
2.4. Autophagic Flux Is Disrupted in Dynein Silencing PC12 Cells
2.5. Discussion
3. Experimental Section
3.1. Reagents and Antibodies
3.2. Cell Culture
3.3. Animals
3.4. Western Blot Analysis
3.5. Cycloheximide(CHX)-Based Protein Chase Experiment
3.6. Immunocytochemistry
3.7. Electron Microscopy Study
3.8. Reverse Transcription PCR (RT-PCR)
3.9. Data Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Eisbach, S.E.; Outeiro, T.F. Alpha-synuclein and intracellular trafficking: Impact on the spreading of Parkinson’s disease pathology. J. Mol. Med 2013, 91, 693–703. [Google Scholar]
- Allan, V.J. Cytoplasmic dynein. Biochem. Soc. Trans 2011, 39, 1169–1178. [Google Scholar]
- Rishal, I.; Fainzilber, M. Retrograde signaling in axonal regeneration. Exp. Neurol 2010, 223, 5–10. [Google Scholar]
- Farrer, M.J.; Hulihan, M.M.; Kachergus, J.M.; Dachsel, J.C.; Stoessl, A.J.; Grantier, L.L.; Calne, S.; Calne, D.B.; Lechevalier, B.; Chapon, F.; et al. DCTN1 mutations in Perry syndrome. Nat. Genet 2009, 41, 163–165. [Google Scholar]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol 2007, 178, 1025–1038. [Google Scholar]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol 2007, 6, 352–361. [Google Scholar]
- Pan, T.; Kondo, S.; Le, W.; Jankovic, J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 2008, 131, 1969–1978. [Google Scholar]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem 2010, 285, 13621–13629. [Google Scholar]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov 2007, 6, 304–312. [Google Scholar]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct 2008, 33, 109–122. [Google Scholar]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol 2012, 196, 407–417. [Google Scholar]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 2012, 135, 2058–2073. [Google Scholar]
- Cai, Z.L.; Shi, J.J.; Yang, Y.P.; Cao, B.Y.; Wang, F.; Huang, J.Z.; Yang, F.; Zhang, P.; Liu, C.F. MPP+ impairs autophagic clearance of alpha-synuclein by impairing the activity of dynein. Neuroreport 2009, 20, 569–573. [Google Scholar]
- Kimura, N.; Inoue, M.; Okabayashi, S.; Ono, F.; Negishi, T. Dynein dysfunction induces endocytic pathology accompanied by an increase in Rab GTPases: A potential mechanism underlying age-dependent endocytic dysfunction. J. Biol. Chem 2009, 284, 31291–31302. [Google Scholar]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010, 5, e9313. [Google Scholar]
- Hu, W.D.; Ke, G.X.; Cheng, Y.B.; Yang, Y.P.; Su, M.; Li, D.H.; Sun, X.; Liu, C.F. Autophagy participates in Rotenone-induced dopaminergic neurotoxicity in old rats. Chin. Pharmacol. Bull 2009, 25. [Google Scholar] [CrossRef]
- Braunstein, K.E.; Eschbach, J.; Rona-Voros, K.; Soylu, R.; Mikrouli, E.; Larmet, Y.; Rene, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P.; Muller, H.P.; et al. A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons. Hum. Mol. Genet 2010, 19, 4385–4398. [Google Scholar]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci 2009, 29, 3365–3373. [Google Scholar]
- Yasuda, T.; Nakata, Y.; Mochizuki, H. Alpha-synuclein and neuronal cell death. Mol. Neurobiol 2013, 47, 466–483. [Google Scholar]
- Morfini, G.; Pigino, G.; Opalach, K.; Serulle, Y.; Moreira, J.E.; Sugimori, M.; Llinas, R.R.; Brady, S.T. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc. Natl. Acad. Sci. USA 2007, 104, 2442–2447. [Google Scholar]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis 2009, 35, 385–398. [Google Scholar]
- Lee, H.J.; Khoshaghideh, F.; Lee, S.; Lee, S.J. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur. J. Neurosci 2006, 24, 3153–3162. [Google Scholar]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar]
- Eschbach, J.; Dupuis, L. Cytoplasmic dynein in neurodegeneration. Pharmacol. Ther 2011, 130, 348–363. [Google Scholar]
- Lo, K.W.; Kogoy, J.M.; Pfister, K.K. The DYNLT3 light chain directly links cytoplasmic dynein to a spindle checkpoint protein, Bub3. J. Biol. Chem 2007, 282, 11205–11212. [Google Scholar]
- Tan, S.C.; Scherer, J.; Vallee, R.B. Recruitment of dynein to late endosomes and lysosomes through light intermediate chains. Mol. Biol. Cell 2011, 22, 467–477. [Google Scholar]
- Ravikumar, B.; Acevedo-Arozena, A.; Imarisio, S.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Brown, S.D.; Rubinsztein, D.C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet 2005, 37, 771–776. [Google Scholar]
- Cai, Q.; Lu, L.; Tian, J.H.; Zhu, Y.B.; Qiao, H.; Sheng, Z.H. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 2010, 68, 73–86. [Google Scholar]
- Xie, R.; Nguyen, S.; McKeehan, W.L.; Liu, L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol 2010, 11, 89. [Google Scholar]
- Su, M.; Shi, J.J.; Yang, Y.P.; Li, J.; Zhang, Y.L.; Chen, J.; Hu, L.F.; Liu, C.F. HDAC6 regulates aggresome-autophagy degradation pathway of alpha-synuclein in response to MPP+-induced stress. J. Neurochem 2011, 117, 112–120. [Google Scholar]
- Clark, J.D.; Gebhart, G.F.; Gonder, J.C.; Keeling, M.E.; Kohn, D.F. Special Report: The 1996 Guide for the Care and Use of Laboratory Animals. ILAR J 1997, 38, 41–48. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, D.; Shi, J.-J.; Mao, C.-J.; Liu, S.; Wang, J.-D.; Chen, J.; Wang, F.; Yang, Y.-P.; Hu, W.-D.; Hu, L.-F.; et al. Alteration of Dynein Function Affects α-Synuclein Degradation via the Autophagosome-Lysosome Pathway. Int. J. Mol. Sci. 2013, 14, 24242-24254. https://doi.org/10.3390/ijms141224242
Li D, Shi J-J, Mao C-J, Liu S, Wang J-D, Chen J, Wang F, Yang Y-P, Hu W-D, Hu L-F, et al. Alteration of Dynein Function Affects α-Synuclein Degradation via the Autophagosome-Lysosome Pathway. International Journal of Molecular Sciences. 2013; 14(12):24242-24254. https://doi.org/10.3390/ijms141224242
Chicago/Turabian StyleLi, Da, Ji-Jun Shi, Cheng-Jie Mao, Sha Liu, Jian-Da Wang, Jing Chen, Fen Wang, Ya-Ping Yang, Wei-Dong Hu, Li-Fang Hu, and et al. 2013. "Alteration of Dynein Function Affects α-Synuclein Degradation via the Autophagosome-Lysosome Pathway" International Journal of Molecular Sciences 14, no. 12: 24242-24254. https://doi.org/10.3390/ijms141224242